
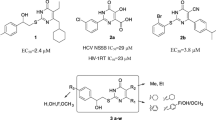
A focused library of heterocyclic compounds including a 2-aminomethyl-1H-benzimidazole (1 – 19), 2-aminomethylindole (20 – 83), benzofuran-2-ylmethylamine (84 – 92), or 2-piperazin-1-ylmethylbenzoxazole (93) fragment was screened for the ability to inhibit in vitro hepatitis C virus (HCV). The synthetic methods were described. The antiviral activity and cytotoxicity data were presented. Most of the compounds carrying a benzoxazol-2-ylmethylamine fragment inhibited Huh7.3 human hepatoma cells infected in vitro with HCV with nanomolar potency but were inactive against the HCV RNA-replicon. The only exception was 9-methyl-N(6)-(3-nitrophenyl)-2,3,4,9-tetrahydro-1H-carbazole-1,6-diamine (67), which demonstrated nanomolar potency against HCV in both models. The most active and selective compounds were (piperazin-1-yl)-[(1Hindol-2-ylmethyl)piperidin-4-yl]-ketones (EC50 0.31 – 2.2 μM, CC50 10.2-110 μM) and 2-(1,2,3a,4,5,6-hexahydropyrazino[3,2,1-jk]carbazol-3-yl)acetamide (EC50 1.69 ± 0.5 μM, CC50 114 ± 42 μM). The two most selective inhibitors (28, TI50 = 52 and 77, TI50 = 68) were selected for further preclinical trials.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Hepatitis C virus (HCV) presents a serious threat to the health and life of mankind because the number of HCV-infected people increases with each year and has reached 500 million according to WHO data. Of these, about 150 million suffer from chronic hepatitis C and 350,000 die yearly from HCV-related liver diseases [http://www.who.int/ mediacentre/factsheets/fs164/en/index.html]. Most modern drugs used for therapy of HCV infections produce side effects and resistant viral strains. Therefore, combination therapy using several drugs with different mechanisms of action is most effective. Hence, the search for HCV inhibitors with novel mechanisms of action remains crucial.
Recently, high-throughput screening of 1200 compounds against HCV NS4B protein by researchers at Stanford University School of Medicine found that the well-known multimodal drug clemizole is a rather active HCV NS4B inhibitor, i.e., it can block this protein and; therefore, eliminate conditions favorable for HCV multiplication without any damage to the infected liver cells. Clemizole is a moderate inhibitor of NS4B with EC50 = 8 μM for the JFH-1 clone of HCV genotype 2a [1].


It is noteworthy that clemizole together with protease inhibitors SCH503034 and VX950 demonstrated high synergism for HCV inhibition. The occurrence of drug-resistant mutants was reduced compared to use of the separate inhibitors. Cross resistance was not observed [2].
The medical community is waiting impatiently for results from clinical trials [NCT00945880] of the safety and tolerability of clemizole to treat hepatitis C in subjects who are treatment-naive.
The present work reports results from screening the activity against HCV of a focused library of heterocyclic compounds containing 2-aminomethyl-1H-benzimidazole (1 – 19), 2-aminomethylindole (20 – 83), benzofuran-2-ylmethylamine (84 – 92), and 2-piperazin-1-ylmethylbenzoxazole (93) fragments.
Tables 1–4 list the substituents in 1 – 94. Most of the tested compounds were commercially available products and were obtained from CDRI (Khimki, Moscow Oblast). The exceptions were 13 – 15, 25 – 28, 58 – 62, and 77, the syntheses of which are described below.
Experimental Chemical Part
PMR spectra of synthesized compounds were recorded from DMSO-d6 solutions on a Bruker DPX-400 spectrometer (400 MHz, 27°C). GC-MS spectra were obtained using a Shimadzu 10Avp HPLC with a Waters XBridge C18 column (3.5 μm, 4.6 × 150 mm) and an API 150 EX mass spectrometer (λ220 and 254 nm). The purity of all synthesized compounds was at least 98.0% according to GC-MS/UV254 GC-MS data and PMR and 13C NMR spectra of the synthesized compounds agreed with their structures.
All reactions were performed in solvents that were purified according to standard procedures. Starting materials were commercially available products and were obtained from Aldrich and CDRI.
2-{3,4-Dihydropyrazino[1,2-a]benzimidazol-2-(1H)-yl}acylamides (13 – 15) were prepared (Scheme 1) via alkylation of 3,4-dihydro-1H-benzo[4, 5]imidazo[1,2-a]pyrazine (1.1) by 2-chloroacetic (1.1.1) or 3-chloropropionic (1.1.2) acid amides.

Scheme 1
2-{3,4-Dihydropyrazino[1,2-a]benzimidazol-2-(1 H)-yl}acylamides (13 – 15), general method. A mixture of 1.1 (173 mg, 1 mmol) that was prepared by the literature method [3], the appropriate ω-chloroacylamide [1.1.1 or 1.1.2 (or its hydrochloride), 1.5 mmol], and K2CO3(414 mg, 3 mmol) in dioxane (10 mL) was stirred at 65°C for 12 h. The reaction was carried out with Cs2CO3 in DMF at 100°C for 72 h for 1.1.2. The solution was cooled, filtered, and evaporated in vacuo. Products 13 – 15 were isolated by column chromatography over silica gel that was treated with Et3N (THF:MeOH eluent, 4:1). Hydrochlorides were prepared by adding a 5% excess of HCl solution (3 M) in dioxane to solutions of 13 – 15 in Me2CO. The precipitates were filtered off, rinsed with i-PrOH and Et2O, and dried in vacuo.
2-{3,4-Dihydropyrazino[1,2-a]benzimidazol-2-(1 H)-yl}acetamide hydrochloride (13•HCl). Yield 22%. GC-MS (ESI) [M + H]+ 231. PMR (DMSO-d6), δ, ppm: 7.87 (m, 1H), 7.82 (m, 1H), 7.70 (br.s, 1H), 7.56 (m, 2H), 7.36 (br.s, 1H), 4.50 (s, 2H), 4.44 (t, J 4.8 Hz, 2H), 3.58 (s, 2H), 3.45 (t, J 4.8 Hz, 2H).
2-{3,4-Dihydropyrazino[1,2-a]benzimidazol-2-(1 H)-yl}-N-[2-(dimethylamino)ethyl]acetamide dihydrochloride (14•HCl). Yield 5%. GC-MS (ESI) [M + H]+ 302. PMR (DMSO-d6), δ, ppm: 10.74 (br.s, 1H), 8.57 (t, J 5.0 Hz, 1H), 7.90 (m, 1H), 7.83 (m, 1H), 7.58 (m, 2H), 4.44 (m, 4H), 3.55 (s, 2H), 3.49 (m, 2H), 3.38 (t, J 4.8 Hz, 2H), 3.16 (m, 2H), 2.75 (d, J 4.4 Hz, 6H).
3-{3,4-Dihydropyrazino[1,2-a]benzimidazol-2-(1 H)-yl}propionamide hydrochloride (15•HCl). Yield 25%. GC-MS (ESI) [M + H]+ 245. PMR (DMSO-d6), δ, ppm: 7.80 (m, 2H), 7.63 (br.s, 1H), 7.49 (m, 2H), 7.06 (br.s, 1H), 4.77 (s, 2H), 4.59 (t, J 5.2 Hz, 2H), 3.79 (t, J 5.2 Hz, 2H), 3.47 (t, J 7.2 Hz, 2H), 2.73 (t, J 7.2 Hz, 2H).
Amides of 1-(1H-indol-2-ylmethyl)piperidinecarboxylic acids of general formulas 25 – 28 and 58 – 62 were prepared

according to Scheme 2 starting from corresponding aldehydes 1.2.1 and 1.2.2, which were prepared by the literature methods [4, 5; respectively]. Aldehydes 1.2.1 and 1.2.2 were transformed via reductive amination into esters 1.2.3, 1.2.4, 1.2.5, and 1.2.6 and then via hydrolysis of the last into acids 1.2.7, 1.2.8, 1.2.9, and 1.2.10, which were converted via amidation by the corresponding amines in the presence of 1,1-carbodiimidazole into products 25 – 28 and 58 – 62.

Scheme 2
1-(4-Chlorobenzyl)-1 H-indole-2-carbaldehyde (1.2.1). NaH (60% in oil, 0.42 g, 10.5 mmol) was rinsed with hexane, stirred and cooled in ice, treated with a solution of methyl 1H-indole-2-carboxylate (1.75 g, 10 mmol) in DMF (20 mL), stirred for 0.5 h, treated with 4-chlorobenzylchloride (1.626 g, 10.1 mmol), stirred for 1 h at 0°C, diluted with benzene (100 mL), and washed with H2O (2×, 100 mL each). The organic layer was dried over Na2 SO4, evaporated in vacuo, and chromatographed over silica gel (hexane:Et2O eluent, 40:1) to afford methyl 1-(4-chlorobenzyl)-1Hindole-2-carboxylate (2.156 g, 72%). PMR (DMSO-d6), δ, ppm: 7.73 (d, J 7.6 Hz, 1H), 7.57 (d, J 8.4 Hz, 1H), 7.39 (s, 1H), 7.33 (m, 3H), 7.16 (t, J 7.4 Hz, 1H), 7.04 (d, J 8.4 Hz, 2H), 5.84 (s, 2H). A solution of the resulting ester (2.15 g, 7.2 mmol) in THF (20 mL) was cooled in ice, treated with LiAlH4 (819 mg, 21.5 mmol) in THF (10 mL), and stirred for 0.5 h at room temperature. The mixture was decomposed by NaOH solution (10%). The precipitate was filtered off. The solution was evaporated in vacuo to afford in quantitative yield 1-(4-chlorobenzyl)-1H-indol-2-yl)methanol. PMR (DMSO-d6), δ, ppm: 7.52 (d, J 7.6 Hz, 1H), 7.33 (d, J 8.4 Hz, 2H), 7.29 (d, J 8.4 Hz, 1H), 7.05 (m, 3H), 6.99 (t, J 7.2 Hz, 1H), 6.45 (s, 1H), 5.47 (s, 2H), 5.31 (br.s, 1H), 4.59 (s, 2H). A mixture of the resulting alcohol (1.162 g, 4.3 mmol) and MnO2 (5.03 g, 58 mmol) in CH2Cl2 (30 mL) was stirred at room temperature until the reaction was finished (24 h, TLC monitoring, hexane:EtOAc eluent, 6:1). The mixture was filtered through Celite and evaporated in vacuo to afford 1.2.1 (1.125 g, 97%). PMR (DMSO-d6), δ, ppm: 9.92 (s, 1H), 7.81 (d, J 8.9 Hz, 1H), 7.61 (d, J 8.4 Hz, 1H), 7.58 (s, 1H), 7.40 (m, 1H), 7.33 (d, J 8.4 Hz, 2H), 7.18 (t, J 7.4 Hz, 1H), 7.07 (d, J 8.4 Hz, 2H), 5.82 (s, 2H).
Ethyl 1-(hetarylmethyl)piperidinecarboxylates (1.2.3 – 1.2.6), general method. A mixture of the appropriate aldehyde (5 mmol, 1.2.1 – 1.2.3), ethyl piperidinecarboxylate (943 mg, 6 mmol), and sodium triacetoxyborohydride (1.6 g, 7.5 mmol) in dichloroethane (20 mL) was stirred at room temperature for 3 d, washed with H2O (2×), dried over Na2SO4, evaporated in vacuo, and chromatographed over silica gel treated with Et3N (hexane:EtOAc eluent, 10:1) to afford the esters (1.2.3 – 1.2.6).
Ethyl 1-[(1-methyl-1 H-indol-2-yl)methyl]piperidine-4-carboxylate (1.2.3). Yield 77%. GC-MS (ESI) [M + H]+ 301. PMR (CDCl3), δ, ppm: 7.57 (d, J 7.6 Hz, 1H), 7.32 (d, J 8.0 Hz, 1H), 7.21 (m, 1H), 7.09 (m, 1H), 6.36 (s, 1H), 4.14 (q, J 7.2 Hz, 2H), 3.81 (s, 3H), 3.62 (s, 2H), 2.89 (m, 2H), 2.30 (m, 1H), 2.05 (t. d, J1 11.4 Hz, J2 2.0 Hz, 2H), 1.87 (m, 2H), 1.72 (m, 2H), 1.26 (t, J 7.2 Hz, 3H).
Ethyl 1-[(1-methyl-1 H-indol-2-yl)methyl]piperidine-3-carboxylate (1.2.4). Yield 82%. GC-MS (ESI) [M + H]+ 301. PMR (CDCl3), δ, ppm: 7.57 (d, J 8.0 Hz, 1H), 7.32 (d, J 8.4 Hz, 1H), 7.20 (m, 1H), 7.09 (m, 1H), 6.37 (s, 1H), 4.11 (m, 2H), 3.79 (s, 3H), 3.64 (s, 2H), 2.95 (m, 1H), 2.70 (m, 1H), 2.54 (m, 1H), 2.35 (m, 1H), 2.13 (m, 1H), 1.89 (m, 1H), 1.71 (m, 1H), 1.55 (m, 2H), 1.22 (t, J 7.2 Hz, 3H).
Ethyl 1-{[1-(4-chlorobenzyl)-1 H-indol-2-yl]methyl}-piperidine-3-carboxylate (1.2.5). Yield 80%. GC-MS (ESI) [M + H]+ 411, 413. PMR (CDCl3), δ, ppm: 7.60 (m, 1H), 7.23 (m, 3H), 7.13 (m, 2H), 6.94 (d, J 8.4 Hz, 2H), 6.45 (s, 1H), 5.49 (s, 2H), 4.08 (m, 2H), 3.54 (m, 2H), 2.89 (m, 1H), 2.64 (m, 1H), 2.35 (m, 1H), 2.28 (m, 1H), 2.07 (m, 1H), 1.85 (m, 1H), 1.65 (m, 1H), 1.50 (m, 1H), 1.39 (m, 1H), 1.21 (t, J 7.2 Hz, 3H).
Ethyl 1-{(1-methyl-1 H-benz[d]imidazol-2-yl)methyl}-piperidine-4-carboxylate (1.2.6). Yield 80%. GC-MS (ESI) [M + H]+ 302. PMR (CDCl3), δ, ppm: 7.74 (m, 1H), 7.35 (m, 1H), 7.28 (m, 2H), 4.13 (q, J 7.2 Hz, 2H), 3.88 (s, 3H), 3.80 (s, 2H), 2.87 (m, 2H), 2.31 (m, 1H), 2.18 (t. d, J1 11.4 Hz, J2 2.4 Hz, 2H), 1.89 (m, 2H), 1.72 (m, 2H), 1.25 (t, J 7.2 Hz, 3H).
1-(Hetarylmethyl)piperidinecarboxylic acids (1.2.7 – 1.2.10), general method. A solution of the appropriate ester of 1-(hetarylmethyl)piperidinecarboxylic acid (3 mmol) in EtOH (15 mL) was treated with a solution of NaOH (240 mg, 6 mmol) in EtOH (10 mL), stirred for 15 h at room temperature, neutralized with AcOH (360 mg, 6 mmol), and extracted with CH2Cl2. The organic layer was dried over Na2SO4 and evaporated in vacuo to afford acids 1.2.7 – 1.2.10.
1-[(1-Methyl-1 H-indol-2-yl)methyl]piperidine-4-carboxylic acid (1.2.7). Yield 78%. GC-MS (ESI) [M + H]+ 273. PMR (DMSO-d6), δ, ppm: 7.46 (d, J 8.0 Hz, 1H), 7.38 (d, J 8.4 Hz, 1H), 7.10 (m, 1H), 6.98 (m, 1H), 6.31 (s, 1H), 3.74 (s, 3H), 3.60 (s, 2H), 2.80 (m, 2H), 2.20 (m, 1H), 2.03 (m, 2H), 1.78 (m, 2H), 1.52 (m, 2H).
1-[(1-Methyl-1 H-indol-2-yl)methyl]piperidine-3-carboxylic acid (1.2.8). Yield 93%. GC-MS (ESI) [M + H]+ 273. PMR (DMSO-d6), δ, ppm: 7.46 (d, J 8.0 Hz, 1H), 7.38 (d, J 8.0 Hz, 1H), 7.10 (m, 1H), 6.98 (m, 1H), 6.32 (s, 1H), 3.73 (s, 3H), 3.62 (m, 2H), 2.84 (m, 1H), 2.64 (m, 1H), 2.39 (m, 1H), 2.20 (m, 1H), 2.08 (m, 1H), 1.77 (m, 1H), 1.63 (m, 1H), 1.41 (m, 2H).
1-{[1-(4-Chlorobenzyl)-1 H-indol-2-yl]methyl}piperidine- 3-carboxylic acid (1.2.9). Yield 82%. GC-MS (ESI) [M + H]+ 383, 385. PMR (DMSO-d6), δ, ppm: 7.51 (d, J 7.6 Hz, 1H), 7.32 (d, J 8.4 Hz, 2H), 7.29 (d, J 8.4 Hz, 1H), 7.06 (m, 1H), 7.01 (m, 3H), 6.43 (s, 1H), 5.50 (s, 2H), 3.54 (m, 2H), 2.81 (m, 1H), 2.60 (m, 1H), 2.15 (m, 2H), 1.99 (m, 1H), 1.72 (m, 1H), 1.54 (m, 1H), 1.29 (m, 2H).
1-[(1-Methyl-1 H-benz[d]imidazol-2-yl)methyl]piperidine-4-carboxylic acid (1.2.10). Yield 74%. GC-MS (ESI) [M + H]+ 274. PMR (DMSO-d6), δ, ppm: 7.76 (m, 2H), 7.42 (m, 2H), 4.74 (s, 2H), 3.98 (s, 3H), 3.63 (m, 2H), 3.28 (m, 2H), 2.57 (m, 1H), 2.05 (m, 2H), 1.99 (m, 2H).
Amides of 1-(hetarylmethyl)piperidinecarboxylic acids (25 – 28 and 58 – 62), general method. Asolution of the appropriate 1-(hetarylmethyl)piperidinecarboxylic acid (1.2.8 – 1.2.11, 1 mmol) and carbodiimidazole (195 mg, 1.2 mmol) in DMF (4 mL) was stirred for 1 h at 65°C, treated with the amine (1.1 mmol), stirred for another 12 h at 65°C, poured into K2CO3 solution (10%), and extracted with CH2Cl2. The extract was dried over Na2SO4 and evaporated in vacuo. The solids were chromatographed over silica gel (EtOAc:Et3N eluent, 40:1). Yields of amides 25 – 28 and 58 – 62, ~80%. Dihydrochlorides of the amides were prepared by adding a 5% excess of HCl solution (3 M) in dioxane to solutions of the bases in Me2CO. The precipitates were filtered off, rinsed with Et2O, and dried in vacuo.
N-(2-Dimethylaminoethyl)-1-(1-methyl-1 H-indol-2-ylmethyl) piperidine-4-carboxamide dihydrochloride (25•2HCl). GC-MS (ESI) [M + H]+ 343. PMR (DMSO-d6), δ, ppm: 10.62 (m, 2H), 8.42 (t, J 5.0 Hz, 1H), 7.59 (d, J 7.2 Hz, 1H), 7.50 (d, J 8.0 Hz, 1H), 7.22 (m, 1H), 7.07 (m, 1H), 6.83 (s, 1H), 4.55 (m, 2H), 3.85 (s, 3H), 3.48 (br.m, 2H), 3.28 (m, 2H), 3.12 (m, 2H), 3.05 (br.m, 2H), 2.74 (d, J 3.2 Hz, 3H), 2.42 (m, 1H), 2.00 (m, 4H).
N-(2-Dimethylaminopropyl)-1-(1-methyl-1 H-indol-2-ylmethyl)piperidine-4-carboxamide dihydrochloride (26•2HCl). GC-MS (ESI) [M + H]+ 357. PMR (DMSO-d6), δ, ppm: 10.51 (m, 2H), 8.23 (t, J 5.2 Hz, 1H), 7.59 (d, J 8.0 Hz, 1H), 7.50 (d, J 8.4 Hz, 1H), 7.22 (m, 1H), 7.08 (m, 1H), 6.82 (s, 1H), 4.54 (m, 2H), 3.85 (s, 3H), 3.48 (br.m, 2H), 3.25 (m, 2H), 3.09 (m, 2H), 2.99 (m, 2H), 2.70 (d, J 4.8 Hz, 3H), 2.38 (m, 1H), 1.96 (m, 4H), 1.78 (m, 2H).
[1-(1-Methyl-1 H-indol-2-ylmethyl)piperidin-4-yl]-(4-methylpiperazin-1-yl)ketone (27). Yield 56%. GC-MS (M + H)+ 355. PMR (DMSO-d6, 400 MHz), δ, ppm: 11.41 (br.s, 1H), 10.93 (br.s, 1H), 7.60 (d, J 7.6 Hz, 1H), 7.51 (d, J = 8.4 Hz, 1H), 7.23 (m, 1H), 7.08 (m, 1H), 6.81 (s, 1H), 4.54 (br.s, 2H), 4.40 (br.m, 1H), 4.12 (br.m, 1H), 3.85 (s, 3H), 3.50 (br.m, 5H), 3.00 (br.m, 4H), 2.87 (br.m, 2H), 2.73 (s, 3H), 1.98 (br.m, 2H), 1.82 (br.m, 2H).
[1-(1-Methyl-1 H-indol-2-ylmethyl)piperidin-4-yl]-(4-methylpiperazin-1-yl)ketone dihydrochloride (27•2HCl). Yield 46%. GC-MS (M + H)+ 355. PMR (DMSO-d6, 400 MHz), δ, ppm: 11.28 (br.s, 1H), 11.14 (br.s, 1H), 7.59 (d, J 8.0 Hz, 1H), 7.50 (d, J 8.0 Hz, 1H), 7.22 (m, 1H), 7.07 (m, 1H), 6.83 (br.m, 1H), 4.57 (br.s, 2H), 4.37 (br.m, 1H), 4.07 (br.m, 1H), 3.87 (s, 3H), 3.55 (br.m, 1H), 3.42 (br.m, 4H), 3.14 (br.m, 2H), 3.02 (br.m, 3H), 2.89 (br.m, 1H), 2.72 (br.m, 3H), 1.98 (br.m, 1H), 1.86 (br.m, 2H), 1.44 (br.m, 1H).
[1-(1-Methyl-1 H-indol-2-ylmethyl)piperidin-4-yl]-(4-propylpiperazin-1-yl)ketone dihydrochloride (28•2HCl). Yield 28%. GC-MS (M + H)+ 383. PMR (DMSO-d6, 400 MHz), δ, ppm: 11.29 (br.s, 1H), 10.80 (br.s, 1H), 7.60 (d, J 8.0 Hz, 1H), 7.51 (d, J 8.0 Hz, 1H), 7.23 (m, 1H), 7.09 (m, 1H), 6.80 (s, 1H), 4.55 (br.s, 2H), 4.39 (br.m, 1H), 4.11 (br.m, 1H), 3.85 (s, 3H), 3.59 (br.m, 1H), 3.51 (br.m, 2H), 3.43 (br.m, 2H), 3.12 (m, 1H), 2.98 (br.m, 5H), 2.86 (br.m, 2H), 2.73 (s, 3H), 1.96 (br.m, 2H), 1.82 (br.m, 2H), 1.72 (m, 2H), 0.90 (t, J 7.2 Hz, 3H).
[1-(1-Methyl-1 H-benzimidazol-2-ylmethyl)piperidin-4-yl]-(4-propylpiperazin-1-yl)ketone dihydrochloride(58•2HCl). GC-MS (ESI) [M + H]+ 384. PMR (DMSO-d6), δ, ppm: 11.39 (br.s, 1H), 7.69 (d, J 7.6 Hz, 1H), 7.64 (d, J 8.0 Hz, 1H), 7.34 (m, 1H), 7.28 (m, 1H), 4.62 (s, 2H), 4.40 (br.m, 1H), 4.13 (br.m, 1H), 3.91 (s, 3H), 3.61 (br.m, 4H), 3.15 (br.m, 3H), 2.98 (br.m, 4H), 2.86 (br.m, 2H), 1.95 (br.m, 2H), 1.88 (m, 2H), 1.73 (m, 2H), 0.90 (t, J 7.0 Hz, 3H).
[1-(1-Methyl-1 H-indol-2-ylmethyl)piperidin-3-yl]-(4-methylpiperazin-1-yl)ketone dihydrochloride (59•2HCl). GC-MS (ESI) [M + H]+ 355. PMR (DMSO-d6), δ, ppm: 11.28 (m, 2H), 7.59 (d, J 8.0 Hz, 1H), 7.50 (d, J 8.0 Hz, 1H), 7.22 (m, 1H), 7.07 (m, 1H), 6.84 (s, 1H), 4.57 (s, 2H), 4.38 (m, 1H), 4.07 (m, 1H), 3.87 (s, 3H), 3.53 (m, 2H), 3.41 (m, 4H), 3.14 (m, 2H), 3.02 (br.m, 4H), 2.89 (br.m, 1H), 2.73 (br.s, 3H), 1.98 (m, 1H), 1.86 (m, 2H), 1.44 (m, 1H).
[1-(1-Methyl-1 H-indol-2-ylmethyl)piperidin-3-yl]-(4-propylpiperazin-1-yl)ketone (60). Yield 47%. GC-MS (M + H)+ 383. PMR (DMSO-d6, 400 MHz), δ, ppm: 11.19 (br.m, 2H), 7.59 (d, J 7.6 Hz, 1H), 7.50 (d, J 8.0 Hz, 1H), 7.22 (m, 1H), 7.07 (m, 1H), 6.83 (br.m, 1H), 4.56 (br.s, 2H), 4.37 (m, 1H), 4.05 (m, 1H), 3.87 (s, 3H), 3.61 (m, 1H), 3.44 (br.m, 5H), 3.14 (br.m, 2H), 2.98 (br.m, 4H), 2.86 (br.m, 1H), 1.98 (br.m, 1H), 1.86 (br.m, 2H), 1.70 (br.m, 2H), 1.44 (br.m, 1H), 0.89 (br.m, 3H).
[1-(1-Methyl-1 H-indol-2-ylmethyl)piperidin-3-yl]-(4-propylpiperazin-1-yl)ketone dihydrochloride (60•2HCl). Yield 47%. GC-MS (M + H)+ 383. PMR (DMSO-d6, 400 MHz), δ, ppm: 11.19 (br.m, 2H), 7.59 (d, J 7.6 Hz, 1H), 7.50 (d, J 8.0 Hz, 1H), 7.22 (m, 1H), 7.07 (m, 1H), 6.83 (br.m, 1H), 4.56 (br.s, 2H), 4.37 (m, 1H), 4.05 (m, 1H), 3.87 (s, 3H), 3.61 (m, 1H), 3.44 (br.m, 5H), 3.14 (br.m, 2H), 2.98 (br.m, 4H), 2.86 (br.m, 1H), 1.98 (br.m, 1H), 1.86 (br.m, 2H), 1.70 (br.m, 2H), 1.44 (br.m, 1H), 0.89 (br.m, 3H).
{1-[1-(4-Chlorobenzyl)-1 H-indol-2-ylmethyl]piperidin-1-yl}-(4-methylpiperazin-1-yl)ketone dihydrochloride (61•2HCl). Yield 79%. GC-MS (M + H)+ 465, 467. PMR (DMSO-d6, 400 MHz), δ, ppm: 11.07 (br.m, 2H), 7.63 (d, J 7.6 Hz, 1H), 7.44 (d, J 7.6 Hz, 1H), 7.34 (d, J 8.0 Hz, 2H), 7.17 (m, 1H), 7.09 (m, 1H), 6.98 (br.m, 1H), 6.93 (d, J 8.0 Hz, 2H), 5.69 (br.s, 2H), 4.50 (br.s, 2H), 4.38 (br.m, 1H), 4.07 (br.m, 1H), 3.49 (br.m, 5H), 3.16 (br.m, 2H), 3.00 (br.m, 3H), 2.89 (br.m, 1H), 2.74 (br.m, 3H), 1.95 (br.m, 1H), 1.86 (br.m, 2H), 1.43 (br.m, 1H).
{1-[1-(4-Chlorobenzyl)-1 H-indol-2-ylmethyl]piperidin-1-yl}-(4-propylpiperazin-1-yl)ketone dihydrochloride (62•2HCl). Yield 78%. GC-MS (M + H)+ 493, 495. PMR (DMSO-d6, 400 MHz), δ, ppm: 11.12 (br.m, 2H), 7.63 (d, J 7.6 Hz, 1H), 7.44 (d, J 8.0 Hz, 1H), 7.34 (d, J 8.0 Hz, 2H), 7.17 (m, 1H), 7.09 (m, 1H), 6.99 (br.m, 1H), 6.93 (d, J 8.0 Hz, 2H), 5.69 (br.s, 2H), 4.51 (br.s, 2H), 4.37 (br.m, 1H), 4.06 (br.m, 1H), 3.59 (m, 1H), 3.41 (br.m, 5H), 3.14 (br.m, 2H), 2.98 (br.m, 4H), 2.85 (br.m, 1H), 1.96 (br.m, 1H), 1.86 (br.m, 2H), 1.70 (br.m, 2H), 1.43 (br.m, 1H), 0.90 (br.m, 3H).
2-(8-Isopropyl-2,3,3a,4,5,6-hexahydro-3 H-pyrazino-[3,2,1-jk]carbazol-3-yl)acetamide (77) was prepared (Scheme 3) via hydrogenation of 1.3.1 over Pd/C followed by alkylation of the resulting 1.3.2 by chloroacetamide.

Scheme 3
2-(8-Isopropyl-2,3,3a,4,5,6-hexahydro-3 H-pyrazino-[3,2,1-jk]carbazol-3-yl)acetamide hydrochloride (77•HCl). A solution of 9-[2-(dibenzylamino)ethyl]-6-isopropyl-3,4-dihydro-2H-carbazol-1(9H)-one (8.45 g, 18.8 mmol) in MeOH (80 mL) was hydrogenated in an autoclave with Pd/C (0.5 g, 10%) at 30 atm for 24 h at 70°C. The solution was filtered through Celite and evaporated in vacuo. The resulting 2-(8-isopropyl-2,3,3a,4,5,6-hexahydro-3H-pyrazino[3, 2, 1-jk]carbazole was refluxed with chloroacetamide (1.93 g, 20.6 mmol) and K2CO3(3.88 g) in MeCN (50 mL) for 12 h. The solvent was vacuum distilled. The solid was worked up with H2O and extracted with CH2Cl2. The extract was dried over Na2SO4 and evaporated. The solid was recrystallized from EtOH to afford 77 (3.27 g, 56%). GC-MS (M + H)+ 312. PMR (CDCl3, 400 MHz), δ, ppm: 7.34 (s, 1H), 7.21 (d, J 8.4 Hz, 1H), 7.10 (dd, J1 8.4 Hz, J2 1.2 Hz, 1H), 7.02 (br.s, 1H), 5.50 (br.s, 1H), 4.18 (dd, J1 11.6 Hz, J2 3.6 Hz, 1H), 3.84 (dt, J1 11.6 Hz, J2 4.8 Hz, 1H), 3.59 (m, 1H), 3.53 (d, J 12.8 Hz, 1H), 3.23 (dd, J1 12.4 Hz, J2 4.8 Hz, 1H), 3.06 (m, 3H), 2.82 (dd, J1 11.6 Hz, J2 6.4 Hz, 1H), 2.70 (m, 1H), 2.22 (m, 2H), 1.86 (m, 1H), 1.53 (m, 1H), 1.32 (d, J 6.8 Hz, 6H). Hydrochloride 77•HCl was precipitated by adding a 5% excess of HCl solution (3 N) in dioxane to a solution of base 77 in CH2Cl2. 77•HCl: PMR (DMSO-d6, 400 MHz), δ, ppm: 10.89 (br.s, 1H), 8.16 (br.s, 1H), 7.75 (br.s, 1H), 7.34 (d, J 8.0 Hz, 1H), 7.31 (c, 1H), 7.10 (d, J 8.0 Hz, 1H), 4.77 (br.m, 1H), 4.46 (d, J 6.4 Hz, 1H), 4.25 (br.m, 1H), 3.95 (br.m, 4H), 2.97 (p, J 6.8 Hz, 1H), 2.73 (br. d, J 15.6 Hz, 1H), 2.58 (br.m, 1H), 2.35 (br.m, 1H), 2.20 (br.m, 1H), 1.78 (br.m, 2H), 1.24 (d, J 6.8 Hz, 6H).
Experimental Biological Part
Antiviral activity of the compounds was studied using HCV-infected Huh7.3 human hepatoma cell line and enzyme immunoanalysis (EIA) for HCV core antigen. The inhibition efficiency of the tested compounds for antigen production (virus replication) was measured in HCV-infected cells (JFH-1 clone). The Huh7.3 cell culture used in this experiment was produced by selection of Huh-7 cells. This cell line possesses increased sensitivity to hepatitis C virus infection and is capable, being infected by HCV, of supporting the full cycle of virus replication [6]. The recombinant HCV virus used in the experiment was obtained by transfection of Huh7 cells with JFH-1 HCV RNA obtained via in vitro transcription. The matrix DNA used for transcription was obtained starting with the JFH-1 DNA primary structure (NCBI, catalog No. AB047639) [7].
Cytotoxicity of the compounds was determined using the MTT assay for Huh7.3 human hepatoma cell line according to the published method [8].
Tables 1 – 4 present the antiviral activity (EC50), cytotoxicity (CC50), and therapeutic index (TI50 = CC50/EC50).
Antiviral activity of the compounds against HCV in Huh7 human hepatoma cell line containing subgenomic HCV RNA-replicon (Table 5) was determined using the previously described procedure [9]. The percent HCV inhibition by all tested compounds at a concentration of 10 μM was determined in the first stage. The concentration dependence of the inhibition was studied and the EC50 values were determined for compounds that showed 50% and greater inhibition and also for 1 (clemizole), 28, and 77, which showed less than 50% inhibition at 10 μM. The cytotoxicity of the tested compounds was estimated in parallel for Huh7 human hepatoma cell culture, as previously described [8].
Results and Discussion
Antiviral activity was studied for HCV-infected Huh7.3 human hepatoma cell line. It was shown that 2-aminomethylbenzimidazoles (1 – 5), 1,2,3,4-tetrahydrobenzo[4, 5]imidazo[1,2-a]pyrazines (6 – 19), and 2-aminomethylindoles (20 – 23) had micromolar activity (EC50 = 3.91 – 7.26) that was practically the same as clemizole (EC50 = 8.96 μM). However, like clemizole, this series of compounds had very low therapeutic indices (TI50 < 20; TI50 of clemizole, 3.8).
The 2-piperidin-1-ylmethyl-1H-indoles (25 – 62, Table 2) turned out to be more attractive. Nanomolar HCV inhibitors (29, 31 – 34, 40, 43, 44, 46, 47, 50, 51, 53, 55) with TI values from 11 to 36 were found among them. The most promising compound in this series was (4-isopropylpiperazin-1-yl)-[1-(1-methyl-1H-indol-2-ylmethyl)piperidin-4-yl] methanone (28) with EC50 = 2.12 ± 1.35 μM and TI50 = 52.
Nanomolar inhibitors (65 – 68) were also observed among substituted 2,3,4,9-tetrahydro-1H-carbazol-2-ylamines (63 – 74). However, their therapeutic indices turned out to be low (10 < TI50 < 21, Table 3). Only inhibitor 70 with EC50 = 2.82 μM and TI50 = 36 was identified in this series.
2,3,3a,4,5,6-Hexahydro-1H-pyrazino[3, 2, 1-jk]carbazoles (75 – 80) also had micromolar activity (Table 3). Inhibitor 77 (EC50 = 1.69 ± 0.55 μM; TI50 = 68) was the most attractive among them. 2,3-Dihydropyrazino[1,2-a]indol-1-ylamine (82, EC50 = 2.25 μM, CC50 = 86.2 μM, TI50 = 38) had activity and cytotoxicity that was slightly inferior to it.
The most active of the substituted N-[2-(aminophenylmethyl) benzofuran-3-yl]amides (84 – 92) were 86 (EC50 = 2.46 μM) and 90 (EC50 = 2.55 μM). However, these compounds had lower TI values (TI50 = 19 and 17, respectively) than inhibitors 82 and 86. Benzoxazole 93 displayed weak activity (EC50 = 9.23 μM) and a low therapeutic index (TI50 = 5.6).
Astudy of the activity of 1 – 93 against HCV RNA-replicon genotypes (1a, 1b, and 2a) showed that most of them were inactive because their activities were less than or comparable with the cytotoxicity (Table 5) whereas AV4025 and tubercidin (antiviral activity and cytotoxicity standards, respectively) showed the expected activity and cytotoxicity. The only exception was 67, which exhibited nanomolar activity against all three HCV genotypes and micromolar cytotoxicity (EC50 = 0.24 – 0.78 μM, CC50 = 9.89 μM, TI50 = 13 – 41).
Thus, compound 67 showed high (nanomolar) activity and specificity for both the HCV replicon model and the in vitro HCV infection model (Table 3). Further optimization of its activity and specificity seem worthwhile.

Thus, the antiviral activity in vitro and cytotoxicity of heterocyclic compounds incorporating 1H-benzimidazol-2- yl- (1 – 19), indol-2-yl- (20 – 83), benzofuran-2-yl- (84 – 92), and benzoxazol-2-ylmethylamine (93) fragments were studied. It was shown that most of the last ones were micromolar inhibitors for in vitro HCV-infected Huh7.3 human hepatoma cell line but were inactive against HCV RNA-replicon. The only exception was 9-methyl-N(6)-(3-nitrophenyl)-2,3,4,9-tetrahydro-1H-carbazole-1,6-diamine (67), which exhibited nanomolar activity in both instances.
The most attractive compounds of those studied were (4-isopropylpiperazin-1-yl)-[(1H-indol-2-ylmethyl)piperidin-4-yl]methanones (28, 34, 47) and 2-(8-isopropyl-1,2,3a,4,5,6-hexahydropyrazino[3, 2, 1-jk]carbazol-3-yl)acet amide (77) according to results from studies of the in vitro antiviral activity and cytotoxicity. Inhibitors 28 and 77 had the highest therapeutic indices (TI50 = 52 and 68, respectively) and were selected for further preclinical development.
References
S. Einav, D. Gerber, P. D. Bryson, et al., Nat. Biotechnol., 26, 1019 – 1027 (2008); Published online, Aug. 31, 2008; doi: 10.1038 / nbt.1490.
S. Einav, D.-S. Dvory-Sobol, E. Gehrig, et al., J. Infect. Dis., 202, 65 – 74 (2010); doi: 10.1086 / 653080, “The Hepatitis C Virus (HCV) NS4B RNABinding Inhibitor Clemizole is Highly Synergistic with HCV Protease Inhibitors.”
O. Cherkaoui, E. M. Essassi, and R. Zniber, Bull. Soc. Chim. Fr., 128, 255 – 259 (1991).
I. Yu. Grishin, N. M. Przhiyalgovskaya, Yu. M. Chunaev, et al., Khim. Geterotsikl. Soedin., No. 7, 907 – 910 (1989).
M. J. Plater, P. Barnes, L. K. McDonald, et al., Org. Biomol. Chem., 7, 1633 – 1641 (2009).
R. Bartenschlager and T. Pietschmann, Proc. Natl. Acad. Sci. USA, 102(28), 9739 – 9740 (2005).
T. Wakita, T. Pietschmann, T. Kato, et al., Nat. Med., 11, 791 – 796 (2005).
A. P. Wilson, Cytotoxicity and Viability Assays in Animal Cell Culture, A Practical Approach, J. R. W. Masters (ed.), Oxford University Press, Oxford (2000), Vol. 1, pp. 175 – 219.
J. T. Guo, V. V. Bichko, and C. Seeger, J. Virol., 75, 8516 – 8523 (2001).
Author information
Authors and Affiliations
Corresponding authors
Additional information
Translated from Khimiko-Farmatsevticheskii Zhurnal, Vol. 49, No. 6, pp. 10 – 19, June, 2015.
Rights and permissions
About this article

Cite this article
Ivachtchenko, A.V., Yamanushkin, P.M., Mit’kin, O.D. et al. New Heterocyclic Hepatitis C Virus (HCV) Inhibitors Containing A 2-Aminomethyl-1H-Indole Fragment. Pharm Chem J 49, 352–361 (2015). https://doi.org/10.1007/s11094-015-1285-x
Received:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s11094-015-1285-x