
Abstract
Many patients with Parkinson’s disease (PD) will develop cognitive impairment. Cross-sectional studies have shown that certain protein levels are altered in the cerebrospinal fluid (CSF) of PD patients with dementia and are thought to represent potential biomarkers of underlying pathogenesis. Recent studies suggest that CSF biomarker levels may be predictive of future risk of cognitive decline in non-demented PD patients. However, the strength of this evidence and difference between specific CSF biomarkers is not well delineated. We therefore performed a systematic review to assess if levels of specific CSF protein biomarkers are predictive of progression to cognitive impairment. Nine articles were identified that met inclusion criteria for the review. Findings from the review suggest a convergence of evidence that a low baseline Aβ42 in the CSF of non-demented PD patients predicts development of cognitive impairment over time. Conversely, there is limited evidence that CSF levels of tau, either total tau or phosphorylated tau, is a useful predictive biomarker. There are mixed results for other CSF biomarkers such as α-synuclein, Neurofilament light chain, and Heart fatty acid-binding protein. Overall the results of this review show that certain CSF biomarkers have better predictive ability to identify PD patients who are at risk for developing cognitive impairment. Given the interest in developing disease-modifying therapies, identifying this group will be important for clinical trials as initiation of therapy prior to the onset of cognitive decline is likely to be more efficacious.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Cognitive impairment in Parkinson’s disease (PD) is common and disabling (Hely et al. 2008). In all comers with PD, the prevalence of dementia is 25 % (Aarsland et al. 2005); in patients who live more than 20 years with motor symptoms, the prevalence rises to 83 % (Reid et al. 2011). However, many patients develop cognitive impairments that are less severe than those seen in patients with dementia. Therefore, the current clinical diagnosis is divided into PD-MCI (Parkinson’s disease-Mild Cognitive Impairment) and PDD (Parkinson’s disease dementia) (Table 1) (Litvan et al. 2012; Emre et al. 2007). Surprisingly, many patients show these milder cognitive deficits very early in the disorder. Approximately 15-40 % of patients meet diagnostic criteria for PD-MCI at the time when motor symptoms start (Caviness et al. 2007; Yarnall et al. 2014; Aarsland et al. 2009), and 3 years later over 50 % of non-demented patients will meet criteria for PD-MCI (Broeders et al. 2013a). While most longitudinal studies have shown that patients first develop PD-MCI prior to the development of PDD, not all PD-MCI patients are destined to develop PDD (Kehagia et al. 2013; Williams-Gray et al. 2009; Robbins and Cools 2014). In those who do convert to PDD, the conversion rate and risk factors predicting conversion are largely unknown (Broeders et al. 2013b). This is in stark contrast to Alzheimer’s disease (AD), where amnestic-MCI is a strong predictor of progression to dementia with a conversion rate of approximately 12 % a year (Fischer et al. 2007).
In addition to uncertainty in predicting progression, the spectrum of cognitive dysfunction in PD is highly variable in its extent and severity and spans multiple cognitive domains affecting executive and visuospatial function, memory, language, attention and working memory (Caviness et al. 2007). This clinical variability in PD underscores a complex, and poorly understood, neuropathological process. An emerging theory is that cognitive dysfunction in PD may represent a heterogeneous collection of syndromes, each with distinct biological and genetic markers (Williams-Gray et al. 2009; Kehagia et al. 2010; Tachibana 2013; Nombela et al. 2014). Whereas loss of dopaminergic neurons in the substantia nigra and aggregation of α-synuclein (α-syn) with the formation of Lewy bodies are the hallmarks of PD, there are likely multiple neurotransmitters and neural circuits involved in the development of cognitive symptoms (Halliday et al. 2014). AD type changes, such as amyloid-beta (Aβ) plaques and tau tangle pathologies, are found in approximately 50 % of PDD patients at autopsy. Aβ plaques are extracellular deposits of abnormal Aβ peptide, and tau tangles are intracellular aggregates of the microtubule-associated protein tau that exhibit hyperphosphorylation and oxidative modifications. From the AD literature, it is thought that spontaneous aggregation and deposition of Aβ is the inciting event in a molecular cascade resulting in cognitive dysfunction (Mattson 2004). These pathologies are found in brain regions involved in learning and memory, as well as behavioral regulation, and in AD these regions display reduced numbers of synapses and damaged neurites (DeKosky and Scheff 1990). It is suggested that plaque and tangle deposition induces oxidative damage, impairs energy metabolism, and perturbs calcium homeostasis (Butterfield et al. 2001). The causal relationship between these AD type pathologies and PD cognitive impairment is not completely clear; however, Aβ plaques and tau tangles have been proposed to influence the spread of α-syn and rate of PD cognitive decline (Irwin et al. 2013).
Early prediction of dementia risk in PD patients would be useful on multiple levels. Cognitively normal PD patients at high risk for developing impairment would be ideal candidates for research clinical trials of potentially disease-modifying therapies. Clinically, patients and family members would benefit from understanding an individual’s risk of cognitive decline, as they plan for retirement and long-term care needs. Indeed, there is an emerging interest in developing biomarkers that accurately stratify patient risk, track cognitive changes over time, and monitor response to treatment. Given the proximity of cerebrospinal fluid (CSF) to the brain, CSF biomarkers have been investigated as representative of pathological changes of cognitive decline in other neurodegenerative disorders. Specifically in amnestic-MCI and AD, low CSF levels of Aβ and elevated levels of tau proteins, both total-tau (t-tau) and phosphorylated (p-tau), have been shown to predict progression of cognitive decline (Diniz et al. 2008). Low Aβ in the CSF is thought to reflect increased deposition in neuritic plaques and perhaps alterations in CSF transport (Motter et al. 1995). This has been supported by population-based autopsy studies that found an inverse correlation between low concentrations of Aβ in the CSF and high numbers of plaques in the neocortex and hippocampus (Strozyk et al. 2003). Conversely, tau levels are elevated in the CSF in AD patients, and the concentration of tau protein is thought to correlate with the intensity of neuronal damage (Blennow and Hampel 2003). Given the notion that AD type changes may contribute to PDD, CSF levels of Aβ and tau have already been investigated in several cross-sectional studies investigating the relationship between these proteins and cognitive impairment in patients with PD. In non-demented PD patients, reduced CSF Aβ has been associated with cognitive impairment in phonemic fluency (Compta et al. 2009), verbal memory (Alves et al. 2010), processing speed (Leverenz et al. 2011), and visual memory (Yarnall et al. 2014), although one study found no association on neuropsychological testing (Kang et al. 2013). Many of these same studies also looked at CSF tau levels and found normal or even decreased levels of CSF tau in PD patients without dementia (Kang et al. 2013; Yarnall et al. 2014; Alves et al. 2010). When looking at cognitively impaired patients, the results have been inconsistent: some studies show mild to moderately increased t-tau levels in PDD patients compared to PD patients or healthy controls (B. Mollenhauer et al. 2006; Parnetti et al. 2008; Compta et al. 2009). In addition to Aβ and tau, CSF α-syn has been investigated as a possible marker of cognitive impairment in PD, since Lewy body pathology alone can be the sole pathology in PDD and CSF α-syn is thought to reflect Lewy body pathology in the brain of PD patients. With this in mind, several cross-sectional studies have investigated total α-syn and its other forms, such as oligomeric or phosphorylated α-syn in PD patients with and without cognitive impairment or dementia. While most of these studies have found no relationship between CSF α-syn and cognitive impairment (Hall et al. 2012; Kang et al. 2013; Yarnall et al. 2014), given the pathological evidence of its role in the pathogenesis of PDD CSF α-syn has remained an appealing candidate biomarker. More recent studies have turned towards other biomarkers implicated in cognitive dysfunction. Levels of Neurofilament light chain (NFL) are elevated in the CSF of AD patients and correlate with degree of brain atrophy (Scherling et al. 2014). Heart fatty acid-binding protein (HFABP) is elevated in patients with Lewy Body dementia (Wada-Isoe et al. 2008) and is thought to interfere with mitochondrial transport and oxidative function (Fournier et al. 1978).
While these correlation studies provide promising evidence that CSF could be a useful biomarker of cognitive impairment in PD, disease-modifying treatments, such as anti-amyloid immunotherapies like monoclonal antibodies, are more likely to be successful if given before clinical symptoms of cognitive decline are manifest. Therefore, it is important to know whether these same biomarkers will have prognostic value in anticipating the progression of cognitive decline in patients with PD. In recent years, there have been an increasing number of publications investigating baseline CSF proteins in longitudinally followed PD patients. Here we undertake a systematic review of these longitudinal studies, specifically investigating the role of CSF biomarkers in predicting risk of cognitive impairment in PD.
Methods
Information Sources/Study Search Strategy
We systematically searched PubMed, including MEDLINE, and Scopus, including Embase, for articles pertaining to CSF biomarkers, cognition, and Parkinson’s Disease, for all publications through October 1, 2015. The MeSH and EMTREE vocabularies were used whenever possible, along with keyword variations of the CSF biomarkers and disease terms. The initial search was run in Pubmed without limits or restrictions and then translated to the other database. There were no search specifications for cognitive outcomes or tasks or longitudinal study design in the initial search given the myriad possible variations of these terms. The following search terms were used including all abbreviations: ‘cerebrospinal fluid’ (including ‘CSF’), and ‘amyloid-β’ (including ‘amyloid’ or ‘abeta’ or ‘Aβ42’ or ‘β-amyloid 1-42’), or ‘α-synuclein’ (including ‘α-syn’), or ‘t-tau’ (including ‘total tau’) or ‘p-tau’ (including ‘phosphorylated at threonine’ or ‘phosphorylated tau’) and ‘Parkinson’s Disease.’ The combined yield of all searches was 347 citations. An additional search in Scopus removing qualifiers for specific proteins, with search terms denoting ‘longitudinal’ (including ‘cohort’ or ‘follow-up’ or ‘long-term’ or ‘prospective’) and ‘cognition’ yielded an additional 49 citations, of which 6 were identified as duplicates. The citations were exported into Endnote X7 (Thomson Reuters) and distributed to the two authors for screening.
Study Selection
We included a study if it satisfied the following criteria: human studies, in English, with full-text available (no conference proceedings, editorials, or abstracts only), that were focused on CSF biomarkers, assessed data longitudinally, and had some measure of cognitive impairment (Fig. 1).

Flowchart illustrates the selection of studies in the systematic review according to PRISMA criteria
Data Collection and Extraction
Two reviewers (K.L., K.P.) independently evaluated each abstract for inclusion. Next, we obtained full publication for further assessment and data extraction. The same reviewers independently reviewed each article and reached a final consensus for inclusion. These reviews abstracted the information from the eligible articles: author, journal, year of publication, number of subjects and clinical characteristics of subjects, measures of cognitive impairment, CSF biomarker levels, and method of measurement of biomarkers. The various CSF biomarkers and cognitive impairment measures were the main areas of interest extracted from the included studies.
Assessment of Methodological Quality
Two reviewers (K.L., K.P.) independently assess the quality of each study according to the Preferred Reporting Items for Systematic Reviews and Meta-Analyses (PRISMA) criteria (Moher et al. 2009).
Results
Study Selection
Numbers of studies screened, assessed for eligibility and included in the review, with reason for exclusion at each stage, are provided in the Figure 1. The search yielded 370 literature citations for potential inclusion. The most common reason for exclusion was absence of longitudinal assessment methodologically (n = 168). We excluded a total of 89 articles because they were review-based articles. 47 articles were not human-based studies and 37 were not CSF-based studies. 10 were excluded as they did not have any measure of cognitive impairment. 7 were deemed not relevant by both reviewers and 4 were abstracts only. A total of 11 full text articles were assessed for eligibility, of which an additional 2 were excluded after further analysis revealed that they did not provide longitudinal data. A total of 9 studies were included in the qualitative synthesis.
Baseline Study Characteristics
The nine included studies were published between 2010 and 2015 (Tables 2 and 3). Four studies were performed in North America (Liu et al. 2015; Siderowf et al. 2010; Stewart et al. 2014; Terrelonge et al. 2015) and five studies in Europe (Alves et al. 2014; Backstrom et al. 2015; Compta et al. 2013; Hall et al. 2015; Parnetti et al. 2014). The studies involved 1218 subjects diagnosed with Parkinson’s disease (PD). PD diagnosis was made using Brain Bank (Alves et al. 2014; Backstrom et al. 2015; Liu et al. 2015; Parnetti et al. 2014; Siderowf et al. 2010; Stewart et al. 2014; Terrelonge et al. 2015; Compta et al. 2013) and National Institute of Neurological Disorder and Stroke (NINDS) (Alves et al. 2014; Hall et al. 2015) diagnostic criteria. Three out of the nine study cohorts included newly diagnosed PD patients, defined as average disease duration since diagnosis of 1.5 months, (Alves et al. 2014), 7.0 months (Terrelonge et al. 2015), and 16 months (Backstrom et al. 2015). Two studies used the Deprenyl And Tocopherol Antioxidative Therapy Of Parkinsonism (DATATOP) cohort (Liu et al. 2015; Stewart et al. 2014). The DATATOP cohort was first assembled in 1987 to study the effectiveness of the monoamine oxidase type B inhibitor deprenyl and the antioxidant alpha-tocopherol in delaying PD progression (Steering 1989). The cohort included 800 newly diagnosed non-demented and unmedicated PD patients who were followed longitudinally with clinical and biological data collected at follow up time points. The longitudinal data were separated into Phase 1 and Phase 2, as demarcated by the initiation of levodopa therapy. The two studies included in this review using the DATATOP cohort assessed data from Phase 1 (newly diagnosed and not yet on levodopa at baseline) and Phase 2 (average disease duration 3.8 years and just started levodopa at baseline). Of the remaining studies the average PD disease duration at baseline was 3 to 10 years, average 6.9 years (Compta et al. 2013; Hall et al. 2015; Parnetti et al. 2014; Siderowf et al. 2010).
Some assessment of cognitive function was obtained at baseline in all studies, and these assessments were used to identify a cohort of PD patients without cognitive impairment; however, the methods for determining whether patients exhibited baseline cognitive impairment differed greatly among studies. Parnetti et al. (2014) used the Mini-Mental state examination (MMSE), with an average score of 27 (20–30; IQR 25.8–30), and the Montreal – Cognitive Assessment (MoCA), with an average score of 25.5 (17–30; IQR 22.8–28). Backstrom et al. (2015) used only the MMSE, with an average score of 29 (no range provided). Hall et al. (2015) also only used the MMSE, with an average score of 29 (27–29). Terrelonge et al. (2015) used the PPMI cohort, which excluded PD patients if they had a “clinical diagnosis of dementia” at baseline. Stewart et al. (2014) and Liu et al. (2015) used the DATATOP cohort, which excluded PD patients with MMSE score < 23. Compta et al. (2013) used a Swedish cohort, which excluded PD patients with MMSE scores ≤ 24. Siderowf et al. (2010) used a cohort with a baseline Mattis Dementia Rating Scale score of at least 133, citing a study that found a mean score of 133 for patients with PD without dementia (Llebaria et al. 2008). Finally, Alves et al. (2014) used the clinical diagnosis of PDD to exclude dementia in their baseline cohort. Follow up time for all studies ranged from 1.5 to 9 years, average of 2.6 years (see Table 1 for details).
Neuropsychological Assessments
Each of the nine studies used different measures to assess cognition, which were administered both at baseline and at follow up. See Table 2 for details of cognitive assessments used in each study.
CSF Biomarkers Predicting Cognitive Decline
In all studies subjects had CSF obtained at baseline, which was then used to predict cognitive outcomes at follow up. We will discuss the findings for each of the CSF biomarkers studied, which is summarized in Table 4.
CSF Biomarker Measurement Method
All of the studies either used xMAP Luminex platform (Luminex Corp, Austin, TX) or sandwich ELISA (Innogenetics, Ghent, Belgium) immunoassays to measure CSF biomarker levels. Given that α-synuclein concentrations are mildly correlated with hemoglobin concentrations, samples of α-synuclein were either adjusted for CSF hemoglobin concentration (Backstrom et al. 2015; Stewart et al. 2014; Terrelonge et al. 2015) or samples with hemoglobin levels > 1000 ng/L were excluded (Hall et al. 2015).
β-Amyloid 1–42 Peptide (Aβ42)
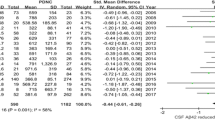
Eight of the nine studies assessed CSF Aβ42 levels at baseline. There was uniform agreement in all but one study (Liu et al. 2015) that low baseline Aβ42 levels in non-demented PD patients predicted the development of cognitive impairment at follow up. Low baseline CSF Aβ42 was associated with various cognitive outcome measures, depending on the study. Three of the studies used the development of PDD, as defined clinically by consensus agreement among two neurologist specialized in movement disorders (Alves et al. 2014; Backstrom et al. 2015; Compta et al. 2013). All three studies found that CSF Aβ42 levels were significantly lower at baseline in patients who developed PDD at follow up as compared to a control group (p < 0.005 in all three studies). The control groups were either healthy controls (Backstrom et al. 2015) or PD patients without cognitive impairment (Alves et al. 2014; Compta et al. 2013). Terrelonge et al. (2015) also assessed a global clinical outcome of “PD with Cognitive Impairment,” as defined by impairment on at least 2 of 6 neuropsychological tests greater than 1.5 standard deviation below mean scores derived from healthy controls. At 2-year follow up, 12.9 % of their cohort met criteria for cognitive impairment and this group had significantly lower baseline CSF Aβ42 levels as compared to non-cognitively impaired PD patients (p < 0.01). Other studies looked at the rate of change over time on cognitive assessments and found that a faster rate of deterioration on certain cognitive tests correlated with baseline low CSF Aβ42 levels. For instance, Siderowf et al. (2010) found that low CSF Aβ42 significantly correlated with the annualized rates of decline on the DRS-2 (β = 0.040). When using a cutoff value of CSF Aβ42 ≤ 192 pg/ml, patients below that value declined an average of 5.85 points more quickly than those above that value (p = 0.002). Similarly, Hall et al. (2015) found that patients with lower CSF Aβ42 levels had a faster rate of decline on cognitive assessments. They dichotomized their cohort using a cutoff value of CSF Aβ42 ≤ 550 pg/ml and found that patients below this level had a more significant deterioration in delayed memory recall (as assessed by the ADAS-cog score) at 2-year follow up (p = 0.002). Parnetti et al. (2014) found that lower baseline CSF Aβ42 significantly correlated with lower scores on the MMSE and MoCA at 3-year follow up (r = −0.52, p < 0.001 for MMSE, r = −0.45, p < 0.01 for MoCA). The range of Aβ42 across studies was 224–730 pg/ml.
Total tau Protein (t-tau) and Phosphorylated tau (p-tau)
All but one study (Stewart et al. 2014) assessed CSF levels of t-tau. Surprisingly, none of the studies found a significant correlation between baseline levels of t-tau and measures of cognitive impairment at follow up. The range of t-tau levels was 40.9–422 pg/ml. These same studies also assessed levels of p-tau and only one found a significant correlation between higher baseline p-tau and rate of cognitive decline on certain neuropsychological assessments at follow up (Liu et al. 2015). While cognitive decline over time was observed in the entire cohort, in those with higher CSF p-tau levels at baseline there was a significantly faster annualized rate of decline on the Selective Reminding Test-Total (r = −0.122, p < 0.002) and Symbol Digit Modalities Test (r = −0.219, p < 0.0001). Interestingly, this relationship was only found in the Phase 2 study (average disease duration 3.8 years), and not in Phase 1 (newly diagnosed, drug naïve). The range of CSF p-tau was 14.9–59 pg/ml.
α-Synuclein
Four of the nine studies assessed α-syn levels at baseline (Backstrom et al. 2015; Hall et al. 2015; Stewart et al. 2014; Terrelonge et al. 2015). Hall et al. (2015) found that higher levels of baseline α-syn were significantly correlated with worsening cognitive speed as measured by A Quick Test of Cognitive Speed at 2 year follow up (r = 0.373, p < 0.007). Stewart et al. (2014) looked at the inverse and found that low CSF α-syn predicted preservation of cognitive function at follow up as measured by Selective Reminding Test-Total and Delayed and New Dot Test (r = −0.124, r = −0.051, and r = −0.032, respectively; all p < 0.002). By contrast, neither Backstrom et al. (2015) nor Terrelonge et al. (2015) found any relationship between high baseline α-syn and development of PDD or PD with cognitive impairment at follow up. The range of baseline CSF α-syn was broad: 0.47 to 720 ng/ml.
Other CSF Markers and Ratios Assessed
In addition to Aβ42, t-tau, p-tau, and α-syn, both Backstrom et al. (2015) and Hall et al. (2015) also measured levels of Neurofilament Light Chain (NFL). Whereas Backstrom et al. (2015) found high NFL at baseline significantly predicted the development of PDD at follow up (p < 0.001), Hall et al. (2015) did not find a significant relationship between high NFL and rate of decline over time on cognitive assessments. The baseline level of NFL in the Backstrom et al. (2015) cohort was 1201 pg/ml, whereas it was 900 pg/ml in the Hall et al. cohort (2015). Backstrom et al. (2015) additionally assessed Heart fatty acid-binding protein (HFABP). They found that high levels at baseline significantly predicted the development of PDD at follow up (p < 0.001).
There were numerous CSF protein ratios assessed in an attempt to use combined CSF biomarkers to find stronger correlations with outcome measures. Of these, Parnetti et al. (2014) was able to find a significant correlation between a low Aβ42/t-tau ratio and increased rate of decline on the MMSE (p < 0.05), but this did not correlate significantly with decline on the Montreal – Cognitive Assessment. Liu et al. (2015) found that a high p-tau/Aβ42 ratio significantly correlated with a faster rate of decline only on the Selective Reminding Test-Total and Symbol Digit Modalities Tests (p < 0.0001) during the Phase 2 study. None of the other ratios assessed had predictive value (see Table 2).
Discussion
This current systematic review reinforces the evidence that baseline levels of certain CSF biomarkers may add critical information to assessing the risk of progression to cognitive impairment in non-demented PD patients. Specifically, the majority of studies suggest the strongest predictive biomarker of future cognitive impairment is low baseline CSF Aβ42. In addition, these studies suggest there may also be a role for other CSF biomarkers, including tau (total and phosphorylated), α-syn, NFL, and HFABP. The identification of biomarkers that precede the development of PD-MCI or PDD is important given the emergence of potential disease-modifying therapies, such as anti-amyloid immunotherapies like monoclonal antibodies as discussed below. Taken together, PD patients without cognitive impairment, but with a ‘pathological’ CSF profile at baseline might be at increased risk for the development of cognitive impairment, and thus represent a potential group for intervention, either from a clinical or research perspective.
CSF Aβ42 is Predictive of Cognitive Decline in non-Demented PD Patients
Our systematic review suggests a convergence of evidence that low baseline CSF Aβ42 is predictive of cognitive decline in non-demented PD patients. This was true across most studies regardless of baseline study characteristics (Table 1). Notably, low CSF Aβ42 was predictive of cognitive decline for both newly diagnosed PD patients as well as PD patients who were over a decade out from initial diagnosis (Compta et al. 2013). This finding was also seen for follow up times ranging from 0.3 years to 9 years, and was consistent in studies using either the xMAP Luminex or ELISA assay methods. Finally, this was seen despite a large variability in outcome measures, including type and range of cognitive assessments used and clinical definitions of cognitive impairment or decline. However, given that only nine studies met inclusion for this review, the relationship between baseline low Aβ42 and worse cognitive outcomes should be viewed with caution and warrants further validation across different PD populations. Specifically, since none of the studies included genetic data other than Apolipoprotein E (Siderowf et al. 2010; Stewart et al. 2014; Liu et al. 2015) this relationship should be investigated further in patients with known genetic risk polymorphisms. In addition, because studies did not use consistent neuropsychological batteries or even tests within the same cognitive domains, there was not enough data to determine if baseline CSF Aβ42 was predictive of all types of cognitive decline or more predictive of decline in specific domains, like episodic memory. Future longitudinal studies of PD cognitive impairment should strive to include broad testing across cognitive domains (Litvan et al. 2012), and ideally use agreed upon cognitive batteries to further decipher this important issue.
The one study that was unable to find a relationship between low CSF Aβ42 and future cognitive impairment did find a correlation between the ratio of p-tau/Aβ42 and subsequent decline on cognitive measures (Liu et al. 2015). One explanation for the absence of predictive value of CSF Aβ42 alone may be that the baseline CSF Aβ42 level in their cohort was among the lowest of all studies included (Aβ42 232 pg/ml compared to range of all studies 224–730 pg/ml). This study looked at the correlation between changes in CSF proteins and decline in cognitive test performance. Possibly, CSF Aβ42 levels at baseline had already declined such that there was a floor effect and further decreases over time did not register a discernable change. Going forward, CSF Aβ42 levels may have a stronger predictive effect when used as a dichotomous cutoff indicating a “low” value, as has been suggested in AD (Shaw et al. 2009), although further work is needed to determine the most predictive cutoff value. Furthermore, whereas Liu et al. (2015) was able to find a predictive effect when combing Aβ42 levels into a ratio with p-tau, possibly implicating p-tau as a predictive marker, this effect was not replicated by Terrelonge et al. (2015), who found no correlation between the ratio of p-tau/Aβ42 and the development of PDD at follow up. This latter finding is consistent with the results of the other studies as detailed below suggesting p-tau as a poor predictive marker for subsequent cognitive decline.
Lastly, a possible explanation for the particularly low baseline CSF Aβ42 in the DATATOP cohort could be if tubes made of glass or polystyrene, as opposed to polypropylene, ever came into contact with the CSF prior to analysis. It is now appreciated that polystyrene has a very high affinity for proteins, and Aβ42 in particular; even a very short exposure of samples to this material can bind proteins, leaving concentrations of proteins lower in the remaining fluid (Bjerke et al. 2010). Given this and other causes of variability during quantification of CSF proteins (Brit Mollenhauer et al. 2015), future studies should employ standardized protocols to minimize confounding factors that could emerge during CSF collection or analysis.
CSF tau is not Predictive of Cognitive Decline in non-Demented PD Patients
Previously, CSF tau levels have been looked at with great interest in PD, given the numerous studies showing tau tangles in up to 50 % of patients with PDD as well as the evidence that CSF tau levels are predictive of progression to MCI and AD (Ewers et al. 2012). In PD, several cross-sectional studies have shown high levels of CSF tau in patients with PDD, as compared to non-demented PD (Compta et al. 2009; B. Mollenhauer et al. 2006; Montine et al. 2010; Vranova et al. 2014). Furthermore, one longitudinal study found that CSF ratios of p-tau/tau and p-tau/Aβ42 at baseline correlated with progression of motor symptoms in PD (Zhang et al. 2013). However, in this review, there is little evidence that CSF levels of t-tau or p-tau are useful predictors of future PD cognitive impairment. Only Liu et al. (2015) was able to correlate higher levels of p-tau at baseline (but not t-tau) with an increased rate of cognitive decline at follow up, and this finding was only in CSF taken at the beginning the Phase 2 study when patients started taking levodopa (duration mean ± SD 3.82 ± 1.58, range 1–8 years), not Phase 1 (duration mean ± SD 1.99 ± 1.33, range 0–6 years). This predictive effect was also only for a small subset of the cognitive assessments administered (Selective Reminding Test-Total and Symbol Digit Modalities Tests). All of the other studies in this review failed to show a relationship between t-tau or p-tau with any follow up cognitive measures. Moreover, Siderowf et al. (2010) found that adding t-tau or p-tau in combined metric with Aβ42 actually diminished the predictive effect. This information is useful for researchers with limited resources and suggests that future studies may benefit from investing in other CSF biomarkers when looking for positive predictors of cognitive decline in early PD patients.
Limitations in Biomarker Predictive Ability
It is important to make the distinction between the ability of a biomarker to reflect a disease state or pathology and the ability to predict a future clinical change or outcome. The clinical heterogeneity in the progression of cognitive impairment in PD makes this distinction especially challenging. Whereas most patients will eventually develop dementia, up to 20 % can go decades with only mild cognitive changes (Hely et al. 2008). This becomes relevant when comparing the data from prior cross-sectional studies with longitudinal studies. It is likely that the levels of CSF biomarkers do not follow a linear projection over time and may vary as the burden of pathology progresses in the brain. The accumulation of amyloid plaques and concomitant decrease in CSF Aβ42 is likely one of the earliest events in the pathogenesis of cognitive impairment in PD and consequently precedes the onset of clinical dementia. Therefore, as is shown within this review, CSF Aβ42 levels serve as a predictive marker across studies despite time from diagnosis and disease duration. Conversely, it is thought that CSF tau levels may rise only after extensive neuronal damage has occurred and the development of PDD is clinically evident. As such, this might explain why CSF tau levels do not appear to prove useful as predictive markers even if pathological levels represent a diseased state. Such temporal considerations should be taken into account when hypothesizing predictive effects for other CSF biomarkers.
Considerations of Other CSF Biomarkers
Although several other biomarkers have shown an association with cognitive impairment in cross-sectional studies, the results in the reviewed longitudinal studies were mixed for the majority of candidates. Only a small subset of studies assessed other CSF biomarkers, such as α-syn, NFL, and HFABP. There may be some evidence that high CSF levels of these proteins may predict progression to cognitive impairment, but outcomes are inconsistent. For example, whereas two studies found a correlation between high baseline levels of α-syn and an increased rate of decline on certain cognitive measures at follow up (Hall et al. 2015; Stewart et al. 2014), two other studies were unable to show that higher levels of a-syn at baseline were predictive either PDD or PD with CI (Backstrom et al. 2015; Terrelonge et al. 2015). Similarly, while one study found that high CSF NFL levels at baseline was predictive of PDD at follow up (Backstrom et al. 2015), another study found no correlation between elevated CSF NFL and rate of decline on cognitive measures (Hall et al. 2015). There was one study that investigated of CSF levels of HFABP (Backstrom et al. 2015). In their cohort, CSF levels of HFABP at baseline predicted development of PDD at follow up. However, more studies are needed. Lack of consistency may be due to a number of factors, including laboratory methodology, patient cohort characteristics, variability of outcome measures, including type and range of cognitive assessments used, and clinical definitions of PDD.
Emerging Therapies for Amyloid Accumulation in Neurodegenerative Disease
The ability to predict progression to cognitive impairment in PD is important given the ongoing interest in developing molecularly-targeted disease-modifying therapies in neurodegenerative diseases (Rafii and Aisen 2015). For instance, amyloid accumulation is integral to the pathogenesis of AD, and recent trials have looked at treatment with anti-amyloid immunotherapy using monoclonal antibodies, such as Solanezumab, Crenezumab, and Gantenerumab (Blennow et al. 2014). These antibodies bind to Aβ and work to either prevent aggregation or accelerate removal. In Phase II trials these antibody therapies failed to show overall difference between treatment and placebo on primary cognitive outcome measures. However, many believe these studies failed because they used a pooled analysis of all patients, including those with advanced disease who were not as likely to benefit from disease-modifying effects. Indeed, some of these studies trended towards significance when looking at sub-group analysis at mild, early AD (Doody et al. 2014). From this, a number of clinical trials have turned their attention towards preclinical AD, or those defined as high risk for the development of AD (either by genetic risk profile or amyloid biomarkers), but without cognitive impairment at time of treatment. There are currently four clinical trials ongoing for the secondary prevention of AD dementia in high risk, but asymptomatic patients. The Anti-Amyloid in Asymptomatic Alzheimer’s (A4) is a Phase III study comparing solanezumab with placebo (Sperling et al. 2012). The Dominantly Inherited Alzheimer’s Network – Therapy Unit (DIAN-TU) trial includes preclinical AD patients who harbor high-risk mutations and will be randomly assigned to receive one of three investigational drugs, while non-carriers will receive placebo (Morris et al. 2012). Finally, the Alzheimer’s Prevention Initiative has two trials. One, the Autosomal Dominant Alzheimer’s Disease (API-ADAD) trial, is studying crenezumab in preclinical PSEN1 E280A mutations carriers, and the other, the ApoE4 (API-APOE4) trial, will test two investigational drugs in preclinical AD patients who are homozygous for apolipoprotein E4 (Tariot et al. 2014).
By contrast, the role of amyloid accumulation in the development of PD cognitive impairment is complex. AD-type pathology, with Aβ plaques and tau tangles, are found in up to 50 % of PDD patients at autopsy (Irwin et al. 2013). However, it is possible that amyloid deposition alone, independent of tau pathology, could hasten the development of PD cognitive impairment. For instance, a recent review found while PD patients had PET Aβ deposition that was below AD thresholds, there was still a correlation between these lower than AD-range levels of neocortical Aβ and poor cognitive function (Lin and Wu 2015). The longitudinal data discussed in this review appear to agree that PD patients with low CSF Aβ42, and thus presumed neocortical Aβ deposition, are indeed at increased risk for progression to cognitive impairment. Further studies with combined CSF Aβ42 and amyloid PET imaging, with longitudinal follow up, are needed to confirm this presumption. Nevertheless, PD with a high likelihood of amyloid pathology, as shown with low CSF Aβ42, could be an ideal target for the same anti-amyloid immunotherapies currently being tested in preclinical AD. The ability to accurately identify these patients is of utmost importance in providing early disease-modifying therapies that could potentially prevent or slow progression of cognitive decline.
Future Directions
Lastly, CSF biomarkers are only one of multiple other types of biomarkers under ongoing investigation. It is likely that genetics, brain network connectivity, and biochemical changes all contribute to cognitive dysfunction in PD (B. Mollenhauer et al. 2014). Predicting risk of cognitive decline will likely best be assessed using a combination of biomarkers, such as gene identification, blood markers, and imaging modalities like functional MRI or PET. These could be further combined with demographic and clinical risk factors to identify those patients at highest risk for PDD as early as possible in their disease. This would be invaluable to the initiation of early treatment and clinical management of this devastating symptom.
References
Aarsland, D., Zaccai, J., & Brayne, C. (2005). A systematic review of prevalence studies of dementia in Parkinson’s disease. Movement Disorders, 20(10), 1255–1263. doi:10.1002/mds.20527.
Aarsland, D., Bronnick, K., Larsen, J. P., Tysnes, O. B., & Alves, G. (2009). Cognitive impairment in incident, untreated Parkinson disease: the Norwegian ParkWest study. Neurology, 72(13), 1121–1126. doi:10.1212/01.wnl.0000338632.00552.cb.
Alves, G., Bronnick, K., Aarsland, D., Blennow, K., Zetterberg, H., Ballard, C., et al. (2010). CSF amyloid-beta and tau proteins, and cognitive performance, in early and untreated Parkinson’s disease: the Norwegian ParkWest study. Journal of Neurology, Neurosurgery, and Psychiatry, 81(10), 1080–1086. doi:10.1136/jnnp.2009.199950.
Alves, G., Lange, J., Blennow, K., Zetterberg, H., Andreasson, U., Forland, M. G., et al. (2014). CSF Abeta42 predicts early-onset dementia in Parkinson disease. Neurology, 82(20), 1784–1790. doi:10.1212/wnl.0000000000000425.
Andersson, M., Wiig, E. H., Minthon, L., & Londos, E. (2007). A quick test for cognitive speed: a measure of cognitive speed in dementia with Lewy bodies. American Journal of Alzheimer’s Disease and Other Dementias, 22(4), 313–318.
Backstrom, D. C., Eriksson Domellof, M., Linder, J., Olsson, B., Ohrfelt, A., Trupp, M., et al. (2015). Cerebrospinal fluid patterns and the risk of future dementia in early, incident Parkinson disease. JAMA Neurology. doi:10.1001/jamaneurol.2015.1449.
Benedict, R.H. (1997). Brief visuospatial memory test--revised: professional manual: PAR.
Benton, A. L., Hamsher, K., Varney, N. R., & Spreen, O. (1983). Judgment of line orientation. New York: Oxford University Press.
Bjerke, M., Portelius, E., Minthon, L., Wallin, A., Anckarsäter, H., Anckarsäter, R., et al. (2010). Confounding factors influencing amyloid beta concentration in cerebrospinal fluid. International Journal of Alzheimer’s disease, 2010.
Blennow, K., & Hampel, H. (2003). CSF markers for incipient Alzheimer’s disease. The Lancet Neurology, 2(10), 605–613.
Blennow, K., Hampel, H., & Zetterberg, H. (2014). Biomarkers in amyloid-β immunotherapy trials in Alzheimer’s disease. Neuropsychopharmacology, 39(1), 189–201.
Bowie, C. R., & Harvey, P. D. (2006). Administration and interpretation of the trail making test. Nature Protocols, 1(5), 2277–2281.
Broeders, M., de Bie, R. M., Velseboer, D. C., Speelman, J. D., Muslimovic, D., & Schmand, B. (2013a). Evolution of mild cognitive impairment in Parkinson disease. Neurology, 81(4), 346–352. doi:10.1212/WNL.0b013e31829c5c86.
Broeders, M., Velseboer, D. C., de Bie, R., Speelman, J. D., Muslimovic, D., Post, B., et al. (2013b). Cognitive change in newly-diagnosed patients with Parkinson’s disease: a 5-year follow-up study. Journal of International Neuropsychological Society, 19(6), 695–708. doi:10.1017/s1355617713000295.
Butterfield, D. A., Drake, J., Pocernich, C., & Castegna, A. (2001). Evidence of oxidative damage in Alzheimer’s disease brain: central role for amyloid β-peptide. Trends in Molecular Medicine, 7(12), 548–554.
Caviness, J. N., Driver-Dunckley, E., Connor, D. J., Sabbagh, M. N., Hentz, J. G., Noble, B., et al. (2007). Defining mild cognitive impairment in Parkinson’s disease. Movement Disorders, 22(9), 1272–1277. doi:10.1002/mds.21453.
Compta, Y., Marti, M. J., Ibarretxe-Bilbao, N., Junque, C., Valldeoriola, F., Munoz, E., et al. (2009). Cerebrospinal tau, phospho-tau, and beta-amyloid and neuropsychological functions in Parkinson’s disease. Movement Disorders, 24(15), 2203–2210. doi:10.1002/mds.22594.
Compta, Y., Pereira, J. B., Rios, J., Ibarretxe-Bilbao, N., Junque, C., Bargallo, N., et al. (2013). Combined dementia-risk biomarkers in Parkinson’s disease: a prospective longitudinal study. Parkinsonism & Related Disorders, 19(8), 717–724. doi:10.1016/j.parkreldis.2013.03.009.
DeKosky, S. T., & Scheff, S. W. (1990). Synapse loss in frontal cortex biopsies in Alzheimer’s disease: correlation with cognitive severity. Annals of Neurology, 27(5), 457–464.
Diniz, B. S., Pinto Junior, J. A., & Forlenza, O. V. (2008). Do CSF total tau, phosphorylated tau, and beta-amyloid 42 help to predict progression of mild cognitive impairment to Alzheimer’s disease? A systematic review and meta-analysis of the literature. The World Journal of Biological Psychiatry, 9(3), 172–182. doi:10.1080/15622970701535502.
Donnelly, K., Donnelly, J., Lichter, D., & Hershey, L. (1995). Longitudinal assessment of verbal and visual memory in Parkinson’s disease patients. Archives of Clinical Neuropsychology, 10(4), 322–323.
Doody, R. S., Thomas, R. G., Farlow, M., Iwatsubo, T., Vellas, B., Joffe, S., et al. (2014). Phase 3 trials of solanezumab for mild-to-moderate Alzheimer’s disease. New England Journal of Medicine, 370(4), 311–321.
Emre, M., Aarsland, D., Brown, R., Burn, D. J., Duyckaerts, C., Mizuno, Y., et al. (2007). Clinical diagnostic criteria for dementia associated with Parkinson’s disease. Movement Disorders, 22(12), 1689–1707. doi:10.1002/mds.21507. quiz 1837.
Ewers, M., Walsh, C., Trojanowski, J. Q., Shaw, L. M., Petersen, R. C., Jack, C. R., Jr., et al. (2012). Prediction of conversion from mild cognitive impairment to Alzheimer’s disease dementia based upon biomarkers and neuropsychological test performance. Neurobiology of Aging, 33(7), 1203–1214. doi:10.1016/j.neurobiolaging.2010.10.019.
Fischer, P., Jungwirth, S., Zehetmayer, S., Weissgram, S., Hoenigschnabl, S., Gelpi, E., et al. (2007). Conversion from subtypes of mild cognitive impairment to Alzheimer dementia. Neurology, 68(4), 288–291.
Folstein, M. F., Folstein, S. E., & McHugh, P. R. (1975). “Mini-mental state”. A practical method for grading the cognitive state of patients for the clinician. Journal of Psychiatric Research, 12(3), 189–198.
Fournier, N., Geoffroy, M., & Deshusses, J. (1978). Purification and characterization of a long chain, fatty-acid-binding protein supplying the mitochondrial β-oxidative system in the heart. Biochimica et Biophysica Acta (BBA)-Protein Structure, 533(2), 457–464.
Gill, D. J., Freshman, A., Blender, J. A., & Ravina, B. (2008). The Montreal cognitive assessment as a screening tool for cognitive impairment in Parkinson’s disease. Movement Disorders, 23(7), 1043–1046.
Golden, C. (1978). Stroop colour and word test. AGE, 15, 90.
Groth-Marnat, G. (2009). Handbook of psychological assessment: Wiley.
Hall, S., Ohrfelt, A., Constantinescu, R., Andreasson, U., Surova, Y., Bostrom, F., et al. (2012). Accuracy of a panel of 5 cerebrospinal fluid biomarkers in the differential diagnosis of patients with dementia and/or parkinsonian disorders. Archives of Neurology, 69(11), 1445–1452. doi:10.1001/archneurol.2012.1654.
Hall, S., Surova, Y., Ohrfelt, A., Zetterberg, H., Lindqvist, D., & Hansson, O. (2015). CSF biomarkers and clinical progression of Parkinson disease. Neurology, 84(1), 57–63. doi:10.1212/wnl.0000000000001098.
Halliday, G. M., Leverenz, J. B., Schneider, J. S., & Adler, C. H. (2014). The neurobiological basis of cognitive impairment in Parkinson’s disease. Movement Disorders, 29(5), 634–650. doi:10.1002/mds.25857.
Heaton, R. K. (1993). Wisconsin card sorting test: computer version 2. Odessa: Psychological Assessment Resources.
Hely, M. A., Reid, W. G., Adena, M. A., Halliday, G. M., & Morris, J. G. (2008). The Sydney multicenter study of Parkinson’s disease: the inevitability of dementia at 20 years. Movement Disorders, 23(6), 837–844. doi:10.1002/mds.21956.
Irwin, D. J., Lee, V. M., & Trojanowski, J. Q. (2013). Parkinson’s disease dementia: convergence of alpha-synuclein, tau and amyloid-beta pathologies. Nature Reviews Neuroscience, 14(9), 626–636. doi:10.1038/nrn3549.
Ivnik, R. J., Smith, G. E., Lucas, J. A., Tangalos, E. G., Kokmen, E., & Petersen, R. C. (1997). Free and cued selective reminding test: MOANS norms. Journal of Clinical and Experimental Neuropsychology, 19(5), 676–691.
Johnson-Greene, D. (2004). Dementia rating scale-2 (DRS-2): by P.J. Jurica, C.L. Leitten, and S. Mattis: psychological Assessment Resources, 2001. Archives of Clinical Neuropsychology, 19(1), 145–147. doi:10.1016/j.acn.2003.07.003.
Kang, J. H., Irwin, D. J., Chen-Plotkin, A. S., Siderowf, A., Caspell, C., Coffey, C. S., et al. (2013). Association of cerebrospinal fluid beta-amyloid 1–42, T-tau, P-tau181, and alpha-synuclein levels with clinical features of drug-naive patients with early Parkinson disease. JAMA Neurology, 70(10), 1277–1287. doi:10.1001/jamaneurol.2013.3861.
Kaplan, E. F., Goodglass, H., & Weintraub, S. (1983). The Boston naming test: experimental edition. Boston: Kaplan & Goodglass (2nd ed.). Philadelphia: Lea & Febinger.
Kehagia, A. A., Barker, R. A., & Robbins, T. W. (2010). Neuropsychological and clinical heterogeneity of cognitive impairment and dementia in patients with Parkinson’s disease. Lancet Neurology, 9(12), 1200–1213. doi:10.1016/s1474-4422(10)70212-x.
Kehagia, A. A., Barker, R. A., & Robbins, T. W. (2013). Cognitive impairment in Parkinson’s disease: the dual syndrome hypothesis. Neurodegenerative Diseases, 11(2), 79–92. doi:10.1159/000341998.
Leverenz, J. B., Watson, G. S., Shofer, J., Zabetian, C. P., Zhang, J., & Montine, T. J. (2011). Cerebrospinal fluid biomarkers and cognitive performance in non-demented patients with Parkinson’s disease. Parkinsonism & Related Disorders, 17(1), 61–64. doi:10.1016/j.parkreldis.2010.10.003.
Lin, C.-H., & Wu, R.-M. (2015). Biomarkers of cognitive decline in Parkinson’s disease. Parkinsonism & Related Disorders, 21(5), 431–443.
Litvan, I., Bhatia, K. P., Burn, D. J., Goetz, C. G., Lang, A. E., McKeith, I., et al. (2003). SIC Task Force appraisal of clinical diagnostic criteria for parkinsonian disorders. Movement disorders, 18(5), 467–486.
Litvan, I., Goldman, J. G., Troster, A. I., Schmand, B. A., Weintraub, D., Petersen, R. C., et al. (2012). Diagnostic criteria for mild cognitive impairment in Parkinson’s disease: movement disorder society task force guidelines. Movement Disorders, 27(3), 349–356. doi:10.1002/mds.24893.
Liu, C., Cholerton, B., Shi, M., Ginghina, C., Cain, K. C., Auinger, P., et al. (2015). CSF tau and tau/Abeta42 predict cognitive decline in Parkinson’s disease. Parkinsonism & Related Disorders, 21(3), 271–276. doi:10.1016/j.parkreldis.2014.12.027.
Llebaria, G., Pagonabarraga, J., Kulisevsky, J., Garcia-Sanchez, C., Pascual-Sedano, B., Gironell, A., et al. (2008). Cut-off score of the Mattis dementia rating scale for screening dementia in Parkinson’s disease. Movement Disorders, 23(11), 1546–1550. doi:10.1002/mds.22173.
Lopes, M., Brucki, S. M. D., Giampaoli, V., & Mansur, L. L. (2009). Semantic verbal fluency test in dementia. Dementia & Neuropsychologia, 3, 315–320.
Mattson, M. P. (2004). Pathways towards and away from Alzheimer’s disease. Nature, 430(7000), 631–639.
Mitrushina, M., Boone, K.B., Razani, J., & D’Elia, L.F. (2005). Handbook of normative data for neuropsychological assessment: Oxford University Press.
Moher, D., Liberati, A., Tetzlaff, J., & Altman, D. G. (2009). Preferred reporting items for systematic reviews and meta-analyses: the PRISMA statement. Journal of Clinical Epidemiology, 62(10), 1006–1012. doi:10.1016/j.jclinepi.2009.06.005.
Mohs, R., Rosen, W., & Davis, K. (1983). The Alzheimer’s disease assessment scale: an instrument for assessing treatment efficacy. Psychopharmacology Bulletin, 19(3), 448.
Mollenhauer, B., Bibl, M., Wiltfang, J., Steinacker, P., Ciesielczyk, B., Neubert, K., et al. (2006). Total tau protein, phosphorylated tau (181p) protein, beta-amyloid(1–42), and beta-amyloid(1–40) in cerebrospinal fluid of patients with dementia with Lewy bodies. Clinical Chemistry and Laboratory Medicine, 44(2), 192–195. doi:10.1515/cclm.2006.035.
Mollenhauer, B., Rochester, L., Chen-Plotkin, A., & Brooks, D. (2014). What can biomarkers tell us about cognition in Parkinson’s disease? Movement Disorders, 29(5), 622–633. doi:10.1002/mds.25846.
Mollenhauer, B., Parnetti, L., Rektorova, I., Kramberger, M. G., Pikkarainen, M., Schulz‐Schaeffer, W. J., et al. (2015). Biological confounders for the values of cerebrospinal fluid proteins in Parkinson’s disease and related disorders. Journal of Neurochemistry. doi:10.1111/jnc.13390.
Montine, T. J., Shi, M., Quinn, J. F., Peskind, E. R., Craft, S., Ginghina, C., et al. (2010). CSF Abeta(42) and tau in Parkinson’s disease with cognitive impairment. Movement Disorders, 25(15), 2682–2685. doi:10.1002/mds.23287.
Morris, J. C., Aisen, P. S., Bateman, R. J., Benzinger, T. L., Cairns, N. J., Fagan, A. M., et al. (2012). Developing an international network for Alzheimer’s research: the dominantly inherited Alzheimer network. Clinical Investigation, 2(10), 975–984.
Motter, N., Vigo‐Pelfrey, C., Kholodenko, D., Barbour, R., Johnson‐Wood, K., Galasko, D., et al. (1995). Reduction of β‐amyloid peptide42 in the cerebrospinal fluid of patients with Alzheimer’s disease. Annals of Neurology, 38(4), 643–648.
Nombela, C., Rowe, J. B., Winder-Rhodes, S. E., Hampshire, A., Owen, A. M., Breen, D. P., et al. (2014). Genetic impact on cognition and brain function in newly diagnosed Parkinson’s disease: ICICLE-PD study. Brain, 137(Pt 10), 2743–2758. doi:10.1093/brain/awu201.
Parnetti, L., Tiraboschi, P., Lanari, A., Peducci, M., Padiglioni, C., D’Amore, C., et al. (2008). Cerebrospinal fluid biomarkers in Parkinson’s disease with dementia and dementia with Lewy bodies. Biological Psychiatry, 64(10), 850–855. doi:10.1016/j.biopsych.2008.02.016.
Parnetti, L., Farotti, L., Eusebi, P., Chiasserini, D., De Carlo, C., Giannandrea, D., et al. (2014). Differential role of CSF alpha-synuclein species, tau, and Abeta42 in Parkinson’s Disease. Frontiers in Aging Neuroscience, 6, 53. doi:10.3389/fnagi.2014.00053.
Rafii, M. S., & Aisen, P. S. (2015). Advances in Alzheimer’s disease drug development. BMC Medicine, 13, 62. doi:10.1186/s12916-015-0297-4.
Randall, K. D., & Kerns, K. A. (2011). Selective reminding test. In Encyclopedia of Clinical Neuropsychology (pp. 2235–2237): Springer.
Redding, G. M., & Gerjets, D. A. (1977). Stroop effect: interference and facilitation with verbal and manual responses. Perceptual and Motor Skills, 45(1), 11–17.
Reid, W. G., Hely, M. A., Morris, J. G., Loy, C., & Halliday, G. M. (2011). Dementia in Parkinson’s disease: a 20-year neuropsychological study (Sydney Multicentre Study). Journal of Neurology, Neurosurgery, and Psychiatry, 82(9), 1033–1037. doi:10.1136/jnnp.2010.232678.
Robbins, T. W., & Cools, R. (2014). Cognitive deficits in Parkinson’s disease: a cognitive neuroscience perspective. Movement Disorders, 29(5), 597–607. doi:10.1002/mds.25853.
Scherling, C. S., Hall, T., Berisha, F., Klepac, K., Karydas, A., Coppola, G., et al. (2014). Cerebrospinal fluid neurofilament concentration reflects disease severity in frontotemporal degeneration. Annals of Neurology, 75(1), 116–126.
Schmidt, M. (1996). Rey auditory verbal learning test: a handbook. Los Angeles: Western Psychological Services.
Sebaldt, R., Dalziel, W., Massoud, F., Tanguay, A., Ward, R., Thabane, L., et al. (2009). Detection of cognitive impairment and dementia using the animal fluency test: the DECIDE study. The Canadian Journal of Neurological Sciences, 36(05), 599–604.
Shaw, L. M., Vanderstichele, H., Knapik‐Czajka, M., Clark, C. M., Aisen, P. S., Petersen, R. C., et al. (2009). Cerebrospinal fluid biomarker signature in Alzheimer’s disease neuroimaging initiative subjects. Annals of Neurology, 65(4), 403–413.
Siderowf, A., Xie, S. X., Hurtig, H., Weintraub, D., Duda, J., Chen-Plotkin, A., et al. (2010). CSF amyloid {beta} 1–42 predicts cognitive decline in Parkinson disease. Neurology, 75(12), 1055–1061. doi:10.1212/WNL.0b013e3181f39a78.
Smith, A. (2000). Symbol digit modalities test: SDMT: Testzentrale.
Sperling, R., Donohue, M., & Aisen, P. (2012). The A4 trial: anti-amyloid treatment of asymptomatic Alzheimer’s disease. Alzheimer’s & Dementia, 8(4), 425–P426.
Steering, D. (1989). DATATOP: a multicenter controlled clinical trial in early Parkinson’s disease. Archives of Neurology, 46(10), 1052–1060.
Stewart, T., Liu, C., Ginghina, C., Cain, K. C., Auinger, P., Cholerton, B., et al. (2014). Cerebrospinal fluid alpha-synuclein predicts cognitive decline in Parkinson disease progression in the DATATOP cohort. [Randomized Controlled Trial Research Support, N.I.H., Extramural]. The American Journal of Pathology, 184(4), 966–975. doi:10.1016/j.ajpath.2013.12.007.
Strozyk, D., Blennow, K., White, L., & Launer, L. (2003). CSF Aβ 42 levels correlate with amyloid-neuropathology in a population-based autopsy study. Neurology, 60(4), 652–656.
Tachibana, H. (2013). Cognitive impairment in Parkinson’s disease. Seishin Shinkeigaku Zasshi, 115(11), 1142–1149.
Tariot, P. N., Ho, C., Langlois, C., Reiman, E. M., Lopera, F., Langbaum, J. B., et al. (2014). The Alzheimer’s prevention initiative. Alzheimer’s & Dementia: The Journal of the Alzheimer’s Association, 10(4), P247.
Terrelonge, M., Jr., Marder, K. S., Weintraub, D., & Alcalay, R. N. (2015). CSF beta-amyloid 1–42 predicts progression to cognitive impairment in newly diagnosed Parkinson disease. Journal of Molecular Neuroscience. doi:10.1007/s12031-015-0647-x.
Tombaugh, T. N., Kozak, J., & Rees, L. (1999). Normative data stratified by age and education for two measures of verbal fluency: FAS and animal naming. Archives of Clinical Neuropsychology, 14(2), 167–177.
Vanderploeg, R. D., Schinka, J. A., Jones, T., Small, B. J., Borenstein Graves, A., & Mortimer, J. A. (2000). Elderly norms for the hopkins verbal learning test-revised. The Clinical Neuropsychologist, 14(3), 318–324.
Vranova, H. P., Henykova, E., Kaiserova, M., Mensikova, K., Vastik, M., Mares, J., et al. (2014). Tau protein, beta-amyloid(1)(−)(4)(2) and clusterin CSF levels in the differential diagnosis of Parkinsonian syndrome with dementia. Journal of Neurological Sciences, 343(1–2), 120–124. doi:10.1016/j.jns.2014.05.052.
Wada-Isoe, K., Imamura, K., Kitamaya, M., Kowa, H., & Nakashima, K. (2008). Serum heart-fatty acid binding protein levels in patients with Lewy body disease. Journal of the Neurological Sciences, 266(1), 20–24.
Warrington, E. K., & James, M. (1991). The visual object and space perception battery. Bury St Edmunds: Thames Valley Test Company.
Williams-Gray, C. H., Evans, J. R., Goris, A., Foltynie, T., Ban, M., Robbins, T. W., et al. (2009). The distinct cognitive syndromes of Parkinson’s disease: 5 year follow-up of the CamPaIGN cohort. Brain, 132(Pt 11), 2958–2969. doi:10.1093/brain/awp245.
Woods, S. P., Delis, D. C., Scott, J. C., Kramer, J. H., & Holdnack, J. A. (2006). The California verbal learning test–second edition: test-retest reliability, practice effects, and reliable change indices for the standard and alternate forms. Archives of Clinical Neuropsychology, 21(5), 413–420.
Yarnall, A. J., Breen, D. P., Duncan, G. W., Khoo, T. K., Coleman, S. Y., Firbank, M. J., et al. (2014). Characterizing mild cognitive impairment in incident Parkinson disease: the ICICLE-PD study. Neurology, 82(4), 308–316. doi:10.1212/wnl.0000000000000066.
Zhang, J., Mattison, H. A., Liu, C., Ginghina, C., Auinger, P., McDermott, M. P., et al. (2013). Longitudinal assessment of tau and amyloid beta in cerebrospinal fluid of Parkinson disease. Acta Neuropathologica, 126(5), 671–682. doi:10.1007/s00401-013-1121-x.
Acknowledgments
This research was supported by grants from the NIH/NIA (AG047366), NIH/NIAAA (AA023165), NIH/NINDS (NS075097, NS091461, NS086085, and NS071675), and Michael J. Fox Foundation for Parkinson’s disease Research.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article

Cite this article
Leaver, K., Poston, K.L. Do CSF Biomarkers Predict Progression to Cognitive Impairment in Parkinson’s disease patients? A Systematic Review. Neuropsychol Rev 25, 411–423 (2015). https://doi.org/10.1007/s11065-015-9307-8
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s11065-015-9307-8