
Abstract
The degradation of perfluorosurfactants (PFS), particularly of PFOS, has been studied in dielectric barrier discharge (DBD) and nano-pulse corona discharge (PCD) reactors. DBD-plasma is generated in two different types of reactors. First, in a suitable falling film reactor with a planar configuration for the treatment of ca. 0.4 L PFS solution, and second, in a horizontal trough reactor for the treatment of ca. 8 L PFS contaminated water. For the comparison, the efficiency of PFS degradation by ozonation and photocatalytic ozonation processes were also examined using a similar falling film reactor, and it was found that these methods are not as efficient as the DBD plasma. The degradation of PFSs by non-thermal plasma was investigated in dependence on PFS concentration and gas atmosphere by HPLC/MS and ion chromatography. Concerning the energy yield, the nano-pulse corona is significantly more efficient than the DBD plasma. For an initial PFOS concentration of 10 mg/L the G50 of the PCD is about 200 mg/kWh, while it is less than 100 mg/kWh for the DBD reactor. Compared to the plasma in He atmosphere, in all reactors the decomposition of PFS under Ar atmosphere results in a deeper mineralization, which is expressed by fluoride recovery.
Similar content being viewed by others

Explore related subjects
Discover the latest articles, news and stories from top researchers in related subjects.Avoid common mistakes on your manuscript.
Introduction
Perfluorooctanesulfonic acid (PFOS) and perfluorooctanoic acid (PFOA) are considered as perfluorosurfactants (PFS) and anthropogenic global pollutants, which are resistant to conventional water and wastewater remediation technologies. Owing to their excellent properties of lowering the surface tension of water, perfluorinated surfactants have found wide applications in water-repellent textiles, surface treatment of paper, and electroplating industries. Moreover, large amounts of PFSs are released into the environment through synthetic firefighting foams. Due to their resistance against chemical and biological degradations, these compounds are found in surface waters and accumulated within the nature and along the food chain [1]. Therefore, PFOS and its salts have been listed as persistent organic pollutants in the Stockholm Convention and the production and the use of these compounds have been severely limited.
A comprehensive overview of the current research on the removal of PFSs is given by Merino et al. [2]. The strong carbon–fluorine bond makes PFS inert to most conventional treatment techniques including ozonation at normal conditions. Anion exchange and adsorption on activated carbon are effective methods for separation of PFSs from process and ground water. Application of advances oxidation processes (AOPs) for decomposition of PFSs has been studied and a review, in particular, on the photocatalytic degradation of PFOA and PFOS has been published by Wang et al. [3]. An alternative approach for the removal of PFS compounds from aqueous samples via decomposition and mineralization to non-harmful products is developed by the non-thermal plasma (NTP) technology. The NTP technology for wastewater treatment has been emerged in the last years. There are numerous publications devoted to the decomposition of individual contaminants using different plasma sources and reactor designs. Recently published reviews [4, 5] have summarized the results of those studies and underlined the necessity of further studies in this area. For any specific application, a comprehensive comparison with other AOPs and choose of a suitable NTP system is indispensable.
NT-plasma-based processes for decomposing PFS contaminants have been investigated by Obo et al. [6] and Stratton et al. [7]. Mededovic-Thagard et al. [8], based on their experiments on NTP-aided degradation of several types of water contaminants, reported that surfactants are preferably decomposed by NTP above the surface of water.
After conducting some initial experiments to test the applicability of NT plasma for the decomposition of PFOS and PFOA and comparing with the results of ozonation and photocatalytic ozonation—similar to the works we have already reported for diclofenac and ibuprofen [9], for 2, 4-dichlorophenoxyacetic acid and 2, 4-dichlorophenol [10], and for methylene blue [11]—the degradation of these PFSs in various NTP reactors were systematically studied.
Experimental
Materials
PFOA (95%) was obtained from Riedel-de Haën and PFOS as potassium salt (95%) was purchased from 3M. The gases (O2, N2, Ar, and He) were obtained from Air Liquide. Unless otherwise indicated, the solutions were prepared using deionized (d.i.) water (≤ 0.1 µS/cm). The real groundwater samples for the comparative suitability studies were collected from a contaminated site in Gerresheim, Germany.
Reactor Design
Two different reactor concepts for plasma treatment of contaminated water have been designed: (1) dielectric barrier discharge (DBD) and (2) nano-pulse corona discharge (PCD).
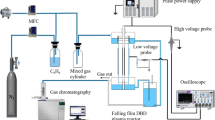
As illustrated in Fig. 1, the DBD reactor is realized as a planar falling film reactor; the R1 with two falling films with an area of 68 × 29 cm as described in details in the previous publication [9], and the R2 with a longer falling film area of 144 × 22 cm flowing only in one-side of the reactor. The high voltage electrode consists of a self-adhesive aluminum foil (R1: 47 × 19 cm, R2: 20 × 132 cm) coated on the inner side of the dielectric ceramic plate, while the ground electrode is made of copper strips stuck on Pilkington Active™ glass inside the falling film. A Minipuls 6 generator (GBS Elektronik, Radeberg, Germany) providing up to 21 kV (RMS) at frequencies of 5–20 kHz was used to supply the high voltage. The power uptake of the plasma was measured by the current and voltage waveforms. The Minipuls 6 device has both an output monitor (1:3000) for output voltage and a current monitor (10 V/A) for output current. The waveforms were then obtained by an oscilloscope (DPO 2002 B, Tektronix, Newark, USA) and integrated to determine the discharge power. The gas volume of the system was reduced from 9 L in R1 to 1 L in R2.

Schematic illustration of the falling film DBD reactors R1 and R2 with α = 90° (left) and R3 with α = 0° and the pulse corona plasma reactors R4 and R5 (right)
The ozonation and photocatalytic ozonation experiments were performed utilizing a falling film photo-reactor in a similar conformation to the DBD reactor as described previously [9]. A set of 7 UV-A lamps (Narva, Germany, model LT 15W/009) with a maximum wavelength at ca. 350 nm was installed in-between the glass plates of the reactor R1, replacing the high voltage electrode. The ozonation and photocataytic ozonation were performed at an oxygen flow rate of 10 L/h and an input ozone content of 90 mg/L. The Pilkington Active™ glass is coated with a TiO2 nano-layer, which acts as a photocatalyst by generating hydroxyl radicals.
The horizontal trough DBD reactor (R3) has the same dimensions as R2; however, the water is not flowing in a thin (ca. 0.2 mm) falling film form, but as a layer with a thickness of about 30 mm. The total volume of solution in R3 is about 8 L. The gap between the water surface and the ceramic dielectric is 6–8 mm that is filled with argon or helium gases. The dielectric plate also serves as a cover insulating the gas space. The high voltage electrode consists of a self-adhesive aluminum foil (114 × 20 cm) and is stuck on the top of the dielectric plate, while the ground electrode is made of self-adhesive copper strips stuck on the bottom of the reactor inside the water. The high voltage for R2 and R3 is provided as for R1.
For the pulse corona reactor (R4), a horizontal trough made of PVC with dimensions of 52 × 24 × 12 cm was utilized (Fig. 1, right side). Usually 7–8 L solution was treated in this reactor. The trough was covered by the high voltage electrode, which consists of a circuit board equipped with ca. 2000 tin-plated brass pins (D = 1.3 mm, L = 7 mm) directed towards the surface of water. The gap between the pin tips and the water surface is 5–10 mm, depending on the volume of loaded solution. The water sample (7–8 L in total) was continuously circulated by a membrane pump at 0.5–4.5 L/min flow rate.
A second pulse corona reactor (R5, 155 × 28 × 6 cm) for the treatment of up to 5 L solution was constructed. The R5 provides a larger surface-to-volume ratio with the same number of pins spread over the larger electrode surface that stays 5–10 mm above the surface of water. The distance between the pins in R5 is 9 mm, while it was 6 mm in R4. Table 1 gives an overview of the reactors used in the present study.
The nano-second high voltage pulses were provided by a FPG 30-2NM20 (FID GmbH, Burbach, Germany) nano-pulse generator. It generates pulses with a positive polarity, a duration of 20 ns, peak voltage of up to 20 kV, and pulse repetition rate of up to 2 kHz. Figure 2 represents the current and voltage waveforms that were used to calculate the power of the plasma. The voltage waveform was measured by a passive high voltage probe (P6015A, Tektronix, Newark, USA) and the current waveform was determined by a current transformer (Stangenes, Palo Alto, USA, model 2–0.1 W, output volts/ampere 0.1) in combination with an oscilloscope (DPO 202 B, Tektronix).

Typical waveforms of pulse voltage and discharge current for the pulse corona discharge in the reactor R4 under He atmosphere at 18 kV
The DBD reactors were regulated, by a self-made LabView script, at an input power of 150 W for R1 and 200 W for R2. However, the nano-pulse reactors were operated at 15–18 kV and 2 kHz, yielding a power of 85–110 W in He and 100–130 W in Ar.
Due to the deformation of the falling film flow and overheating by the discharge, the DBD experiments were performed in a burst mode, the plasma was applied periodically as 1 s plasma and 1 s pause. The energy yield G50 in which, defined as the amount of PFS decomposed per kWh at 50% conversion, is calculated according to the Eq. 1 [12]:
where C0 is the initial concentration (mg/L), V indicates liquid volume (L), kobs stands for the first-order removal rate constant (min−1), and P demonstrates the plasma power (kWh/min).
Except the preliminary experiments (“Comparative Suitability Studies” section), which were done only for orientation, all experiments were repeated at least 3 times and the average error of the results is about 10%.
Analysis
The concentrations of PFOS, PFOA and their decomposition by-products were determined by HPLC/MS, according to DIN 38407-F42:2011-03. The separation and quantification of the compounds were done according to their retention times and mass peaks. Where necessary, samples were concentrated and purified prior to the measurements by solid-phase extraction methods using a weak anion exchange polymer (Oasis Wax, Waters). HPLC analyses were performed using analytical columns containing polar modified RP materials such as Zorbax Eclipse XDB-C18 (Agilent), and a mixture of methanol/water solution was used as a mobile phase. The characteristic masses of the individual PFSs, as shown in the supplementary information (Table S2), were used for identification and quantification of the corresponding substances. The limit of determination depends on the utilized volume of the sample. In the actual cases, using 60 and 200 ml of sample the determination limits were 1.0 and 0.01 µg/L, respectively.
The concentrations of fluoride and sulfate, as the mineralization products, were determined by ion chromatography using a Dionex DX 500 with a CD20 conductivity detector connected to an IonPac TAC-LP1 (4 × 35 mm) anion concentrator column, an IonPac AG14 (4 × 50 mm) guard column, and an IonPac AS14 (4 × 250 mm) analytical column. The flow rate of the eluent, consisting of NaHCO3 (1.0 mM) and Na2CO3 (3.5 mM), was fixed at 1.2 mL/min. For each analysis, a sample volume of 1 mL was injected into the IonPac TAC-LP1 anion concentrator column.
The fluoride and sulfate concentrations, C (µMol/L), were measured to calculate the corresponding recoveries, as following:
Results and Discussion
Comparative Suitability Studies
In the previous work [9], it has been shown that the application of NT-plasma for the degradation of water contaminants is only reasonable when it is compared with other established AOPs and its predominance over them is indicated. Therefore, the degradation of PFOS and PFOA in a ground water sample, from a contaminated site, was studied by the ozonation, photocatalytic ozonation, and DBD plasma at comparable conditions using the falling film reactor R1. This reactor was specially designed to compare the performance of different AOPs [9].
Molecular dynamic simulations demonstrated that PFOA and PFOS enrich at the surface of the liquid film, due to their surface-active properties, where the perfluorinated carbon chain protruding from the surface [8, 13, 14]. The falling film reactor has a large surface-to-volume ratio, thus, it is beneficial, especially, for the treatment of those surfactants at the gas–liquid interface. Furthermore, due to the small thickness of the falling liquid film layer the absorption of UV irradiation by the medium can be neglected. Therefore, this design of the reactor is suitable for the direct comparison of PFS decomposition by ozonation, ozone/UV irradiation, and non-thermal plasma.
In each experiment, 0.8 L of solution was continuously circulated at the rate of 1 L/min through the reactor R1 and treated for 1 h by different methods. The results, given in Table 2 and Fig. 3, in general demonstrate the applicability of these methods for the decomposition of PFS. Ozone is a very strong oxidizing agent (E0 = 2.07 V/SHE) against organic substances, which has already been applied for the treatment of industrial wastewater containing PFOS and PFOA [15]. It has been pointed out that these perfluoroalkyl acids under the alkaline conditions can be efficiently decomposed over several hours of a batch ozonation process. In the experiments, ozonation and photocatalytic ozonation surprisingly showed almost identical decomposition yields for PFOS and PFOA. The degradation yields, however, show large differences between PFOS and PFOA decomposition after 1 h treatment. Since the direct ozone reaction mechanisms (cyclo addition, electrophilic and nucleophilic reactions) are not feasible, decomposition of these perfluoro compounds is expected to proceed via radical chain reactions. Analyzing the publications on photocatalytic degradation of PFS, Wang et al. [3] concluded that the direct photolysis of the PFS compounds does not play a role in the decomposition reaction, as there is only little UV absorption by PFS at wavelength > 220 nm.

Degradation of PFOS under Ar and He in R2 at 200 W plasma power (0.4 L solution with the initial concentration of 10 mg/L and the circulation rate of 1 L/min; the reaction time is corrected for the burst mode)
The performance of plasma treatment of PFS-containing water samples in terms of reaction kinetics, and consequently, degradation efficiency is much more promising, As almost complete decomposition was observed after 1 h treatment (cf. Fig. S1 in supporting information). Thus, following the plasma treatment of PFOS with DBD and pulse-corona reactors will be further discussed.
DBD Plasma Reactors
For a systematic investigation of the plasma treatment, a larger DBD reactor (R2) with 0.4 L solution capacity was constructed that possesses an expanded falling film area, resulting in a longer residence time of solution in the reactor. Furthermore, in this reactor the gas space was reduced to ensure an efficient gas exchange. To compare the efficiency of the DBD and PCD reactors, the experiments were limited to PFOS decomposition, since it is proved that PFOS is more resistant against decomposition. The initial concentration of PFOS was increased to 10 mg/L to be able to study the degradation products well. Figure 3 shows the concentration profiles of PFOS in the improved DBD reactor (R2) under helium and argon plasma gases. After 30 min treatment by DBD plasma, more than 95% of PFOS was decomposed. The decrease in the PFOS concentration follows the first-order kinetics with the rate constants of 0.09 min−1 and 0.11 min−1 using argon and helium, respectively.
Different decomposition mechanisms for the treatment of PFS by plasma technology have been discussed. Hayashi et al. [16] proposed the removal of sulfo group (SO3H−) as the first steps of PFOS degradation, which is followed by consecutive decarboxylation and shortening of the perfluoroalkyl chain owing to the reaction with positively charged ions generated by the plasma (M+), according to the Eqs. 2–7. Consequently, shorter perfluoroalkyl acids as secondary by-products of PFOS decomposition are formed in the solution. Moreover, the C–C bonds cleavage by UV-irradiation, generated by the plasma discharge, may contribute to the decomposition of PFS.
and so on:
A look at the concentration profiles (Fig. 4) of the shorter PFS, which are detected as intermediates, proves the assumed step-by-step decarboxylation mechanism of PFOS. The concentration of PFOA, as the first reaction by-product, rises and reaches to the peak value after approx. 15 min of the treatment when subsequently the consumption and decomposition of PFOA to perfluoroheptanoic acid (PFHpA) and perfluorohexanoic acid (PFHxA) is exceeding its formation rate from PFOS decay. At this point, the differences between Ar and He gases as plasma atmosphere are more pronounced not only by reducing the concentration of PFOS (shown in Fig. 3) but this is also reflected in the fluoride recovery that was 25% after 30 min treatment by plasma in Ar, while it was only 14% in He. In conclusion, it seems that the degradation in argon results in a deeper mineralization than in helium.

Development of intermediate concentrations during the degradation of PFOS (the reaction time is corrected for the burst mode; for the operational conditions see Fig. 3)
Looking at the absolute amounts of the analyzed secondary by-products indicates that roughly one-tenth (10%) of the initial PFOS concentration can be traced and explained by this reaction mechanism. The concentration of fluoride, determined by ion chromatography, along with the LC/MS analysis show a much deeper mineralization degree than it was expected by considering only the concentration profiles of the formed by-products of PFS degradation.
To benefit from the surface active properties of the PFS, the third DBD reactor (R3) was developed in which the liquid was not flowed as a falling film, where the surfactant cannot enrich at the surface, but as a trough with a larger liquid volume (ca. 8 L). The intention of this approach is that the surfactants accumulate at the water surface and the plasma can impact on a concentrated PFS layer. The results of 1 and 10 mg/L PFOS degradation are shown in Fig. 5. For the degradation of 10 mg/L PFOS, a direct comparison with the falling film reactor (R2, Fig. 3) is possible. For 10 mg/L PFOS degradation, a direct comparison with the falling film reactor (R2, Fig. 3) is possible. Considering the larger sample volume of R3 with 8 L, compared to 0.4 L in the falling film reactor R2, the amounts of decomposed PFOS in both reactors are comparable. Obviously, the applied liquid circulation flow rate prevents an effective accumulation of the surfactant on the liquid surface.

PFOS degradation (1 and 10 mg/L in 8 L solution) in R3 at 200 W plasma power under He atmosphere and a liquid circulation rate of 1 L/min
Nano-pulse Corona Reactors
To the best of our knowledge, no direct comparison between the DBD and the pulse corona plasma for decontamination of water has been reported. Comparing with the DBD reactor, the developed nano-pulse corona reactor has the following advantages:
-
No need to operate in the burst mode due to applying short (ca. 20 ns) pulses, which prevents thereby the formation of sparks
-
Generating a much more stable plasma without the risk of sparks generation, even under oxygen atmosphere
-
The continuous recharging of the dielectric in the DBD reactor leads to heating the dielectric up, which consequently results in the energy losses.
Therefore, it was of special interest to examine the performance of a nano-pulse reactor in the degradation of PFS. The treatments of a PFOS solution with an initial concentration of 10 mg/L under argon and helium plasma gases are compared in Fig. 6. While in the DBD reactor the PFOS degradation in He is slightly preferred (cf. Fig. 3), in the pulse corona reactor the situation seems to be conversed.

Normalized degradation of PFOS (nominal initial concentration 10 mg/L) and recovery of fluoride in the nano-pulse corona reactor (R4) at ca. 110 W in He and 130 W in Ar (the relative fluoride concentration represents the ratio of fluoride recovery to the expected total fluoride content of the solution)
The trend toward a deeper mineralization under argon gas, as expressed for the fluoride recovery, is explicitly pronounced for both of the DBD and pulse corona reactors, and the fluoride recovery significantly exceeds the recovery of sulfate. This suggests that the removal of the sulfonate group is not the first step in PFOS decomposition as oftentimes is proposed. The first attack is probably on the perfluoroalkyl chain, which is accumulated at the interface and directed towards the gas phase. In addition, Obo et al. [6] postulated that some non-identified degradation by-products are still containing the sulfonate group, which may be the reason for loss of fluorine mass balance. However, further sulfonates, as PFOS decomposition by-products, were not identified by LC/MS analyses. Actually, trace amounts of perfluorohexanesulfonic acid (PFHxS) and perfluorobutanesulfonic acid (PFBS), which were present in the initial PFOS source as impurities, undergo the same decomposition route during the plasma treatment.
The results obtained by the PCD reactor are compared with those of the DBD reactors in Table 3. The energy yields obtained by the PCD reactor, which are influenced by not only the plasma power but also the initial concentration, liquid volume, and the degradation rate, are significantly higher than those obtained by the DBD reactors.
Based on the promising results of R4, a second PCD reactor (R5) was developed in which the ratio between the solution surface and its volume as well as the plasma area is significantly larger than in R4, although the number of HV pins remains unchanged. Figure 7 compares decomposition of 1 mg/L PFOS under He atmosphere in R4 and R5 reactors at comparable conditions. The results, which are reported in Table 3, indicate that the degradation of PFOS in R5 is significantly faster than in R4. The pseudo-first order rate constant obtained by R5 is more than four times larger than that obtained by R4, although the volume of sample in two reactors does not differ significantly (5 L in R5 compared to 7.5 L in R4).

Normalized degradation of PFOS (nominal initial concentration 1 mg/L, in He atmosphere) in R4 at ca. 100 W and in R5 at ca. 90 W
Obviously, the design of R5 provides a better interaction of the plasma with PFS than it is in R4. The main parameters that influence the performance of the PCD reactor seem to be the ratio of the surface of water under the discharge to the solution volume, which is affected by the density of HV pins. If the pins are too close to each other, their sphere of influence may overlap. Further studies are required to find the optimal conditions of PFS degradation by a nano-PCD reactor.
Using direct current plasma generated within bubbles in water, Obo et al. [6] obtained an efficient degradation of PFOS. Applying a 120 W plasma input power for degradation of a PFOS solution with an initial concentration of 50 mg/L they found approx. 50% degradation by 150 min treatment. Observing these data, one can estimate an energy yield G50 of 80 mg PFOS/kWh. The energy yield is a parameter that is dependent on the initial concentration of solution (cf. Eq. 1). Considering the lower initial concentration of PFOS in the present study ([PFOS]0 = 10 mg/L), the energy yield (G50) of the nano pulse corona reactor (R4) is significantly higher (ca. 200 mg PFOS/kWh) than that found by Obo et al.
On the other hand, Stratton et al. [7] developed an efficient plasma-based water treatment (PWT) process using a laminar jet with bubbling (LJB) reactor by which treated a PFOA solution at comparable conditions (C0 = 8.28 mg/L, 77 W) and attained a higher degradation efficiency with an energy yield of about 500 mg PFOA/kWh. In a high removal efficiency mode at only 4.1 W they obtained even higher efficiencies.
The degradation of PFSs is also possible by photocatalytic reactions, especially by the photocatalytic ozonation [3]. However, a direct comparison between the results is difficult, as the operational conditions vary significantly. As an example, Huang et al. [17] reported on the degradation of PFOA by the photocatalytic ozonation using TiO2 photocatalyst. From the reported data, one can estimate an energy yield of 360 mg/kWh for the treatment of a PFOA solution with an initial concentration of 10 mg/L; however, this value does not take the energy consumption of ozone production into the account.
To elucidate the influence of dissolved salts and organic compounds on the decomposition of PFS, a groundwater (GW) sample spiked with PFOS and PFOA was treated in the PCD R5. The concentrations of dissolved ions are given in the supplementary information (Table S1). Organic substances such as humic acids that might act as radical quencher are oxidized in the plasma. The chemical oxygen demand (COD) of this water sample is 120 mg/L and the conductivity is 631 µS/cm. The degradation of PFOS in the surface water sample is only slightly retarded in comparison with the experiments done in deionized water. The results are summarized in Table 3. Similar results were also obtained by Stratton et al. [7] using their PWT process for the decontamination of groundwater samples containing 0.2–3.1 nM PFS.
The presence of other compounds in the water may compete with PFS molecules to react with reactive species. Nevertheless, it seems that the nano-pulse corona plasma is an effective way to remove PFS not only from synthetic samples, but also from real polluted water.
Conclusion
It has been shown that, in comparison with the ozonation and photocatalytic ozonation processes, the non-thermal plasma is the preferred method to decompose PFSs in water. The comparison of various NTP reactors indicates obvious advantages of the nano-pulse corona discharge reactor over the DBD reactor. Both the energy yield and the degree of mineralization, especially using argon gas as the plasma atmosphere, are promising for further studies. Comparing with the results of other studies (cf. 3.3.), in which different reactor designs and plasma generations have been applied, underlines the compelling necessity of optimizing the NTP generation before any practical application. The advantages of a NTP water treatment are the absence of any chemical addition and the possibility to reach an extensive mineralization at moderate conditions. Nevertheless, the other issue that must be taken into account is the costs of electric equipment and the control systems. It is, therefore, necessary to analyze for each case whether and what NTP water treatment is suitable to solve the Problem.
References
Houde M, Martin JW, Letcher RJ, Solomon KR, Muir DC (2006) Biological monitoring of polyfluoroalkyl substances: a review. Environ Sci Technol 40(11):3463–3473
Merino N, Qu Y, Deeb RA, Hawley EL, Hoffmann MR, Mahendra S (2016) Degradation and removal methods for perfluoroalkyl and polyfluoroalkyl substances in water. Environ Eng Sci 33:615–649
Wang S, Yang Q, Chen F, Sun J, Luo K, Yao F, Wang X, Wanga D, Li X, Zeng G (2017) Photocatalytic degradation of perfluorooctanoic acid and perfluorooctane sulfonate in water: a critical review. Chem Eng J 328:927–942
Jiang B, Zheng J, Qiu S, Wu M, Zhang Q, Yan Z, Xue Q (2014) Review on electrical discharge plasma technology for wastewater remediation. Chem Eng J 236:348–368
Magureanu M, Bradu C, Parvulescu VI (2018) Plasma processes for the treatment of water contaminated with harmful organic compounds. J Phys D Appl Phys 51:313002
Obo H, Takeuchi N, Yasuoka K (2015) Decomposition of perfluorooctanesulfonate (PFOS) by multiple alternating argon plasmas in bubbles with gas circulation. Int J Plasma Environ Sci Technol 9:62–68
Stratton GR, Dai F, Bellona CL, Holsen TM, Dickenson ERV, Mededovic-Thagard S (2017) Plasma-based water treatment: efficient transformation of perfluoroalkyl substances in prepared solutions and contaminated groundwater. Environ Sci Technol 51:1643–1648
Mededovic-Thagard S, Stratton GR, Dai F, Bellona CL, Holsen TM, Bohl DG, Eunsu P, Dickenson ERV (2017) Plasma-based water treatment: development of a general mechanistic model to estimate the treatability of different types of contaminants. J Phys D Appl Phys 50:1–13
Hama Aziz KH, Miessner H, Mueller S, Kalass D, Moeller D, Khorshid I, Rashid MAM (2017) Degradation of pharmaceutical diclofenac and ibuprofen in aqueous solution, a direct comparison of ozonation, photocatalysis, and non-thermal plasma. Chem Eng J 313:1033–1041
Hama Aziz KH, Miessner H, Mueller S, Mahyar A, Kalass D, Moeller D, Khorshid I, Rashid MAM (2018) Comparative study on 2,4-dichlorophenoxyacetic acid and 2,4-dichlorophenol removal from aqueous solutions via ozonation, photocatalysis and non-thermal plasma using a planar falling film reactor. J Hazard Mater 343C:107–115
Hama Aziz KH, Mahyar A, Miessner H, Mueller S, Kalass D, Moeller D, Khorshid I, Rashid MAM (2018) Application of a planar falling film reactor for decomposition and mineralization of methylene blue in the aqueous media via ozonation, Fenton, photocatalysis and non-thermal plasma: a comparative study. Process Saf Environ 113:319–329
Malik MA (2010) Water purification by plasmas: which reactors are most energy efficient? Plasma Chem Plasma Process 30:21–31
Simister EA, Lee EM, Lu JR, Thomas RK, Ottewill RH, Rennie AR, Penfold J (1992) Adsorption of ammonium perfluorooctanoate and ammonium decanoate at the air/solution interface. J Chem Soc Faraday Trans 88:3033–3041
Zhang L, Wu P, Wei Q, Shen JW, Liu Z, Ren T, Zhang W, Wang X (2016) The effect of spacer on the structure of surfactant at liquid/air interface: a molecular dynamics simulation study. J Mol Liq 222:988–994
Lin AYC, Panchangam SC, Chang CY, Hong PKA, Hsueh HF (2012) Removal of perfluorooctanoic acid and perfluorooctane sulfonate via ozonation under alkaline condition. J Hazard Mater 243:272–277
Hayashi R, Obo H, Takeuchi N, Yasuoka K (2015) Decomposition of perfluorinated compounds in water by DC plasma within oxygen bubbles. Electr Eng Jpn 190(3):9–16
Huang J, Wang X, Pan Z, Li X, Ling Y, Li L (2016) Efficient degradation of perfluorooctanoic acid (PFOA) by photocatalytic ozonation. Chem Eng J 296:329–334
Acknowledgements
This work was funded by the German Federal Ministry of Economic Affairs and Energy (BMWi, Grant Number ZF4296101CR6). Our special thanks go to Vladimir Efanov and FID GmbH for providing the nano-pulse generator.
Author information
Authors and Affiliations
Corresponding author
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Electronic Supplementary Material
Below is the link to the electronic supplementary material.
Rights and permissions
About this article

Cite this article
Mahyar, A., Miessner, H., Mueller, S. et al. Development and Application of Different Non-thermal Plasma Reactors for the Removal of Perfluorosurfactants in Water: A Comparative Study. Plasma Chem Plasma Process 39, 531–544 (2019). https://doi.org/10.1007/s11090-019-09977-6
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s11090-019-09977-6