
Abstract
The first principle methods are employed to determine the electronic properties, half-metallic ferromagnetism and exchange splitting in Ba1−xTixO compound based on titanium (Ti)-doped BaO at concentration x = 0.125. The generalised gradient functional of Wu and Cohen is utilized to characterize the structural parameters of both BaO and Ba0.875Ti0.125O materials, whereas the Tran-Blaha-modified Becke-Johnson potential is used to calculate the magnetic properties and electronic structures with perfect band gaps. The variations of lattice constants and bulk modulus of Ba0.875Ti0.125O were investigated. In Ba0.875Ti0.125O, the ferromagnetic arrangement is mainly endorsed by the direct exchange splitting. The Ba0.875Ti0.125O compound is true half-metallic ferromagnetic and 100% spin polarization with a half-metallic gap of 0.803 eV. Consequently, the Ba0.875Ti0.125O can be considered as potential material for spintronics.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
1 Introduction
Spintronics is an attractive field of research into spin-based physical phenomena and electronics, it is known as spin electronics because the spin of electron plus is used to improve the performance of spin-based electronic devices (Wolf et al. 2001; Liu et al. 2019). Recently, the diluted magnetic semiconductors (DMS) are realized from the rare earth and transition metal atoms doped semiconductors, they have been widely investigated as promising candidates for several spintronics applications due to their intermediate properties, which combine magnetic and semiconductor behaviours (Pan et al. 2008; Poornaprakash et al. 2020; Doumi et al. 2013). The half-metallic ferromagnets DMS have been considered an important class of materials for spintronics applications because their electronic structures reveal a semiconductor feature in one spin directions and a metallic character in the other spin channel (Hirohata et al. 2020; Korichi et al. 2020). However, spintronics utilizes both spin and charge of electron to generate new functionalities in spin-based devices. The advantages of spintronics devices with respect to the conventional electronics are the non-volatility, reduced energy consumption, high transistor density and fast data processing. (Kaminska et al. 2008; Goktas 2020; Khandy et al. 2019).
The alkaline-earth-metal binary oxides have been considered as potential materials for spintronics and optoelectronics applications (Kenmochi et al. 2004a, b; Albanese and Pacchioni 2017). The barium oxide (BaO) is one of the alkaline-earth-metal with wide band gap (Yang et al. 2016), it has several technological applications for example its use as a bumper layer on silicon in the epitaxial growth of perovskite oxides (Yang et al. 2016; McKee et al. 2001). The half-metallic ferromagnetic property was predicted in the new class of DMS based on C-doped MgO, SrO and BaO (Kenmochi et al. 2004a) and CaO doped with C and N atoms (Kenmochi et al. 2004b). Albanese and Pacchioni (2017) have investigated magnetic interaction of nitrogen N-doped BaO using first-principle approaches. The BaO is a potential candidate for spintronics according to first-principle study, revealing a half-metallic ferromagnetic behavior in the BaO under the effect of chromium (Cr) doping (Lakhdari et al. 2019).
In this work, we have studied the structural parameters, electronic properties, induced ferromagnetism, half-metallic behavior, exchange splitting and crystal field energy in the Ba1−xTixO compound with concentration x = 0.125 of titanium (Ti). The calculations were carried out by using the first-principle methods of full-potential linearized augmented plane-wave (FP-LAPW) approach (Singh and Nordstrom 2006) and density functional theory (DFT) (Hohenberg and Kohn 1964; Kohn and Sham 1965).
2 Method of calculations
The structural, magnetic properties and electronic structures of Ba1−xTixO were determined by utilizing the first-principle concepts of the FP-LAPW approach and the DFT implemented in WIEN2k code (Blaha et al. 2001). The structural properties of Ba1−xTixO at x = 0.125 and BaO compound are determined by the use of the GGA-WC functional (Wu and Cohen 2006), whereas the Tran–Blaha-modified Becke–Johnson exchange potential (TB-mBJ) combined with the local density approximation (Tran and Blaha 2009; Koller et al. 2011) is employed to compute the magnetic moments, exchange constants, exchange splitting, crystal field energy and electronic structures with perfect bands gaps of Ba0.875Ti0.125O.
In the interstitial sites of crystal, the wave functions have been extended to plane waves by means of a cut-off of kmax RMT = 8.0, where the kmax corresponds to the largest k vector in the plane wave and the RMT represents the mean radius of the sphere of muffin-tin. The charge density is Fourier extended up to Gmax = 14 (a.u.)−1, where Gmax corresponds to the largest vector in the Fourier expansion, while the maximum value of the partial waves inside the atomic sphere is lmax = 10. The core and valence states are separated by the cut-off of − 6 Ry. The integration of the Brillouin zone is achieved by employing the Monkhorst–Pack technique (Monkhorst and Pack 1976), where the meshes of (4 × 4 × 2) and (4 × 4 × 4) are employed respectively for Ba1−xTixO and BaO. The self-consistent is achieved by setting the convergence of the total energy at 0.1 mRy.
The calculations are based on the conventional BaO lattice with NaCl structure and space group of (\(Fm\overline{3}m\)) number 225, where the barium is located at the (0, 0, 0) position and oxygen at (0.5, 0.5, 0.5). We Ba8O8 supercell of 16 atoms is used to create the Ba7TiO8 supercell by the substitution of the Ba cationic site at the (0, 0, 0) position with the titanium (Ti) magnetic atom. Therefore, we get the Ba0.875Ti0.125O (x = 0.125) supercell with tetragonal structure and space group of P4/mmm number 123.
3 Results and discussions
3.1 Structural properties
We have determined the thermodynamic stability of Ba0.785Ti0.125O compound in the ordered NaCl (B1) structure from the energy of formation, which is given by the following expression (Bai et al. 2011; Doumi et al. 2015a):
where the Etotal(Ba8−zTizO8) is the minimum total energy of Ba8−zTizO8 per atom, while the minimum total energies per atom of bulks Ba, Ti and O correspond to the E(Ba), E(Ti) and E(O), respectively. In the case of the Ba0.785Ti0.125O material, z equal to 1, which refers to the substituted Ti atom in the Ba8−yTiyO8 structure. The calculated formation energy of − 5.12 eV is negative, which means that the Ba0.785Ti0.125O compound is thermodynamically stable and it can be synthesized experimentally.
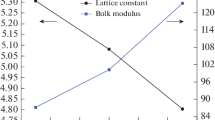
The lattice constants (a), bulk modules (B) and their pressure derivatives (B′) of BaO and Ba0.785Ti0.125O compounds are calculated by the fit of the empirical Murnaghan’s equation (Murnaghan 1944). The computed values of structural parameters are shown in Table 1 with other experimental values (Liu and Bassett 1972; Kaneko et al. 1982; PARK and Sivertsen 1977; Chang and Graham 1977) and theoretical results (Yang et al. 2016; Lakhdari et al. 2019; Nejatipour and Dadsetani 2015; Cinthia et al. 2015; Santana et al. 2016). We have noticed that there are changes in the values of lattice constant and bulk modulus of Ba0.875Ti0.125O doping material compared to the BaO. This is due to the difference in the sizes of ionic radii of Ba and Ti elements. However, the lattice constant of Ba0.785Ti0.125O decreases with respect to the BaO, leading to the increase of bulk modulus of Ba0.785Ti0.125O. Therefore, the Ba0.785Ti0.125O becomes harder than that BaO. The behaviours of these two parameters of Ba0.785Ti0.125O material were observed for CaO doped with Ti (Korichi et al. 2020) and BaO doped with chromium (Cr) (Lakhdari et al. 2019). It should be noted that the Ba0.785Ti0.125O material has not been studied theoretically and experimentally, so our calculations of structural properties of Ba0.785Ti0.125O cannot be compared to other studies. Besides, the calculated standard deviations of 0.089 and 0.066 Å respectively for BaO and Ba0.785Ti0.125O are very low, indicating that our results for the lattice parameters are very close to the average of the values of our calculations, other theoretical and experimental data.
Moreover, the results of a and B parameters of BaO are consistent to the experimental results (Kaneko et al. 1982; Liu and Bassett 1972; PARK and Sivertsen 1977) and theoretical calculations (Lakhdari et al. 2019) found by GGA-WC (Wu and Cohen 2006). Also, these parameters are better than the theoretical values (Yang et al. 2016; Lakhdari et al. 2019; Nejatipour and Dadsetani 2015; Cinthia et al. 2015; Santana et al. 2016) calculated with generalised gradient functional of Perdew-Burke-Ernzerhof (GGA-PBE) (Perdew et al. 1996) and the local density approach (LDA) (Perdew and Zunger 1981) with respect to the experimental values, meaning that the GGA-WC (Wu and Cohen 2006) provides good values for the structural properties due to its the performance for determining structural properties compared to other approximations (Wu and Cohen 2006; Doumi et al. 2015c; Sajjad et al. 2015; Bourega et al. 2019). The performance of the GGA-WC approximation for computing the structural parameters results from the fourth-order gradient expansion of exchange and correlation of its gradient functional (Wu and Cohen 2006; Doumi et al. 2015b, 2020).
3.2 Electronic structures
Firstly, we have determined the electronic properties of the BaO and Ba0.875Ti0.125O compounds, and then we have studied the effect of the titanium (Ti) magnetic atoms on the half-metallic property and the origin of ferromagnetic arrangement in Ba0.875Ti0.125O. Figures 1 and 2 show the band structures of BaO and Ba0.875Ti0.125O, respectively. The projected total and partial densities of states (DOS) of Ba0.875Ti0.125O are displayed by the Figs. 3 and 4 respectively. The Fig. 1 shows that there is a band gap around Fermi level because all electron states are bound to the valance band, which means that BaO bulk has semiconductor nature. The maximum of the valence band and the minimum of the conduction band occur at the same X high symmetry point, revealing direct gap (EXX) for BaO. For the Ba0.875Ti0.125O doping system, the band structures show different behavior compared to the BaO bulk due to the magnetic character of injected Ti atoms. The minority-spin bands keep the semiconductor feature, but the majority-spin bands become metallic as shown in Fig. 2.

Band structures for BaO

Spin-polarized band structures for Ba0.875Ti0.125S, a Majority spins (up) and b Minority spins (dn)

Spin-polarized total densities of states (DOS) of Ba, O and Ti atoms and total-DOS for Ba0.875Ti0.125O

Spin-polarized partial densities of Ti_d, Ti-d_t2g, Ti-d_eg, Ba_p and O_p states for Ba0.875Ti0.125O
From Fig. 3, the metallic character of majority spins results from the localized magnetic spins of 3d (Ti) states around EF, while the minority spins have unoccupied 3d (Ti) states sited at the lower part of the conduction band. Consequently, the Ba1−xTixO reflects a perfect half-metallic ferromagnetic behavior. The direct band gap of minority-spin bands located at the X high symmetry point is called the half-metallic ferromagnetic (HMF) gap (GHMF), whereas the minimum energy located between the Fermi energy (EF) (0 eV) and the maximum of valence band corresponds to the half-metallic (HM) gap (GHM). In the case of the minority spins of Ba0.875Ti0.125O material, the HM gap describes the absolute energy of maximum of valence band with respect to the EF. The Table 2 summarizes the calculated HM ferromagnetic band gap and HM band gap of minority spins of Ba0.875Ti0.125O and direct band gap (EXX) of BaO, with other theoretical (Yang et al. 2016; Lakhdari et al. 2019; Nejatipour and Dadsetani 2015; McLeod et al. 2010; Teli and Sirajuddeen 2019) and experimental (Zollweg 1958; Saum and Hensley 1959; Kaneko and Koda 1988) values. The Ba0.875Ti0.125O material has a ferromagnetic half-metallic gap (GHMF) and a half-metallic gap (GHM) of 2.701and 0.803 eV, respectively. The Ba0.875Ti0.125O has direct half-metallic ferromagnetic gap (GHMF) positioned at \(\Gamma\) point, although the half-metallic gap (GHM) arises between the Fermi level and maximum of valence bands. The direct wide gap of 3.5 eV of BaO calculated with the TB-mBJ is more improved with respect to the theoretical values (Yang et al. 2016; Lakhdari et al. 2019; Nejatipour and Dadsetani 2015; McLeod et al. 2010; Teli and Sirajuddeen 2019) found by the GGA-WC (Wu and Cohen 2006), GGA-PBE (Perdew et al. 1996) and LDA (Perdew and Zunger 1981). The better gap of BaO is due to the performance of TB-mBJ for predicting values of band gaps of semiconductors and insulators (Koller et al. 2011; Bhattacharjee and Chattopadhyaya 2017; Chattopadhyaya and Bhattacharjee 2017).
The Ba0.875Ti0.125O material is doped by the titanium (Ti) transition atom, which is characterized by the 3d electronic levels localized in the gap around EF of majority spins as seen in Fig. 4. The main part of the total-DOS of majority spins at EF is originated from the contributions of the Ba and Ti atoms. The metallic nature of majority spins results from the p-d hybridization of electronic states of 3d of titanium and p of oxygen as shown in Fig. 4. In addition, the 3d levels of titanium are splitted into t2g partially occupied states and eg empty states due to the octahedral crystal field environment originated form neighbouring oxygen ions. From Fig. 3, we can see clearly that the total-DOS is metallic for the majority spins but a semiconductor character occurs around EF for the minority spins. Consequently, the Ba0.875Ti0.125O material has half-metallic ferromagnetic feature and it can be considered as a potential compound for spintronics applications.
3.3 Magnetic properties
3.3.1 Magnetic moments
The origin of magnetism in the Ba0.875Ti0.125O compound is due to the localization of the partially occupied 3d states of Ti of the majority spins around EF. The Fig. 4 of majority spins shows that the eg states are unoccupied, while the t2g states are two-thirds filled. Thus, the t2g levels are partially occupied by two electrons, which induce a total magnetic moment of 2 μB. The results of computed total and local magnetic moments of Ba0.875Ti0.125O compound are given in Table 3. We have found that the magnetic moment of Ti of 1.778 μB is reduced below the value of 2 μB and little moments are induced at the Ba, O sites. The negative moment of O atom indicates that the interaction between the (Ba, Ti) and O magnetic spins is anti-ferromagnetic. The Ba and Ti positive moments generate ferromagnetic interaction.
3.3.2 Exchange couplings
The exchange interactions between the conduction or the valence bands and electronic states of 3d states of titanium are explained by the N0α and N0β exchange parameters. The N0α describes the exchange coupling between the s-type of conduction bands and 3d levels of titanium, whereas the N0β determines the exchange coupling between the p-type of valence bands and 3d levels of titanium. The N0α and N0β constants are computed by using the mean-field theory, given by the following relations (Sanvito et al. 2001; Raebiger et al. 2004):
where the \(\Delta E_{c} = E_{c}^{ \downarrow } - E_{c}^{ \uparrow }\) and \(\Delta E_{v} = E_{v}^{ \downarrow } - E_{v}^{ \uparrow }\) correspond respectively to the conduction and valence band-edge spin-splittings at Γ high symmetry point of Ba0.875Ti0.125O. The \(\langle s\rangle\) represents the half total magnetic moment per Ti and x = 0.125 is the concentration of Ti atom (Sanvito et al. 2001). Table 4 summarizes the calculated N0α and N0β exchange parameters of Ba0.875Ti0.125O compound. Its shows that the N0α is positive and N0β is negative, revealing ferromagnetic and anti-ferromagnetic exchange couplings respectively between conduction bands and d (Ti) orbitals and between the valence bands and d electronic levels of titanium.
3.3.3 Exchange splittings
The magnitude of ferromagnetism is measured by the use of two factors such as direct exchange and crystal field splittings. The energy that separate the d (Ti) empty levels \((d \downarrow )\) of minority spins and the d (Ti) partially filled levels \((d \uparrow )\) of majority spins corresponds to the direct exchange splitting \((\Delta_{x} (d) = d \downarrow - d \uparrow )\). The crystal field splitting \((\Delta E_{CF} = E_{eg} - E_{t2g} )\) is described by the splitting between the Eeg and Et2g energies respectively for eg of t2g states. It has been reported that the ferromagnetic state in Ca0.875Ti0.125O compound is mainly predominated by the crystal field energy (Korichi et al. 2020). For our material, the Fig. 4 depicted that the eg peaks are located at high levels with respect to the unoccupied \((d \downarrow )\) minority-spin states, but the peaks of \((d \uparrow )\) and t2g partially occupied states of majority spins occur at the same level with respect to EF. Therefore, the \((\Delta E_{CF} )\) crystal field splitting is mainly contributed over the \(\Delta_{x} (d)\) splitting. This result is revealed by the smaller value of 3.755 eV of the \(\Delta_{x} (d)\) splitting compared to the \((\Delta E_{CF} )\) energy of 4.136 eV as seen in Table 4. This kind of process reveals that the crystal field contribution endorses mostly the ferromagnetic state compared to the indirect exchange splitting.
4 Conclusion
We have used the concepts of DFT and FP-LAPW methods to determine the electronic properties, structural parameters, half-metallic ferromagnetic feature, exchange constants, crystal field energy and exchange splittings in the Ba1−xTixO doped with titanium (Ti) impurity at the concentration x = 0.125 such as Ba0.875Ti0.125O compound. The calculated structural parameters of BaO bulk agree with theoretical and experimental results owing to the accurate GGA-WC functional for predicting structural parameters. The lattice parameter of Ba0.875Ti0.125O decreases, leading to the variation of the bulk modulus towards higher value compared to the BaO. These changes result from the size distinction of ionic radii of the Ba and Ti elements. The calculations of TB-mBJ potential revealed that the BaO has a semiconductor character with wide direct band gap, whereas the Ba0.875Ti0.125O doping compound has a integral total magnetic moment with spin polarization of 100% and a half-metallic ferromagnetic behavior. The crystal field contribution endorses mainly the ferromagnetic state in Ba0.875Ti0.125O. Consequently, the Ba0.875Ti0.125O is an accurate half-metallic ferromagnetic and it can be considered as a promising material for possible applications in semiconductors spintronics.
Data availability
The authors declare that all data and material are included and available in the manuscript.
References
Albanese, E., Pacchioni, G.: Ferromagnetism in nitrogen-doped BaO: a self-interaction corrected DFT study. Phys. Chem. Chem. Phys. 19(4), 3279–3286 (2017)
Bai, J., Raulot, J.-M., Zhang, Y., Esling, C., Zhao, X., Zuo, L.: The effects of alloying element Co on Ni–Mn–Ga ferromagnetic shape memory alloys from first-principles calculations. Appl. Phys. Lett. 98(16), 164103 (2011)
Bhattacharjee, R., Chattopadhyaya, S.: Effects of barium (Ba) doping on structural, electronic and optical properties of binary strontium chalcogenide semiconductor compounds—a theoretical investigation using DFT based FP-LAPW approach. Mater. Chem. Phys. 199, 295–312 (2017)
Blaha, P., Schwarz, K., Madsen, G.K.H., Kvasnicka, D., Luitz, J.: WIEN2K: an augmented plane wave and local orbitals program for calculating crystal properties. In: Vienna, K. (ed.) University of Technology, Schwarz, Austria (2001)
Bourega, A., Doumi, B., Mokaddem, A., Sayede, A., Tadjer, A.: Electronic structures and magnetic performance related to spintronics of Sr0.875Ti0.125S. Opt. Quant. Electron 51(12), 385 (2019)
Chang, Z., Graham, E.: Elastic properties of oxides in the NaCl-structure. J. Phys. Chem. Solids 38(12), 1355–1362 (1977)
Chattopadhyaya, S., Bhattacharjee, R.: Theoretical study of structural, electronic and optical properties of BaxPb1−xS, BaxPb1−xSe and BaxPb1−xTe ternary alloys using FP-LAPW approach. J. Alloy. Compd. 694, 1348–1364 (2017)
Cinthia, A.J., Priyanga, G.S., Rajeswarapalanichamy, R., Iyakutti, K.: Structural, electronic and mechanical properties of alkaline earth metal oxides MO (M= Be, Mg, Ca, Sr, Ba). J. Phys. Chem. Solids 79, 23–42 (2015)
Doumi, B., Tadjer, A., Dahmane, F., Mesri, D., Aourag, H.: Investigations of structural, electronic, and half-metallic ferromagnetic properties in (Al, Ga, In)1–x MxN (M= Fe, Mn) diluted magnetic semiconductors. J. Supercond. Novel. Magn. 26(3), 515–525 (2013)
Doumi, B., Mokaddem, A., Ishak-Boushaki, M., Bensaid, D.: First-principle investigation of magnetic and electronic properties of vanadium-and chromium-doped cubic aluminum phosphide. Sci. Semicond. Process. 32, 166–171 (2015a)
Doumi, B., Mokaddem, A., Sayede, A., Dahmane, F., Mogulkoc, Y., Tadjer, A.: First-principles investigations on ferromagnetic behaviour of Be1−xVxZ (Z= S, Se and Te)(x= 0.25). Superlattice. Microstruct. 88, 139–149 (2015b)
Doumi, B., Mokaddem, A., Temimi, L., Beldjoudi, N., Elkeurti, M., Dahmane, F., Sayede, A., Tadjer, A., Ishak-Boushaki, M.: First-principle investigation of half-metallic ferromagnetism in octahedrally bonded Cr-doped rock-salt SrS, SrSe, and SrTe. Eur. Phys. J. B 88(4), 93 (2015c)
Doumi, B., Mokaddem, A., Tadjer, A., Sayede, A.: Recent insights into electronic performance, magnetism and exchange splittings in the Cr-substituted CaO. Front. Chem. 8, 526 (2020)
Goktas, A.: Role of simultaneous substitution of Cu2+ and Mn2+ in ZnS thin films: defects-induced enhanced room temperature ferromagnetism and photoluminescence. Phys. e. 117, 113828 (2020)
Hirohata, A., Yamada, K., Nakatani, Y., Prejbeanu, L., Diény, B., Pirro, P., Hillebrands, B.: Review on spintronics: principles and device applications. J. Magn. Magn. Mater. 509, 166711 (2020)
Hohenberg, P., Kohn, W.: Inhomogeneous electron gas. Phys. Rev. 136(3B), B864-871 (1964)
Kaminska, M., Twardowski, A., Wasik, D.: Mn and other magnetic impurities in GaN and other III–V semiconductors–perspective for spintronic applications. J. Mater. Sci. Mater. El 19(8–9), 828–834 (2008)
Kaneko, Y., Koda, T.: New developments in IIa–VIb (alkaline-earth chalcogenide) binary semiconductors. J. Cryst. Growth 86(1–4), 72–78 (1988)
Kaneko, Y., Morimoto, K., Koda, T.: Optical properties of alkaline-earth chalcogenides. I. Single crystal growth and infrared reflection spectra due to optical phonons. J. Phys. Soc. Jpn. 51(7), 2247–2254 (1982)
Kenmochi, K., Ann Dinh, V., Sato, K., Yanase, A., Katayama-Yoshida, H.: Materials design of transparent and half-metallic ferromagnets of MgO, SrO and BaO without magnetic elements. J. Phys. Soc. Jpn. 73(11), 2952–2954 (2004a)
Kenmochi, K., Seike, M., Sato, K., Yanase, A., Katayama-Yoshida, H.: New class of diluted ferromagnetic semiconductors based on CaO without transition metal elements. Jpn. J. Appl. Phys. 43(7A), L934–L936 (2004b)
Khandy, S.A., Islam, I., Gupta, D.C., Laref, A.: Full Heusler alloys (Co2TaSi and Co2TaGe) as potential spintronic materials with tunable band profiles. J. Solid. State. Chem. 270, 173–179 (2019)
Kohn, W., Sham, L.J.: Self-consistent equations including exchange and correlation effects. Phys. Rev. 140(4A), A1133–A1138 (1965)
Koller, D., Tran, F., Blaha, P.: Merits and limits of the modified Becke–Johnson exchange potential. Phys. Rev. B 83(19), 195134 (2011)
Korichi, K., Doumi, B., Mokaddem, A., Sayede, A., Tadjer, A.: Ferromagnetism, half-metallicity and spin-polarised electronic structures characterisation insights in Ca1−xTixO. Philos. Mag. 100(9), 1172–1190 (2020)
Lakhdari, H., Doumi, B., Mokaddem, A., Sayede, A., Araújo, J.P., Tadjer, A., Elkeurti, M.: Investigation of the substituting effect of chromium on the electronic structures and the half-metallic ferromagnetic properties of BaO. J. Supercond. Novel. Magn. 32(6), 1781–1790 (2019)
Liu, L.G., Bassett, W.A.: Effect of pressure on the crystal structure and the lattice parameters of BaO. J. Geophys. Res. 77(26), 4934–4937 (1972)
Liu, W., Wong, P.K.J., Xu, Y.: Hybrid spintronic materials: growth, structure and properties. Prog. Mater. Sci. 99, 27–105 (2019)
McKee, R., Walker, F., Chisholm, M.: Physical structure and inversion charge at a semiconductor interface with a crystalline oxide. Science 293(5529), 468–471 (2001)
McLeod, J., Wilks, R., Skorikov, N., Finkelstein, L., Abu-Samak, M., Kurmaev, E., Moewes, A.: Band gaps and electronic structure of alkaline-earth and post-transition-metal oxides. Phys. Rev. B 81(24), 245123 (2010)
Monkhorst, H.J., Pack, J.D.: Special points for Brillouin-zone integrations. Phys. Rev. B 13(12), 5188–5192 (1976)
Murnaghan, F.: The compressibility of media under extreme pressures. Proc. Natl. Acad. Sci. U.s.a. 30(9), 244–247 (1944)
Nejatipour, H., Dadsetani, M.: Excitonic effects in the optical properties of alkaline earth chalcogenides from first-principles calculations. Phys. Scr. 90(8), 085802 (2015)
Pan, F., Song, C., Liu, X., Yang, Y., Zeng, F.: Ferromagnetism and possible application in spintronics of transition-metal-doped ZnO films. Mat. Sci. Eng. R 62(1), 1–35 (2008)
PARK, K.O., Sivertsen, J.: Temperature dependence of the bulk modulus of BaO single crystals. J. Am. Ceram. Soc. 60(11–12), 537–538 (1977)
Perdew, J.P., Zunger, A.: Self-interaction correction to density-functional approximations for many-electron systems. Phys. Rev. B 23(10), 5048–5079 (1981)
Perdew, J.P., Burke, K., Ernzerhof, M.: Generalized gradient approximation made simple. Phys. Rev. Lett. 77(18), 3865–3868 (1996)
Poornaprakash, B., Chalapathi, U., Kumar, M., Rajitha, B., Reddy, B.P., Vattikuti, S.P., Park, S.-H.: Tailoring the optical, magnetic, and photocatalytic properties of ZnS quantum dots by rare-earth ion doping. Chem. Phys. Lett. 137609 (2020)
Raebiger, H., Ayuela, A., Nieminen, R.: Intrinsic hole localization mechanism in magnetic semiconductors. J. Phys. Condens. Matter. 16(41), L457–L462 (2004)
Sajjad, M., Manzoor, S., Zhang, H., Noor, N., Alay-e-Abbas, S., Shaukat, A., Khenata, R.: The half-metallic ferromagnetism character in Be1-xVxY (Y= Se and Te) alloys: an ab-initio study. J. Magn. Magn. Mater. 379, 63–73 (2015)
Santana, J.A., Krogel, J.T., Kent, P.R., Reboredo, F.A.: Cohesive energy and structural parameters of binary oxides of groups IIA and IIIB from diffusion quantum Monte Carlo. J. Chem. Phys. 144(17), 174707 (2016)
Sanvito, S., Ordejón, P., Hill, N.A.: First-principles study of the origin and nature of ferromagnetism in Ga1−xMnxAs. Phys. Rev. B 63(16), 165206 (2001)
Saum, G.A., Hensley, E.B.: Fundamental optical absorption in the IIA-VIB compounds. Phys. Rev. 113(4), 1019–1022 (1959)
Singh, D.J., Nordstrom, L.: Planewaves, pseudopotentials, and the LAPW method. Springer (2006)
Teli, N.A., Sirajuddeen, M.M.S.: First-principles calculations of the electronic, magnetic and optical properties of rhenium-doped alkaline earth oxides. Phys. Scr. 95(2), 025801 (2019)
Tran, F., Blaha, P.: Accurate band gaps of semiconductors and insulators with a semilocal exchange-correlation potential. Phys. Rev. Lett. 102(22), 226401 (2009)
Wolf, S., Awschalom, D., Buhrman, R., Daughton, J., von Molnár, V.S., Roukes, M., Chtchelkanova, A.Y., Treger, D.: Spintronics: a spin-based electronics vision for the future. Science 294(5546), 1488–1495 (2001)
Wu, Z., Cohen, R.E.: More accurate generalized gradient approximation for solids. Phys. Rev. B 73(23), 235116 (2006)
Yang, X., Wang, Y., Yan, H., Chen, Y.: Effects of epitaxial strains on spontaneous polarizations and band gaps of alkaline-earth-metal oxides MO (M = Mg, Ca, Sr, Ba). Comput. Mater. Sci. 121, 61–66 (2016)
Zollweg, R.: Optical absorption and photoemission of barium and strontium oxides, sulfides, selenides, and tellurides. Phys. Rev. 111(1), 113–119 (1958)
Acknowledgements
There are no acknowledgments for our manuscript.
Funding
There is no funding statement for our manuscript.
Author information
Authors and Affiliations
Contributions
All authors contributed to the study conception and design. Material preparation, data collection and analysis were performed by all authors. The first draft of the manuscript was written by author: MH and all authors commented on previous versions of the manuscript. All authors read and approved the final manuscript.
Corresponding authors
Ethics declarations
Conflict of interest
The authors declare that they have no conflict of interest.
Human and animal rights
The authors declare that the manuscript does not contain Research involving Human Participants and/or Animals.
Informed consent
There is no informed consent to declare for our manuscript.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
About this article

Cite this article
Hachemaoui, M., Meskine, M., Mokaddem, A. et al. Theoretical characterization of induced ferromagnetism, half-metallic behavior, electronic properties in new Ti-doped BaO. Opt Quant Electron 53, 296 (2021). https://doi.org/10.1007/s11082-021-02960-6
Received:
Accepted:
Published:
DOI: https://doi.org/10.1007/s11082-021-02960-6