
Abstract
Model alloys Ni–(5, 10, 20, 25, 30 wt%)Cr were exposed to Ar–20CO2 and Ar–20O2 at 700 °C. Samples were electropolished before reaction to avoid cold-working effects and, as a result, to reveal the effect of alloy bulk diffusion. All alloys underwent breakaway oxidation in CO2, forming an external Ni-rich oxide scale and other oxides internally. In oxygen, the Ni–(5, 10, 20)Cr alloys formed a nonprotective Ni-rich scale plus an internal oxidation zone, but Ni–(25, 30)Cr formed a protective Cr2O3 scale. Carbide precipitates were found in Ni–20Cr reacted in CO2. It is shown that Wagner’s diffusion theory accounts satisfactorily for the difference in alloy scaling behavior in the two gases.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Technological solutions are being developed to reduce CO2 emission from power plants burning fossil fuels. Recently, a promising technology, oxyfuel combustion, has been tested in laboratories and industry to verify its efficiency. In this process, fuels are burned in pure oxygen mixed with recirculated flue gas instead of air. The resulting flue gas, rich in CO2 and water vapor, offers a simpler and cheaper way to capture CO2 gas. Selecting suitable materials for heat exchangers to handle this hot gas mixture is a principal challenge because it is so corrosive.
Corrosion resistance of chromium-containing alloys usually depends on formation of a chromia scale. This protective scale on steels, however, was found to grow significantly faster in CO2 than in dry air, resulting in greater chromium depletion in the underlying alloy [1]. Consequently, critical Cr concentrations to form and maintain Cr2O3 scale growth on Cr-bearing steels are higher in CO2 corrosion [1]. Thus, Fe–9Cr and Fe–20Cr–20Ni alloys formed a thin, protective Cr2O3 scale in dry air at 818 °C, but a thick iron-rich oxide scale in CO2. As the higher temperatures are needed for increased efficiency, Ni-based alloys are required for their superior creep strength [2, 3].
The aim of the present study was to investigate the effect of Cr concentration on the corrosion resistance at 700 °C of Ni–Cr model alloys in Ar–20CO2 and, for comparison, Ar–20O2. To allow subsequent interpretation using alloy bulk diffusion coefficients, cold worked surface regions were removed by electropolishing.
Experimental Procedures
Five model alloys Ni–(5, 10, 20, 25, 30 wt%)Cr were prepared by induction melting, using pure metals Cr (99.995%) and Ni (99.95%). The alloy grain size after annealing was 2.6 ± 2 mm. Rectangular alloy coupons with dimensions of (1.5 ± 0.4) mm × (7.9 ± 2) mm × (14.5 ± 0.5) mm were surface ground to a 1200-grit finish, electropolished to remove deformed surface material, and then ultrasonically cleaned in alcohol prior to reaction.
All specimens were reacted at 700 °C in an Ar–20CO2 or Ar–20O2 (vol%) mixture with a linear flow rate of 2 cm s−1 and a total pressure of 1 atm. After reaction, the specimens were moved out of the furnace and cooled rapidly under Ar gas. Weight changes were measured using an analytic balance (Precisa 180A) with an accuracy of 0.1 mg. In kinetic plots shown here, each point represents a separate sample. Corrosion products were characterized by X-ray diffractometry (XRD; PANanalytical Xpert MPD) with Cu-Kα radiation and scanning electron microscopy (SEM, Hitachi S3400; acceleration voltage 15 kV) with an energy dispersive X-ray spectrometer (EDX, Bruker). The etchant used to reveal carbides was HNO3: HCl = 2 ml: 10 ml at room temperature.
Results
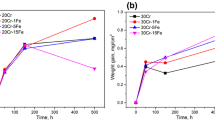
Weight gain kinetics for Ni–Cr alloys in dry CO2 gas are shown in Fig. 1a. Weight uptakes of Ni–(5, 10)Cr increased approximately parabolically with time, and those of Ni–5Cr were larger. The Ni–30Cr alloy had the slowest weight gain rate, followed by Ni–25Cr. Weight uptakes of Ni–20Cr were close to those of Ni–10Cr. No apparent scale spallation was observed for any alloy in CO2 gas. Extents of reaction after 500 h in CO2 and O2 are compared in Fig. 1b. Weight gains of Ni–(10, 20)Cr were larger, but those of Ni–(25, 30)Cr were significantly smaller in oxygen. Scale spallation after reaction in oxygen was extensive for Ni–5Cr, but limited for Ni–10Cr. Scales grown on Ni–(20, 25, 30)Cr in oxygen remained intact during cooling.

a Weight gain kinetics of Ni–(5, 10, 20, 25, 30)Cr in Ar–20CO2 and b weight gains of Ni–Cr alloys exposed to Ar–20CO2 and Ar–20O2 for 500 h
In dry CO2, Ni–(5, 10, 25)Cr alloy formed a Ni-rich oxide scale after 20 h (not shown). Cross sections of Ni–(5, 10)Cr alloys after reaction for 500 h in dry CO2 are shown in Fig. 2a, b. The porous scales were thick and identified by XRD and EDX analyses (not shown) as consisting of an outer NiO layer and an inner (Ni, Cr) oxide layer. An internal oxidation zone (IOZ) containing Cr-rich oxide precipitates was observed beneath the scales (Fig. 2c, d). The IOZ of Ni–5Cr was wider than that of Ni–10Cr.

BSE-SEM cross sections of a Ni–5Cr and b Ni–10Cr reacted in Ar–20CO2 for 500 h. High-magnification images of IOZs of c Ni–5Cr and d Ni–10Cr
Unlike Ni–(5, 10)Cr, the Ni–20Cr alloy formed a scale of NiO alone, with an underlying IOZ in CO2 (Fig. 3a, b). The IOZ consisted of a layer containing a very finely distributed precipitates (IOZ 1) beneath the scale, and a second layer containing somewhat coarser precipitates (IOZ 2). Both IOZ layers contained Cr-rich oxide precipitates. Particles of almost pure Ni were distributed randomly at the scale-IOZ interface.

a BSE-SEM cross section of Ni–20Cr reacted in Ar–20CO2 for 500 h and b high-magnification image of IOZ
The Ni–30Cr alloy formed a Cr2O3 scale (Fig. 4a–c) and thicker nodules after 20 h in CO2 (Fig. 4a). The nodule was found by EDX to consist of an outer NiO layer and an inner Cr-rich oxide layer (Fig. 4b). When the reaction continued to 500 h, this alloy formed a scale made up of an outer NiO layer, an intermediate (Ni, Cr) oxide layer, and an inner Cr2O3 layer (Fig. 5). Scales on Ni–25Cr after 500 h in CO2 (not shown) had the same structure as those shown in Fig. 5 for Ni–30Cr, but a thinner chromia layer.

a BSE-SEM cross section of Ni–30Cr reacted in Ar–20CO2 for 20 h, b EDX line profiles along A–B in (a) and c high-magnification image of the thin scale in (a)

BSE-SEM cross section of Ni–30Cr reacted in Ar–20CO2 for 500 h
Reaction in CO2 resulted in carburization of only one alloy, Ni–20Cr, which formed intergranular Cr-rich carbide precipitates beneath the scale (Fig. 6).

a Intergranular carbides in Ni–20Cr (etched) after reaction in Ar–20CO2 for 500 h, b high-magnification image of carbide precipitates and c EDX line profiles along A–B in (a)
In oxygen, Ni–(5, 10)Cr formed a uniform, thick scale made up of an outer NiO layer and an inner (Ni, Cr) oxide layer (Fig. 7a, b). Beneath the scale was a narrow IOZ (Fig. 7c, d). The Ni–20Cr alloy formed the same scale as Ni–(5, 10)Cr, but its IOZ width varied across the surface (Fig. 8a). The Ni–(25, 30)Cr alloys formed a uniform Cr2O3 scale (Fig. 8b).

BSE-SEM cross sections of a Ni–5Cr and b Ni–10Cr reacted in Ar–20O2 for 500 h. High-magnification images of IOZs of c Ni–5Cr and d Ni–10Cr

BSE-SEM cross sections of a Ni–20Cr and b Ni–30Cr reacted in Ar–20O2 for 500 h
Scale growth kinetics for Ni–(5, 10, 20)Cr are shown in Fig. 9. Plotted values are averages, and standard deviations are shown for measurements after 500 h of reaction. The inner scale grown on Ni–20Cr in O2 was extremely variable, and data are not plotted. After 500 h of reaction, the maximum inner layer thickness was ~11 µm and the minimum ~2 µm. The scales formed in oxygen were thicker than in CO2. Both outer and inner scales on Ni–5Cr grew faster than those on Ni–10Cr and Ni–20Cr in both gases. Assuming the NiO–(Ni, Cr) oxide interface is the original surface, internal oxide penetration depths measured from this original surface are shown in Fig. 10. Depths of internal oxide precipitation were in order of relative size, Ni–10Cr < Ni–20Cr < Ni–5Cr for oxidation in CO2, and Ni–20Cr < Ni–10Cr < Ni–5Cr in oxygen.

Average thicknesses of a outer and b inner scales on Ni–(5, 10, 20)Cr in Ar–20CO2 and Ar–20O2

Average penetration depths of IOZs measured from the original alloy surface of a Ni–(5, 10)Cr and b Ni–20Cr in Ar–20CO2 and Ar–20O2
Discussion
Effect of Chromium Concentration on Corrosion Resistance in CO2 and O2
The Ni–(5, 10, 20)Cr alloys underwent breakaway oxidation in both CO2 and O2 gases (Figs. 2, 3, 7, 8a). The higher Cr alloys, Ni–(25, 30)Cr, formed a Cr2O3 layer but did not stop NiO growth in CO2 (Fig. 5), while these alloys succeeded in forming a protective Cr2O3 in oxygen (Fig. 8b). Clearly, CO2 gas was more corrosive than oxygen. It should be noted that all these results reflect the behavior of bulk alloys, in the absence of surface deformation. However, if the Ni-based alloys had not been subjected to an electropolishing treatment, the results would be quite different. In that case, the presence of a surface deformation layer would lead to a fine grained structure which would accelerate alloy diffusion and hence chromia scale formation [4]. Failure of the Cr2O3 scale may be caused by insufficient Cr supply from the underlying alloy for scale growth. Wagner’s theory is now used to predict the ability of Ni–30Cr to form and maintain a protective chromia scale.
External chromia scale growth can only occur if the chromium level in the alloy is above a critical value, \(N_{\text{Cr}}^{(1)}\), required to favor external scaling over internally precipitated oxide [5]:
Here g is the volume fraction of oxide precipitate (generally approximated as 0.3 [6]), υ the stoichiometric coefficient of CrOυ, \(V_{\text{m}}\) = 7.2 cm3 mol−1 and \(V_{{{\text{CrO}}_{\upsilon } }}\) = 14.6 cm3 mol−1, the molar volumes of alloy and oxide, respectively, \(N_{\text{O}}^{{ ( {\text{S)}}}}\) the oxygen solubility (mole fraction) in the alloy in equilibrium with the oxygen activity at the scale-alloy interface,\(D_{\text{O}}^{{}}\) = 2 × 10−12 cm2 s−1 the diffusion coefficient of oxygen in the alloy [7], and \(\tilde{D}_{\text{Cr}}\) = 1.38 × 10−14 cm2 s−1 the alloy interdiffusion coefficient extrapolated from [8] for diffusion of Cr in Ni–35Cr.
For dissolution of oxygen in the alloy:
and the value of \(N_{\text{O}}^{{({\text{S}})}}\) is calculated from Sievert’s equation:
Here K is the equilibrium constant for (3) and available from [9], and \(P_{{{\text{O}}_{2} }}\) is set by the interfacial equilibrium
leading to a value of \(N_{\text{O}}^{{ ( {\text{S)}}}}\) = 9.4 × 10−5 at 700 °C.
In addition, the alloy must provide sufficient Cr supply via alloy diffusion to the scale–alloy interface to maintain exclusive chromia scale growth. This criterion leads to the requirement [10]:
if the scale is pure Cr2O3. Here \(k_{\text{p}}\) is the parabolic rate constant for chromia thickening
with X the scale thickness and t time. Chromia scale thicknesses on Ni–30Cr reacted for 20 h in CO2 (Fig. 4c) and 500 h in oxygen (Fig. 8b) were used to estimate \(k_{\text{p}}\) values, as 4.6 × 10−15 cm2 s−1 (CO2) and 1.8 × 10−15 cm2 s−1 (oxygen).
The calculated critical values are \(N_{\text{Cr}}^{ ( 1 )}\) = 0.046, and \(N_{\text{Cr}}^{(2)}\) = 0.357 for CO2 and 0.223 for oxygen. Thus, all alloys tested (maximum Cr 30 wt%) are predicted to fail via NiO formation in CO2, just as observed. However, for reaction in O2, the theory indicates Ni–(25, 30)Cr should form and maintain a scale of Cr2O3 alone, again, as was observed. Thus, all predictions are in agreement with the experimental results, indicating that the difference is accounted for simply by the faster scaling induced by CO2.
Internal Oxidation in CO2 and O2
The relative position of the original alloy surface with respect to the inner/outer scale layer interface is of interest. Oxidation of Ni–1Cr in air at 1000 °C [11] formed an outer NiO scale, an inner (Ni, Cr) oxide scale and an IOZ-containing Cr2O3 precipitates. Inert marker experiments for this alloy [11] showed that the outer layer grew by outward diffusion of Ni and the inner layer by inward transport of oxygen. Thus, the original alloy surface may be suggested to correspond approximately to the outer/inner scale layer interface.
The scales and IOZs of Ni–(5, 10, 20)Cr alloys grew according to approximately parabolic kinetics (Figs. 9, 10), showing diffusion controlled growth. These alloys formed an outer NiO scale, an inner (Ni, Cr) scale and an IOZ in both gases (Figs. 2, 7), except Ni–20Cr in CO2 (Fig. 3). The double-layered IOZ of Ni–20Cr in CO2 is spectacular and will be investigated in future. In the other cases, the IOZ was partly consumed by inner scale growth, while new internal oxide precipitates were formed simultaneously at the IOZ front during the scale growth. Rates of internal oxidation beneath an external scale are described by [12].
Here X i and X c are distances of the IOZ front and inner scale–alloy interface measured from the original alloy surface (Figs. 9, 10). The function F(u) is defined as
Experimental results from Fig. 10 show that u ≫ 1, consequently F(u) ≈ 1. Equation (7) is then simplified as
According to Eq. (10), if \(N_{\text{O}}^{{ ( {\text{S)}}}} D_{\text{O}}^{{}}\) values are the same for all alloys, X i (X i − X c ) is inversely proportional to \(N_{\text{Cr}}^{{ ( {\text{O)}}}}\) at time t. Experimental results in Table 1 indicate that X i (X i − X c ) does increase as alloy Cr concentration decreases from 10 to 5% in CO2, but is not proportional to the inverse of \(N_{\text{Cr}}^{{ ( {\text{O)}}}}\) and is unexpectedly high for Ni–20Cr. Moreover, the calculation fails entirely in the oxygen case. There are several likely reasons for this mismatch. Firstly, \(N_{\text{O}}^{{ ( {\text{S)}}}}\) varies with the scale–alloy interfacial equilibrium; secondly, \(D_{\text{O}}^{{}}\) is variable because of effect of internal precipitate number density and shape on interfacial diffusion of oxygen; finally, the IOZ in Ni–20Cr reacted with CO2 is unlike any other.
Analysis of Ni–20Cr reacted in CO2 by TEM and SAD (selected area diffraction) (not shown) confirms that the IOZ 1 (Fig. 3b) consisted of Cr2O3 + NiCr2O4 + Ni, but Cr2O3 + Ni for IOZ 2. The growth of a 3-phase zone is thermodynamically impossible for the ternary Ni–Cr–O system because it is compositionally invariant. However, carbon together with oxygen from CO2 could penetrate into the alloy, forming a new Ni–Cr–O–C system, in which a 3-phase IOZ could develop. No such zone formed when this alloy reacted with O2 to form a true ternary system.
Conclusions
The oxidation behavior in CO2 of model Ni–Cr alloys has been determined. To reveal the effect of alloy bulk diffusion, all samples were electropolished to remove cold worked surface regions. The experimental results showed that CO2 gas was more corrosive than dry oxygen at 700 °C. Wagner’s theory is successful in explaining the effect of Cr concentration on corrosion resistance. The higher scaling rate in CO2 required higher alloy chromium concentrations to maintain Cr2O3 scale growth than in oxygen. It is predicted that 25 wt% Cr is sufficient for reaction in oxygen, but even 30 wt% Cr is not enough in CO2. Diffusion analysis was used to evaluate internal oxidation kinetics as a function of Cr concentration, but failed in both cases. The underlying assumption that oxygen permeability is independent of alloy composition is incorrect, and more detailed analysis of boundary values and interfacial diffusion is required.
References
T. D. Nguyen, J. Zhang and D. J. Young, Corrosion Science 76, 2013 (231).
V. Viswanathan, R. Purgert and P. Rawls, Advanced Materials and Processes 166, (8), 2008 (47).
S. J. Patel, J. J. deBarbadillo, B. A. Baker, and R. D. Gollihue, Procedia Engineering 55, 246 (2013).
H. V. Atkinson, Oxidation of Metals 24, 1985 (177).
C. Wagner, Zeitschrift Elektrochemie 63, 1959 (772).
R. A. Rapp, Acta Metallurgica et Materialia 9, 1961 (730).
R. Barlow and P. J. Grundy, Journal of Materials Science 4, 1969 (797).
J. Askill, Physica Status Solid 8, 1971 (587).
D. J. Young, High Temperature Oxidation and Corrosion of Metals, (Elsevier, UK, 2008).
C. Wagner, Journal of the Electrochemical Society 99, 1952 (369).
C. K. Kim and L. W. Hobbs, Oxidation of Metals 45, 1996 (247).
F. Maak, Zeitschrift für Metallkunde 52, 1961 (538).
Acknowledgement
Financial support from the Australian Research Council’s Discovery Program is gratefully acknowledged.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article

Cite this article
Nguyen, T.D., Xie, Y., Ding, S. et al. Oxidation Behavior of Ni–Cr Alloys in CO2 at 700 °C. Oxid Met 87, 605–616 (2017). https://doi.org/10.1007/s11085-017-9721-9
Received:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s11085-017-9721-9