
A search for new anticonvulsants led to a series of 2-(4-oxo-2-thioxo-1,4-dihydro-3(2H)-quinazolinyl)- acetamides that were synthesized by reacting (4-oxo-2-thioxo-1,4-dihydro-3(2H)-quinazolinyl)acetic acid with the appropriate amines in the presence of N,N′-carbonyldiimidazole. The tested compounds showed weak and moderate anticonvulsant effects in a pentylenetetrazole-induced seizure model in mice. Several features of the structure—activity relationships are discussed.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
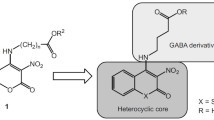
Previously, a series of pyrimidin-4-ones were synthesized by us [1] as potential anticonvulsant analogs of barbiturates and its condensed derivatives [2]. In continuation of experimental modification of already known anticonvulsants and based on modern pharmacophore modelling principles for designing anticonvulsant drugs [3], in particular, considering the presence of a minimum of one hydrophobic ring, the basic structure (scaffold) was a benzannelated pyrimidin- 4(3H)-one, namely, quinazolin-4(3H)-one (Fig. 1). A literature analysis confirmed that this heterocycle had high anticonvulsant potential [4, 5]. Attempts to modify the structure of methaqualone, a soporific and anticonvulsant drug [6], to design a more efficacious and less toxic analog [7, 8], began shortly after its creation.

Modifications and main pharmacophores of target quinazoline derivatives.
The goals of the present investigation were to synthesize novel N-substituted 2-(4-oxo-2-thioxo-1,4-dihydro-3(2H)- quinazolinyl)acetamides and to study their anticonvulsant activity.
Experimental Chemical Part
Commercial reagents (Sigma-Aldrich, USA) were used in the work. Reactions were monitored using TLC (EtOAc– hexane eluent, 1:2) on Sorbfil UV-254 plates. Chromatograms were detected in UV light on a UFS 254/365 chromatographic irradiator (254-nm mode). Melting points (°C) were determined in capillaries on an IA9100X1 digital melting point apparatus (Bibby Scientific Ltd., Staffordshire, Great Britain). Elemental microanalyses used an EA3000 analyzer (EuroVector S.p.A., Redavalle, Italy). Elemental analyses were within ±0.4% of the theoretical values. PMR and 13C NMR spectra were recorded in DMSO-d6 with TMS internal standard on a Mercury-400 spectrometer (400 and 100 MHz, respectively; Varian Inc., Palo Alto, CA, USA). LCMS spectra were recorded using a PE SCIEX API 150EX liquid chromatograph-mass spectrometer.
(4-Oxo-2-thioxo-1,4-dihydro-3(2H)-quinazolinyl)acetic acid (III). Glycine (II, 7.5 g, 0.1 mol) was dissolved in a mixture of H2O (150 mL) and Et3N (14 mL, 0.1 mol), stirred, added to a warm solution of methyl 2-isothiocyanatobenzoate (I, 19.2 g, 0.1 mol) in i-PrOH (200 mL), refluxed for 30 min, acidified with HCl (10 – 12.5 mL) to pH 2, and left for 12 h. The resulting precipitate was filtered off, rinsed with H2O, refluxed with stirring in Me2CO (200 mL), and cooled. The precipitate was filtered off and rinsed twice with Me2CO (20 mL).
N-R-2-(4-Oxo-2-thioxo-1,4-dihydro-3(2H)-quinazolinyl) acetamides (VII) (general method). A suspension of III (0.1 mol) in anhydrous dioxane (200 mL) was treated with carbonyldiimidazole (IV, 0.12 mol) and refluxed for 2-4 h. The course of the reaction was monitored by TLC. When the reaction was finished, the mixture was treated with the appropriate amine (0.12 mol), refluxed for another 2 h, cooled, treated with H2O (500 mL), and left for 12 h. The resulting precipitate was filtered, rinsed with i-PrOH (50 mL), and recrystallized (if necessary) from a mixture of DMF (50 mL) and i-PrOH (200 mL).
Experimental Biological Part
Screening of 10 synthesized compounds for anticonvulsant activity employed a basic pentylenetetrazole convulsion model in 73 laboratory white mice of both sexes (19 – 24 g) [9, 10]. Experiments with animals were conducted in compliance with international rules (Directive 2010/63/EU of the European Parliament and Committee of the European Union dated Sept. 22, 2010, on protection of animals used for scientific purposes).
Mice were divided randomly into 12 groups, each of which consisted of animals of both sexes: control group (n = 8), model seizure induced by pentylenetetrazole (Sigma, USA) at a dose of 90 mg/kg s.c. as an aqueous solution (1); reference drug group (n = 5), sodium valproate (Depakine, Sanofi-Aventis, France) at a dose of 300 mg/kg through a stomach catheter 30 min before pentylenetetrazole injection (2); test animal groups (n = 6) that received the tested compounds at a dose of 100 mg/kg through a stomach catheter as a suspension in Tween-80 30 min before pentylenetetrazole injection.
Animals were observed for 60 min. The duration of the seizure latent period and number of clonic and tonic convulsions were recorded. The percent of animals with clonic and tonic convulsions was calculated. The duration of seizures (from the first to last episode) and lifetimes until death (for mice with lethal outcomes) were determined. The severity of seizures was assessed in balls on the following scale: tremor (1), circus movement (2), clonic seizures (3), clonic-tonic convulsions from the side (4), tonic extension (5), tonic extension with death (6). Results were statistically processed using Statistica 8.0 for Windows. The statistical significance of intergroup differences was evaluated using the parametric Student t-criterion with a normal distribution and the Mann—Whitney U-criterion without one. Quantitative data are given as means and its standard error. Results were calculated in an alternative form (lethality) as percent of the observed effect in a group. The statistical significance in the last instance was determined using the Fisher angular transformation. Differences were considered statistically significant for p < 0.05.
Results and Discussion
The method for synthesizing 4-oxo-2-thioxoquinazolines that was proposed by us [11] was used to afford the key building block (4-oxo-2-thioxo-1,4-dihydro-3(2H)-quinazolinyl) acetic acid (III) and was based on reactions of 2-isothiocyanobenzoic acid esters (I) with primary amines such as amino acids, in particular, glycine (II). The reactions were performed in refluxing i-PrOH for 30 min in the presence of Et3N (Scheme 1).

Synthesis of (4-oxo-2-thioxo-1,4-dihydro-3(2H)-quinazolinyl)acetic acid (III).
N,N′-Carbonyldiimidazole (IV) was selected for the subsequent amidation not only for its relative availability, lack of chemical side reactions, and satisfactory yields of final products (Table 1), but also for the formation of the innocuous side products CO2 and imidazole. This made this method for preparing the amides most acceptable and fruitful for synthesizing the active pharmaceutical ingredients [12] and was consistent with green-chemistry principles.
Target carboxamides VII were synthesized in one pot without isolating intermediate imidazolides V by reacting III with IV followed by addition of appropriate amines VI and refluxing in dioxane (Scheme 2).

Synthesis of N-substituted 2-(4-oxo-2-thioxo-1,4-dihydro-3(2H)-quinazolinyl)acetamides (VIIa-r).
Synthesized VIIa-r were white or slightly yellowish crystalline compounds with sharp melting points that were readily soluble in i-PrOH, dioxane, and DMF and insoluble in H2O (Table 1).
The structures and molecular masses of the synthesized compounds were confirmed by PMR and 13C NMR spectroscopy and chromatography—mass-spectrometry, respectively. Compositions were determined by elemental analyses; purities, by TLC and chromatography—mass-spectrometry (Tables 1 and 2).
PMR spectra contained all required proton resonances and corresponded to the compound structures with respect to integration and multiplicity. Heterocyclic NH protons appeared at weak field (13.05 – 12.80 ppm), as a rule, as broad singlets. Quinazoline aromatic protons appeared as two doublets (H-5, H-8) and two triplets (H-6, H-7) characteristic of ABCD systems. It is noteworthy that the amide NH resonances for VIId and VIIg overlapped the aromatic proton multiplet at 8.02 – 7.9 ppm. 13C NMR spectra were characterized by a C=S resonance at weak field (175.5 – 176.8 ppm). Furthermore, two resonances for heterocyclic and amide C=O groups (159.3 – 170.0 ppm), aromatic fragments (115.3 – 149.3), and aliphatic groups (11.2 – 55.8) were observed. The positions and numbers of them agreed fully with the compound structures.
Primary screening used a pentylenetetrazole-induced convulsion model in mice (Table 3) because it was the gold standard in screening tests for potential new antiepileptic drugs [13]. None of the tested compounds had statistically significant anticonvulsant activity according to the integral protective criterion, i.e., decreased lethality as compared to the control. Two compounds (VIIg and -i) exhibited weak anticonvulsant activity, decreasing the lethality to 66.7 – 83.3% vs. 87.5% in the control, and improved statistically significantly by one indicator the severity. In particular, VIIg with a cyclohexane substituent increased statistically significant (p < 0.05) the lifetime of animals before death whereas VIIi with an ethylphenyl radical decreased the number of clonic-tonic convulsions per animal (p < 0.05).
Compounds VIIa, h, l, m, and p turned out to be inactive for pentylenetetrazole-induced convulsions, did not have statistically significant effects on any their indicators with 50 – 100% deaths, and were statistically significantly inferior to the reference drug sodium valproate for this indicator. Compounds VIIj, q, and r showed moderate anticonvulsant properties: deaths were 100%, all animals had tonic extension that halted breathing. The convulsion severity was 6.00 ±0.00 balls.
Compound VIIj with a 2,4-dichlorobenzyl substituent halved the lifetime of animals before death as compared to the control (p < 0.05). These results were unfavorable despite the fact that VIIj decreased statistically significantly the latent period of seizures by 1.5 times and reduced the number of clonic-tonic convulsions by 1.7 times because the lifetime before death shortened, indicating that the first seizures were lethal. This was also consistent with the duration of the convulsions that was 2.5 times shorter. Many reports claim that a Cl atom in an aryl radical increases the anticonvulsant effect of compounds [14, 15]. Furthermore, the structure of the anti-epileptic drug lamotrigine includes a 2,3-dichloroaryl substituent. However, the results of the present work for the effects of the compounds on increased animal deaths correlate very well with our previous results using pyrimidin-4-one acetamides as examples [16]. Deaths increased and the anticonvulsant effects of the compounds decreased if the number of Cl atoms in the aryl fragment was increased.
Thus, synthesized VIIa-r had weak anticonvulsant effects, indicating that further structural modification is needed to reach the required pharmacological profile. In particular, addition of more aryls to the quinazoline ring and alkylation of the NH would increase the lipophilicity and possibly increase the anticonvulsant effect. Moreover, further testing of this group of compounds in the maximum electroshock model in rodents and in the psychomotor (6 Hz) convulsion model in mice is definitely promising because these methods characterize different mechanisms of anticonvulsant activity, in accordance with contemporary approaches to the discovery of new antiepileptic drugs [17].
References
A. I. Severina, O. O. Skupaya, V. A. Georgiyants, et al., Med. Obraz. Sib., No. 3, (2013); http://ngmu.ru/cozo/mos/article/annotacyfull.php?id=1034.
A. I. Severina, V. A. Georgiyants, S. Yu. Shtrygol, et al., Pharma Chem., 7(11), 43 – 48 (2015).
H. N. Khana, S. Kulsoomb, and H. Rashid, Epilepsy Res., 98(1), 62 – 71 (2012).
H. Georgey, N. Abdel-Gawad, and S. Abbas, Molecules, 13, 2557 – 2569 (2008).
H. A. Abuelizz, R. El Dib, M. Marzouk, et al., Molecules, 22, 1094 – 1107 (2017).
G. McCarthy, B. Myers, and N. Siegfried, Cochrane Database Syst. Rev., 2, No.: CD004146 (2005).
M. F. Zayed, S. K. Ihmaid, H. E. A. Ahmed, et al., Molecules, 22, 188 – 198 (2017).
V. Jatav, P. Mishra, S. Kashaw, et al., Eur. J. Med. Chem., 43, 1945 – 1954 (2008).
H. G. Vogel, Drug Discovery and Evaluation: Pharmacological Assays. Chapter E: Psychotropic and Neurotropic Activity, Berlin (2008), pp. 459 – 493.
A. N. Mironova, N. D. Buntaytan, A. N. Vasil’eva, et al. (eds.), Handbook for Preclinical Drug Trials [in Russian], Part 1, Grif i K, Moscow (2012), p. 944.
A. V. Ivachtchenko, S. M. Kovalenko, and O. G. Drushlyak, J. Comb. Chem., 5(6), 775 – 788 (2003).
J. S. Carey, D. Laffan, C. Thomson, et al., Org. Biomol. Chem., 4, 2337 – 2347 (2006).
R. L Krall, J. K. Penry, B. G. White, et al., Epilepsia, 19(4), 409 – 428 (1978).
M. M. W. Habib, M. A. O. Abdelfattah, and A. H. Abadi, Arch. Pharm., 348, 868 – 874 (2015).
M. Z. Hassan, S. A. Khan, and M. Amir, Eur. J. Med. Chem., 58, 206 – 213 (2012).
H. Severina, O. Skupa, A. Khairulin, et al., J. Appl. Pharm. Sci., 9(2), 012 – 019 (2019).
W. Loscher, Seizure, 20, 359 – 368 (2011).
Author information
Authors and Affiliations
Corresponding author
Additional information
Translated from Khimiko-Farmatsevticheskii Zhurnal, Vol. 54, No. 1, pp. 3 – 8, January, 2020.
Rights and permissions
About this article

Cite this article
Bunyatyan, N.D., Severina, H.I., Mokhamad, E.K.W. et al. Synthesis and Anticonvulsant Activity of New 2-(4-Oxo-2-Thioxo-1,4-Dihydro-3(2h)-Quinazolinyl)Acetamides. Pharm Chem J 54, 1–6 (2020). https://doi.org/10.1007/s11094-020-02147-5
Received:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s11094-020-02147-5