
Abstract
α2-Adrenoreceptors (ARs) are main Gi-protein coupled autoreceptors in sympathetic nerve terminals and targets for dexmedetomidine (DEX), a widely used sedative. We hypothesize that α2-ARs are also potent regulators of neuromuscular transmission via G protein-gated inwardly rectifying potassium (GIRK) channels. Using extracellular microelectrode recording of postsynaptic potentials, we found DEX-induced inhibition of spontaneous and evoked neurotransmitter release as well as desynchronization of evoked exocytotic events in the mouse diaphragm neuromuscular junction. These effects were suppressed by SKF-86,466, a selective α2-AR antagonist. An activator of GIRK channels ML297 had the same effects on neurotransmitter release as DEX. By contrast, inhibition of GIRK channels with tertiapin-Q prevented the action of DEX on evoked neurotransmitter release, but not on spontaneous exocytosis. The synaptic vesicle exocytosis is strongly dependent on Ca2+ influx through voltage-gated Ca2+ channels (VGCCs), which can be negatively regulated via α2-AR – GIRK channel axis. Indeed, inhibition of P/Q-, L-, N- or R-type VGCCs prevented the inhibitory action of DEX on evoked neurotransmitter release; antagonists of P/Q- and N-type channels also suppressed the DEX-mediated desynchronization of evoked exocytotic events. Furthermore, inhibition of P/Q-, L- or N-type VGCCs precluded the frequency decrease of spontaneous exocytosis upon DEX application. Thus, α2-ARs acting via GIRK channels and VGCCs (mainly, P/Q- and N-types) exert inhibitory effect on the neuromuscular communication by attenuating and desynchronizing evoked exocytosis. In addition, α2-ARs can suppress spontaneous exocytosis through GIRK channel-independent, but VGCC-dependent pathway.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
There are arguments for a role of sympathetic nervous system in the regulation of skeletal muscle motor innervation, stability of neuromuscular junctions (NMJs) and neuromuscular communication [1,2,3]. This regulation is dependent on catecholamines and neuropeptides released from sympathetic nerve terminals, which were found in close vicinity to NMJs [4,5,6].
Catecholaminergic control of NMJ structure and function relies on adrenoceptors (ARs), whose different sub-types (namely, α1, α2, β1 and β2) can have a specific relevance for the interactions between presynaptic nerve terminal, perisynaptic Schwann cell and muscle fiber [7,8,9,10,11]. In previous skeletal muscle studies, most attention was focused on the effects of β2-adrenomimetics, since treatments with β2-adrenergic agonists can improve skeletal muscle function in several disorders (congenital myasthenic syndromes, myasthenia gravis, amyotrophic lateral sclerosis and Pompe disease) associated with severe NMJ defects [11,12,13,14,15,16]. However, α2-adrenomimetics, widely used as a sedatives, can inhibit abnormal muscle hypertrophy, attenuate tourniquet-induced structural and functional impairment of the skeletal muscles, oppose opioid-induced muscle rigidity and lipopolysaccharide-induced muscle wasting, as well as prevent diaphragmic fiber atrophy during mechanical ventilation and skeletal muscle ischemia-reperfusion injury [17,18,19,20,21,22]. α2-Agonist DEX worsens mechanical ventilation-induced contractile dysfunction of the diaphragm and can cause muscle flaccidity [17, 23, 24]. The α2-adrenergic regulation of NMJ function may be implicated in the protective and (or) negative effects of α2-AR agonist DEX. However, little is known about the role of α2-ARs in neuromuscular transmission, which defines both the efficacy of translation of motor commands to muscle fibers and the integrity of NMJs. The latter can be compromised by excessive acetylcholine release or its deficit [25,26,27,28].
Previously, we showed that DEX suppresses spontaneous release and decrease the probability of quantum release upon stimulation at low Ca2+ and high Mg2+ in the extracellular solution [9]. Whether α2-AR activation has an inhibitory effect at the physiological Ca2+ level and its underlying mechanism remain unclear. In the present study, we examined the hypothesis that presynaptic α2-ARs may negatively regulate neurotransmitter release via G protein-gated inwardly rectifying potassium (GIRK) channels, thereby hindering activation of the voltage-gated Ca2+ channels (VGCCs) that regulate both evoked and spontaneous neurotransmitter release [29, 30].
Herein, we found that pharmacological stimulation of α2-ARs suppresses evoked neurotransmitter exocytosis and synchronous release (i.e., desynchronizes the quantal release) via a GIRK channel / VGCC (mainly, P/Q- and N-types)-dependent mechanism. While the α2-AR-mediated inhibition of spontaneous exocytosis was GIRK channel-independent and relied on inhibition of P/Q-, L-, or N-type VGCCs.
Materials and Methods
Experimental Animals and Solutions
Experiments were performed on isolated mouse phrenic nerve-hemidiaphragm preparations. BALB/c mice were maintained at a 12-h light/12-h dark cycle; water and food were provided ad libitum. Mice (3–4 months of age) were decapitated with a guillotine; diaphragm muscle with phrenic nerve stubs was quickly excised. The experimental protocols (#23 − 1/23 − 2/3, February 11, 2023 and #23/7; May 12, 2023) were approved by the Bioethics Committee of Kazan Federal Scientific Centre and met the requirements of the EU Directive 2010/63/EU as well as European Convention for the Protection of Vertebrate Animals used for Experimental and other Scientific Purposes (Council of Europe No 123, Strasbourg, 1985).
The diaphragm muscle was dissected into hemidiaphragms with nerve stubs. Neuromuscular preparation was placed in an experimental chamber (total volume of 5 ml) with a Sylgard-coated transparent base and the motor nerve was loosely drawn into a suction electrode connected to an isolated stimulator. The chamber was continuously perfused at ∼3 ml/min and the solution level in the chamber was kept constant. The recording solution was modified Krebs-Ringer Solution (HEPES-buffered): 150.0 mM NaCl, 5.0 mM KCl, 2.0 mM CaCl2, 1.0 mM MgCl2, 11.0 mM glucose, and 5.0 mM HEPES, pH 7.4 with NaOH. The temperature was maintained at 24.0 ± 0.3 °C. The muscle contractions were blocked by incubating the preparations with muscle-type Nav1.4 channel blocker GIIIB µ-conotoxin (Peptide Institute Inc., Japan) at 0.5 µM for 15–20 min followed by thorough rinsing with toxin-free buffer solution [31]. In some experiments, low Ca2+/ high Mg2+ physiological solution containing 150.0 mM NaCl, 5.0 mM KCl, 0.5 mM CaCl2, 5.0 mM MgCl2, 5 mM HEPES, and 11.0 mM glucose was used [9].
Electrophysiological Recordings
The motor nerve was stimulated with supra-threshold stimuli with a 0.1-ms duration at a frequency of 0.5 Hz The method of extracellular recordings and data acquisition and processing was described in detail earlier [9, 32]. Briefly, spontaneous miniature end-plate potentials (MEPPs), presynaptic action potentials, and evoked end-plate potentials (EPPs) were acquired in the junctional region. Extracellular saline-filled micropipettes with a tip diameter of 2–3 μm and input resistance of 2–3 MΩ were placed under the microscope (Olympus BX51WI) control in the junctional zone, where three-phase presynaptic action potentials were observed. The precise position of micropipettes and the stability of presynaptic action potential amplitude and duration were monitored throughout the experiment. The acquired signals were filtered between 0.03 Hz and 10 kHz, digitized at 3 µs intervals by an analog-to-digital converter, fed into the computer and analyzed. MEPP and EPP amplitude, rise time (between 20 and 80% of maximum amplitude), and the time constants of the exponential decay (tau), as well as a ratio of EPP to MEPP rise time (Rt/rt) were estimated. For MEPPs signal-to-noise ratio was > 4.5:1 and threshold for spontaneous signal detection was set at level of 0.2 mV. The frequency of MEPPs was evaluated in each experiment after recording 150–200 spontaneous signals. As an indicator of evoked neurotransmitter release was used a ratio (m) of the mean EPP amplitude and the mean MEPP amplitude (EPP/MEPP amplitude ratio). For this analysis 150–200 MEPPs and EPPs were recorded before and after application of drugs.
Extracellular microelectrode recording at low Ca2+ and high Mg2+ in external solution was used for a precise evaluation of synchronicity of exocytotic events and synaptic delays. Under these conditions, the probability of neurotransmitter release upon stimulus is less than one and uni-quantal EPPs are observed. 200–300 uni-quantal EPPs were recorded in control and after drug application. The probability (m”) of evoked neurotransmitter release was calculated as m”=lnN/N0 (“method of failures”), where N is the total number of impulses and N0 is the number of synaptic failures [33]. To analyze MEPP frequency, 150–200 MEPPs were recorded at the resting conditions. To estimate the kinetics of single exocytotic events upon action potentials, the synaptic delays of uni-quantal EPPs were calculated as a period from the peak of sodium current spike to the point corresponding to 20% of the EPP rise phase. A measure of synaptic delay dispersion, Pcorr (in ms) was estimated from the synaptic delay cumulative distribution curve. Pcorr is the interval between the minimal synaptic delay and the point at which 90% of all uni-quantal EPPs had occurred [34, 35]. Mode and mean value of the synaptic delay distribution were calculated.
Drug Applications
Dexmedetomidine hydrochloride (10 µM DEX; Cat. # SML0956 Sigma) and SKF86466 hydrochloride (10 µM SKF; Cat. # 86129-54-6, Santa Cruz) were used as α2-AR agonist and antagonist, respectively [9, 36]. Our preliminary experiment showed that at lower concentrations (0.5-5 µM) DEX had no significant effects on the parameters of MEPPs and EPPs. Specific channel antagonists were used to block different types of VGCCs: ω-agatoxin IVA (25 nM; Cat. # 4256-s, Peptide Institute, Inc.) for P/Q-type channels (Cav2.1); nitrendipine (10 µM; Cat. # 39562-70-4; Tocris) for L-type channels (Cav1.2), SNX 482 (30 nM; Cat. #4363-s, Peptide Institute, Inc.) for R-type channels (Cav2.3); ω-conotoxin GVIA (10 nM; Cat. # C-300, Alomone Labs) for N-type channels (Cav2.2) [37,38,39,40]. To activate and inhibit GIRK channels, 10 µM ML297 (Cat. # 1443246-62-5, Tocris) and 100 nM tertiapin-Q (TPQ; Cat. # 910044-56-3, Tocris) were applied [41, 42]. Postsynaptic responses were acquired in control and 20 min after the application of the drug-containing solution. To estimate the effect of one drug in the presence of another reagent, the latter reagent was added to the bathing saline 20 min before exposure to the former drug and remained in the perfusion throughout the experiment, excluding the cases of irreversible inhibitors (ω-agatoxin IVA and ω-conotoxin GVIA), which were applied for 20 min followed by washout. The parameters of MEPPs and EPPs did not change after 20 min incubation in the absence of drugs (Suppl. Figure 1a, b).
Statistics
Origin Pro software was used for statistical analysis. Data are presented as mean ± standard deviation (SD). The sample size (n) is the number of independent experiments on separate neuromuscular preparations (muscles) from individual animals; n is indicated in each figure legend, supplementary figure legend and supplementary table. There were no exclusions of outliers; the sample size was determined based on reasonable value of SD. Normality (Shapiro-Wilk test) and variance homogeneity (two sample F-test for variance) were tested. Significance was assessed by a two-tailed, paired t-test or Mann-Whitney U-test. * P ˂ 0.05, ** P ˂ 0.01 and *** P ˂ 0.001 were considered as statistically significant.
Results
Inhibitory Action of α2-adrenergic Receptor Stimulation on Spontaneous and Evoked Neurotransmitter Release
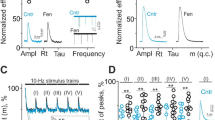
Dexmedetomidine (10 µM; DEX), a selective α2-adrenergic agonist, did not change the MEPP amplitude (Fig. 1a), but decreased the EPP amplitude (Fig. 1b). As a result, the ratio of EPP/MEPP amplitudes (an indicator of evoked neurotransmitter release) decreased (Fig. 1c). In addition, the pharmacological stimulation of α2-ARs reduced the frequency of MEPPs (Fig. 1c).
In the presence of DEX, the kinetics of MEPPs did not change (Fig. 1a) while the rise and decay time of EPPs increased (Fig. 1b). A ratio of the EPP to MEPP rise times (Rt/rt) was 1.8 ± 0.2 in control, and it increased to 2.1 ± 0.3 (n = 9, P = 0.022) upon the application of DEX (Fig. 1c). The DEX-mediated increase in the rise time of EPPs can be a reflection of presynaptic changes. Particularly, the elevation of Rt/rt as well as an increase in the EPP decay time as opposed to the unchanged temporal parameters of MEPPs is an indicator of reduced synchronization of neurotransmitter release [43]. Indeed, the analysis of uni-quantal EPPs (unitary exocytotic events upon nerve stimulation) at low external Ca2+ levels showed a shift of the synaptic delay distribution towards higher values (Fig. 1d). The mode of the distribution and the mean value of synaptic delay increased by 12 ± 13% (P = 0.008) and 11 ± 11% (P = 0.011, n = 12), respectively. At low [Ca2+]out DEX decreased the amplitude of the uni-quantal EPPs as well as the decay time of both MEPPs and uni-quantal EPPs (Suppl. Figure 2a, b).

Effects of selective α2-adrenergic agonist (10µM dexmedetomidine) and antagonist (10 µM SKF 86,466) on spontaneous and evoked neurotransmitter release. (a-c) – Influence of dexmedetomidine (DEX) on amplitude-temporal parameters (Amp, amplitude; rt or Rt, rise time; tau, decay time) of MEPPs (a) and EPPs (b) as well as EPP/MEPP amplitude ratio (m), MEPP frequency (f) and the ratio of Rt/rt (indicator of synchrony of evoked exocytic events constituting multi-quantal EPPs) (c). Inserts, the representative traces of the postsynaptic responses in control (Cntr) and after exposure to DEX. (d) – Distribution of real synaptic delays of evoked exocytosis events recorded at low external Ca2+ level. Under these conditions, uni-quantal EPPs were recorded. Shown averaged curves (n = 12 animals/group). (e-i)- Effects of SKF 86,466 itself and DEX in the presence of SKF 86,466 on the parameters of MEPPs (e) and EPPs (f) as well as EPP/MEPP amplitude ratio (g), frequency of MEPPs (h) and Rt/rt ratio (i). Inserts, the representative postsynaptic responses. (a-c), (e-f) – Data are expressed as average values in individual animals and SD (whiskers); shown changes (in %), where values before 20-min application of drug (DEX, SKF or DEX in SKF-pretreated muscles) were set as 100%. n = 8–9 animals/group. * P < 0.05, ** P < 0.01, *** P < 0.001 – by a paired, two-tailed Student’s t-test compared to control; * P˂0.05 – by Mann-Whitney U-test between independent groups (i)
Thus, pharmacological α2-adrenergic stimulation with DEX can decrease both evoked and spontaneous release while the delay of quantal release increases and varies towards the asynchrony. All this decreases the efficacy of neuromuscular communication. At decreased extracellular Ca2+ concentration, α2-AR agonist also exerted postsynaptic effects, decreasing currents through nicotinic acetylcholine receptors (nAChRs).
The selective α2-AR antagonist SKF 86,466 (10 µM) did not affect the amplitude of either MEPPs (Fig. 1e) or EPPs (Fig. 1f). As a result, the EPP/MEPP amplitude ratio was unchanged (Fig. 1g). Also, SKF failed to affect the MEPP frequency (Fig. 1h). Probably, the levels of endogenous catecholamines, which are released in ex vitro neuromuscular preparations, are not enough to stimulate the presynaptic α2-ARs, whose activation can suppress evoked and spontaneous release (Fig. 1c).
SKF increased the decay time of both MEPPs and EPPs as well as the rise time of EPPs at both normal and low extracellular Ca2+ levels (Fig. 1e, f and Suppl Fig. 2c, d). At normal [Ca2+]out, the ratio of Rt/rt was also elevated in SKF-treated muscles (Fig. 1i), but experiments at low external Ca2+ did not reveal the changes in synaptic delay distribution of uni-quantal EPPs (Suppl. Figure 2e). Accordingly, inhibition of α2-ARs at the basal conditions can mainly modulate neurotransmission at the postsynaptic level, probably releasing nAChR from a tonic influence of muscle fiber α2-ARs. Indeed, at low [Ca2+]out, the antagonist and agonist of α2-ARs increased (Suppl. Figure 2c, d) and decreased (Suppl. Figure 2a, b) decay time of the postsynaptic responses (MEPPs and uni-quantal EPPs), respectively.
In the presence of α2-antagonist SKF, DEX lost its ability to affect EPP rise and decay times (Fig. 1f) as well as the EPP/MEPP amplitude ratio and MEPP frequency (Fig. 1g, h). However, under these conditions, the DEX-mediated increase in the Rt/rt ratio was preserved (Fig. 1i). Simultaneously, at low external Ca2+ level the inhibition of α2-ARs completely prevented the shift of the synaptic delay distribution (including the mode and mean values) upon DEX administration (Suppl Fig. 2f). Overall, an inhibitor of α2-ARs precluded the inhibitory effect of DEX on both spontaneous and evoked release.
GIRK Channel as a Modulator of Neurotransmitter Release and Target for α2-adrenergic Regulation
One of the targets for the α2-AR-dependent regulation could be G protein-coupled inwardly rectifying K+ (GIRK) channels, which are directly modulated by pertussis toxin-sensitive G proteins [44, 45]. These channels are important regulators of both postsynaptic excitability and neurotransmitter release [46].
ML297 (10 µM), a selective activator of GIRK channels [41], had no marked influence on amplitude-temporal parameters of MEPPs, excluding a slight increase in the decay time (Fig. 2a). At the same time, the EPP amplitude and the ratio of EPP/MEPP amplitudes were reduced (Fig. 2b, c); the Rt/rt ratio was increased (Fig. 2c). Also, ML297 suppressed the MEPP frequency (Fig. 2c). Hence, pharmacological activation of GIRK channels can decrease both evoked and spontaneous neurotransmitter release as well as desynchronize exocytotic events upon action potential (Fig. 2a-c). These effects are the same as those of α2-AR agonist DEX (Fig. 1a-c).
To test the involvement of GIRK channels in the effects of α2-AR activation, tertiapin Q (TPQ, 100 nM), a blocker of inwardly rectifying K+ channels [47], was used. TPQ itself did not change the amplitude or the kinetic parameters of MEPPs or EPPs at normal extracellular Ca2+, excluding an increase in EPP rise time (Fig. 2d). As a result, TPQ did not affect the EPP/MEPP amplitude ratio (m), but caused an elevation of the RT/rt ratio (Fig. 2e). However, the latter can be attributed to a postsynaptic action, since at low external Ca2+ level TPQ increased the rise time of both MEPPs and uni-quantal EPPs (Suppl. Figure 3b, c) and did not desynchronize exocytotic events. Instead, at low external Ca2+ neurotransmitter quanta were released more synchronously after TPQ application (Suppl. Figure 3f). TPQ decreased MEPP frequency at low and normal [Ca2+]out (Fig. 2e; Suppl. Figure 3e). At normal external Ca2+ level, the effect of TPQ on MEPP frequency was prevented by α2-AR antagonist SKF 86,466 (Suppl. Figure 3a). Thus, the inhibitor of inwardly rectifying K+ channels had no marked influence on number of quanta released upon presynaptic action potential, but suppressed spontaneous release. The latter effect may require a tonic activation of α2-ARs.

Effects of GIRK channel activator (10 µM ML297) and blocker (100 nM tertiapin-Q, TPQ) on spontaneous and evoked neurotransmitter release. (a-c) – Influence of ML297 on amplitude-temporal parameters (Ampl, amplitude; rt or Rt, rise time; tau, decay time) of MEPPs (a) and EPPs (b) as well as EPP/MEPP amplitude ratio (m), MEPP frequency (f) and Rt/rt ratio (c). Inserts, the typical postsynaptic responses. (d) – Changes in amplitude-temporal parameters of MEPPs and EPPs upon TPQ application. (e) – Influence of TPQ itself and DEX in the presence of TPQ on EPP/MEPP amplitude ratio, MEPP frequency and Rt/rt ratio. The effect of DEX in control (from Fig. 1c) is also shown. (a-e) – Data are expressed as average values in individual animals and SD (whiskers); shown changes (in %), where values before the 20-min application of drug (ML297, TPQ, DEX or DEX in TPQ-pretreated muscles) were set as 100%. n = 7–9 animals/group. * P < 0.05, ** P < 0.01, *** P < 0.001 – by a paired, two-tailed Student’s t-test compared to control
The analysis of ML297 and TPQ action suggests that GIRK channels can bidirectionally modulate spontaneous exocytosis (Fig. 2c vs. Fig. 2e). Indeed, despite the reduction of MEPP frequency upon 20-min application of the GIRK channel activator ML297 (Fig. 2c), in the initial 5-min period of ML297 administration the MEPP frequency was transiently elevated (Suppl. Figure 3a, insert). This can reflect different roles of GIRK channel subpopulations (e.g., in presynaptic nerve terminal and presynaptic Schwann cell).
The blockage of GIRK channels by TPQ completely prevented the inhibitory effect of DEX on the EPP/MEPP amplitude ratio (Fig. 2e). At the same time, DEX-induced decrease in MEPP frequency persisted in the presence of GIRK channel antagonist (Fig. 2e). Accordingly, pharmacological stimulation of α2-ARs can suppress evoked (but not spontaneous) neurotransmitter release via GIRK channel activation.
With regard to the kinetics of quantal release, after blockage of GIRK channels the application of DEX did not modulate the ratio of RT/rt (Fig. 2e) and Pcorr (a measure of synaptic delay dispersion; Supp Fig. 2f) at normal and low external Ca2+ level, respectively. This can reflect a major contribution of GIRK channels in α2-AR-dependent desynchronization of quantal neurotransmitter release at both normal and reduced extracellular Ca2+ levels.
It is worth noting that, at low external Ca2+ levels, inhibition of GIRK channels cannot prevent DEX-mediated decrease in both the frequency of spontaneous exocytosis and probability of single evoked exocytotic events (Suppl Fig. 3d, e). Accordingly, α2-ARs can suppress the probability of evoked exocytotic events, at least at reduced [Ca2+]out, via GIRK channel-independent mechanism. Furthermore, pretreatment with GIRK channel activator ML297 (which itself has effects similar to DEX) prevented only DEX-mediated desynchronization of neurotransmitter release, but not a reduction of evoked and spontaneous neurotransmitter release at normal extracellular Ca2+ level (Suppl Fig. 4).
Taken together, these data suggest that pharmacological activation of both GIRK channels and α2-ARs can suppress spontaneous and evoked neurotransmitter release as well as desynchronize neurotransmitter quanta release. At the same time, α2-AR-mediated attenuation of spontaneous exocytosis is GIRK channel independent, whereas α2-AR-induced decrease in evoked neurotransmitter release (partially) and desynchronization of exocytotic events rely on GIRK channel activation.
The voltage-gated Ca2+ Channels tune the Acetylcholine Release
Evoked neurotransmitter release and its timing as well as spontaneous exocytosis are dependent on Ca2+ influx into the axoplasm through VGCCs [29, 30, 43]. Presynaptic VGCCs, whose activity can be controlled by GIRK channel-dependent hyperpolarization and direct interaction with Gβγ [42, 45, 48, 49], can be key elements in α2-adrenergic regulation of neurotransmitter release in the NMJs.
Herein, selective blockers of the main axonal VGCCs were used (Fig. 3a). Particularly, P/Q-type, L-type, N-type and R-type VGCCs were inhibited with 25 nM ω-agatoxin IVA, 10 µM nitrendipine, 1 µM ω-conotoxin GVIA and 30 nM SNX 482, respectively. The effective concentrations for partial channel blockage were chosen based on previous studies [37,38,39,40]. None of the VGCC blockers affected the amplitude-temporal parameters of MEPPs (Suppl. Table 1), indicating no marked postsynaptic effects of the VGCC inhibition. At the same time, all blockers had effects on the parameters of EPPs, decreasing their amplitude and(or) increasing rise time. The antagonist of R-type VGCCs also increased EPP decay time (Suppl. Table 1).
The antagonists of P/Q- and R-types decreased both the EPP/MEPP amplitude ratio and MEPP frequency as well as increased the ratio of RT/rt, a sign of quantal release desynchronization (Fig. 3b-d). Inhibition of N-type channels had the same effects, but a decrease in the EPP/MEPP amplitude ratio did not reach statistical significance (P = 0.052). The antagonists of P/Q- (a main type in the mammalian NMJs [50]) and R-type channels acting in close nanomolar concentrations affected evoked and spontaneous neurotransmitter release with comparable efficacy. This points to a strong dependence of both evoked and spontaneous synaptic vesicle exocytosis on the activity of P/Q and R-type VGCCs in the NMJs.

Effects of selective voltage-gated Ca2+ channel (VGCC) antagonists on evoked and spontaneous neurotransmitter release. (a) – schematic representation of blockage of VGCCs with AgaTx - ω-agatoxin IVA (25 nM), Nitr – nitrendipine (10 µM), CgTx - ω-conotoxin GVIA (10 nM), SNX - SNX 482 (30 nM). (b-d), Shown changes (in %) in EPP/ MEPP amplitude ratio (b), MEPP frequency (c), and Rt/rt ratio (d) in response to application of specific VGCC blockers. Data are expressed as values in individual animals ± SD (whiskers). n = 6–9 animals/group. * P < 0.05, ** P < 0.01, *** P < 0.001 – by paired, two-tailed Student t-test compared to control
Nitrendipine, an antagonist of L-type VGCCs, had no effect on the EPP/MEPP amplitude ratio, but decreased MEPP frequency and increased the RT/rt ratio (Fig. 3b-d). Hence, a decrease in spontaneous release and desynchronization of evoked exocytotic events can occur at inhibition of any type VGCCs. It should be noted that the effects of VGCC inhibitors (especially, P/Q and R-type) resemble the influence of pharmacological stimulation of α2-ARs in the NMJs. This suggests the existence of an α2-AR-dependent mechanism of inactivation of the VGCCs.
Engagement of voltage-gated Ca2+ Channels in Presynaptic Action of α2-adrenergic Receptor Stimulation
To test the involvement of VGCCs in the effects of α2-AR agonist DEX, DEX was added into the bathing solution after inhibition of P/Q-, N-, L-, and R- types of VGCCs with selective antagonists (Fig. 4).

Role of voltage-gated Ca2+ channels (VGCCs) in the effects of α2-adrenergic agonist dexmedetomidine (DEX). (a) – scheme illustrating a putative contribution of different VGCCs in α2-adrenergic control of evoked (left) and spontaneous (right) neurotransmitter release. (b-d) – Shown the effects of DEX on EPP/MEPP amplitude ratio (b), MEPP frequency (c), and Rt/rt ratio (d) in control (Cntr; from Fig. 1c) and after pre-treatment with selective VGCC antagonists. Data are expressed as values in individual animals ± SD (whiskers). n = 6–9 animals/group. * P < 0.05, ** P < 0.01, *** P < 0.001 – by paired, two-tailed Student t-test compared to values before DEX application. AgaTx, ω-agatoxin IVA; Nitr – nitrendipine; CgTx - ω-conotoxin GVIA; SNX - SNX 482
The blockage of any type of VGCCs (even L-type) prevented α2-AR agonist-mediated decrease in the EPP/MEPP amplitude ratio (Fig. 4a, b). There was only a trend to DEX-mediated reduction of the EPP/MEPP amplitude ratio (P = 0.11) in the presence of R-type channel blocker. Hence, a decrease in activity of P/Q-, N-, R- or L-type VGCCs is enough to suppress the effect of α2-AR stimulation on the evoked neurotransmitter release. It is consistent with a hypothesis that α2-AR-dependent GIRK channel opening could nonspecifically hinder the activation of VGCCs upon presynaptic action potential.
The inhibitory effect of α2-AR agonist on MEPP frequency, which persisted in the presence of GIRK channel inhibitor (Fig. 2f), was prevented by antagonists of P/Q-, N- and L- types of VGCCs (Fig. 4a, c). This suggests that GIRK channel-independent and VGCC-dependent pathway mediates the suppression of spontaneous neurotransmitter release upon α2-AR activation. So, α2-AR-driven regulation of evoked and spontaneous exocytosis occurred via similar VGCCs, but engaged different downstream pathways.
Finally, the increase in RT/rt ratio upon DEX was precluded by antagonists of only P/Q and N-type of VGCCs (Fig. 4a, d). Although, inhibition of any type of VGCCs (including L- and R-types) was able to increase the RT/rt ratio to similar degree (Fig. 3d). Hence, α2-AR-induced desynchronization of neurotransmitter quanta release can be mediated by P/Q- and N-type VGCCs. Furthermore, inhibition of these two types of VGCCs prevented the effects of α 2-AR agonist on both spontaneous and evoked release. This suggests the key role of P/Q- and N-type VGCCs in the presynaptic action of α2-adrenergic agonist.
Discussion
The main finding of the present study are: (i) pharmacological activation of α2-ARs can negatively regulate neuromuscular communication by decreasing neurotransmitter release and desynchronizing quantal release; (ii) GIRK channel opener has the same presynaptic action as the α2-AR activation; (iii) α2-AR-dependent suppression of evoked release depends on GIRK channels as well as P/Q- and N-type VGCCs, whereas decrease in spontaneous exocytosis upon α2-AR stimulation relies on P/Q-, N- and L-type VGCCs, but not GIRK channels; (iv) at low external Ca2+ levels, α2-ARs attenuates the postsynaptic currents via nAChRs.
Skeletal muscles are characterized by extensive sympathetic innervation and a close contact between sympathetic nerve endings and NMJs [2, 4,5,6]. Along with β1- and β2-ARs, α2-ARs of different subtypes (α2A, α2B, α2C) are expressed in the skeletal muscles [8,9,10]. Although, α2-ARs can act as autoreceptors on the sympathetic varicosities in the skeletal muscles [10], our pharmacological analysis together with evidence about suppression of skeletal muscle activity upon DEX treatment [17, 23, 24] suggest direct inhibitory role of α2-ARs in the neuromuscular transmission. Indeed, DEX decreased both evoked and spontaneous neurotransmitter release as well as desynchronized the action potential-elicited exocytotic events. The latter causes a reduction of the peak EPP amplitude, thereby decreasing the safety factor of neuromuscular transmission [43]. DEX-mediated inhibition of neurotransmitter release was also expressed at low Ca2+ / high Mg2+ in the extracellular solution [9]. Furthermore, at low external Ca2+ levels, DEX and α2-AR antagonist SKF decreased and increased, respectively, decay time of both MEPPs and uni-quantal EPPs. SKF increased decay time of MEPPs and EPPs at normal external Ca2+. This points to suppression of currents via postsynaptic nAChRs upon α2-AR activation. Inhibition of nAChR currents by DEX was found in rat superior cervical ganglion neurons [51].
Hypothetically, α2-AR-dependent inhibition of neuromuscular transmission can be a mechanism protecting the muscle from overactivation, especially under stressful conditions. Taking into account that excessive ACh can have detrimental effects on NMJ integrity [25,26,27], the ability of DEX to alleviate muscle injury [18, 19, 21, 22] can be partially dependent on the limitation of spontaneous or (and) evoked exocytosis. At the same time, previously we did not reveal inhibitory action of DEX on neurotransmitter release at high frequency activity, but DEX suppressed “kiss-and-run” mode of exocytosis and increased the dependence of neurotransmission on neurotransmitter refiling during intense activity [5].
α2-AR-induced presynaptic inhibition can be mediated by a direct interaction of Gβγ with VGCCs, GIRK channels or SNARE complex as well as suppression of adenylate cyclase activity [48, 52,53,54,55,56]. In the NMJs, mechanisms underlying inhibitory action of α2-AR were not investigated. Recently, we have found that inhibition of GIRK channels can synchronize exocytotic events with presynaptic action potential and mediate effects of muscarinic AChRs on neurotransmitter release in the frog NMJs [42]. In mammalian NMJs, the role of GIRK channels is unknown. Herein, we revealed that (i) a selective GIRK channel activator ML297 [41] has the same effects on the neurotransmission as α2-AR agonist, and (ii) GIRK channel antagonist TPQ prevents the DEX-induced reduction of evoked neurotransmitter release and desynchronization of quantal release. However, DEX retained the ability to decrease spontaneous exocytosis after both inhibition or activation of GIRK channels. Accordingly, α2-ARs can regulate evoked neurotransmitter release in a GIRK channel-dependent manner, whereas the suppression of spontaneous exocytosis upon DEX application is a GIRK channel-independent. At the same time, pharmacological GIRK activation can reduce spontaneous release.
Probably, there are subpopulations of presynaptic GIRK channels, which differ in coupling to Gβγ and regulate mainly evoked or spontaneous exocytosis. Similarly, an activation of KATP channels negatively regulates both spontaneous and evoked exocytosis in the frog NMJs, but KATP channel activity modulates inhibitory effects of adenosine selectively on the evoked exocytosis [57]. This is consistent with the hypothesis that separate sites and regulatory mechanisms for evoked and spontaneous exocytosis can exists [58, 59]. Furthermore, GIRK channel activation can transiently enhance spontaneous exocytosis in Ca2+-independent manner in the frog NMJs [42]. Herein, we also demonstrated that before a sustainable reduction in the MEPP frequency, GIRK channel opener ML297 caused an elevation of spontaneous neurotransmitter release. In addition, TPQ, a blocker of GIRK channels, decreased the MEPP frequency and inhibition of α2-AR prevented this effect of TPQ. Hence, GIRK channels can regulate spontaneous neurotransmitter release in the opposite ways, and the positive regulation of spontaneous exocytosis can be related to α2-ARs. This complexity may reflect a different function of GIRK channels and α2-ARs in the nerve terminals and perisynaptic Schwann cells. Indeed, an activation of G proteins of perisynaptic Schwann cells inhibits neuromuscular release [60]. Finally, GIRK channels of muscle fibers can contribute to neuromuscular transmission in certain conditions. At low external Ca2+ levels, GIRK channel blocker increased the rise time of both MEPPs and uni-quantal EPPs, indicating on alteration of nAChR currents.
Presynaptic K+ currents through GIRK channels can lead to a reduction of the membrane excitability [61,62,63], hindering activation of VGCCs. The latter are the main regulators of evoked and spontaneous exocytosis as well as the kinetics of neurotransmitter release [29, 30, 43]. In the mice NMJs, more than a dozen VGCCs is located in close vicinity to the exocytotic site and the docked synaptic vesicle [64]. Although P/Q-type VGCCs represent a main entry gate for Ca2+ in the active zone region [50, 65], N-, L- and R- type channels can also contribute to control of neurotransmitter release in the adult NMJs [66]. These channels can compensate defects of P/Q-type channel function [67,68,69,70,71], control the synaptic vesicle recycling [72] and the timing of neurotransmitter release [35, 73].
Using selective P/Q-, N-, L- and R-type VGCC antagonists, we found that: P/Q and R-type channels control the evoked neurotransmitter release; activity of all tested VGCCs is important to synchronize evoked exocytotic events as well as to maintain spontaneous exocytosis. First, this is consistent with the ability of R-type VGCCs to compensate a disturbance in evoked neurotransmitter release due to a reduction of the currents via P/Q-type VGCCs in the NMJs of lethargic and tottering mice or mice with deletion of the Cacna1a allele [67,68,69, 71]. It seems that R-type channels lay close to Ca2+ sensor(s) for evoked exocytosis, and, hence, can replace P/Q-type VGCCs in the triggering of exocytosis in the adult NMJs [74]. Second, synchronization of evoked exocytotic events can require a cooperative activity of many VGCCs to provide a higher Ca2+ influx into the nerve terminal. Modeling of quantal neurotransmitter release suggests that in the presence of fixed Ca2+ buffers a decrease in Ca2+ influx desynchronizes exocytotic events in the NMJs [75]. Another modeling also proposes a more synchronous release in response to increasing extracellular Ca2+ concentration and, hence, a higher Ca2+ entry in the NMJs [76]. Third, VGCCs are important sources of Ca2+ entry for regulation of spontaneous exocytosis, and an opening of VGCCs can occur at near resting membrane potential [29]. Indeed, stochastic opening of P/Q-, N-, L- and R-type VGCCs in presynaptic nerve terminals can enhance spontaneous exocytosis [42, 77]. Taken together, our data indicate that the activity of all tested types of VGCCs contributes to the enhancement of spontaneous exocytosis and synchronization of evoked exocytotic events, whereas the activity of P/Q- and R-type channels mainly defines the number of quanta released upon action potential.
Like GIRK channel blocker, antagonists of P/Q- and N-type VGCCs prevented the DEX-mediated reduction and desynchronization of evoked neurotransmitter release. This suggests that α2-ARs could decrease evoked neurotransmission via a GIRK-channel, P/Q- and N-type VGCC-dependent mechanism. Previously it has been found that an activation of presynaptic GIRK channels mediates inhibitory effects of histamine H3 receptors, GABA (B) receptors, D3 dopamine receptors and somatostatin receptors on P/Q-type VGCCs [61, 62, 78, 79]. Inhibition of P/Q-, N- and L-type VGCCs suppressed the α2-AR-mediated reduction of spontaneous release. This effect of DEX was not modulated by GIRK channel blocker. Probably, the attenuation of spontaneous exocytosis in response to α2-AR activation can be related to inhibition these VGCCs via direct interactions of Gβγ to the channels or decrease in the channel phosphorylation [52,53,54, 80].
Inhibition of L- and R-type VGCCs also precluded the decrease in evoked exocytosis (but not the desynchronization of neurotransmitter release) upon α2-AR activation. Hence, the α2-AR-mediated modulation of evoked neurotransmitter release disappeared when any type of the VGCCs was blocked. Theoretically, there may be a presynaptic mechanism, which is sensitive to the channel functional state and it can prevent the negative regulation of VGCCs if one of the VGCCs is suppressed. Alternatively, inhibition of L- or R-type VGCCs might cause compensatory “overactivity” of other VGCCs.
Conclusions
Here, we found that the activation of α2-ARs with DEX, a widely used sedative drug, inhibits neuromuscular transmission via the presynaptic mechanism in the main respiratory muscle. Particularly, α2-AR-dependent activation of GIRK channels and inhibition of VGCCs (P/Q- and N-types) led to a decrease in evoked neurotransmitter release and desynchronization of exocytotic events. In addition, a pharmacological activation of α2-ARs suppressed spontaneous neurotransmitter exocytosis due to VGCC inhibition (P/Q-, N-, L-types) in a GIRK channel-independent manner. A limitation of the present work is use of a pharmacological approach mainly targeting α2ARs. Further molecular and genetic studies are needed to uncover the detail mechanism underlying the α2-AR-mediated regulation of neuromuscular transmission.
Data Availability
All mentioned data are represented in the main manuscript figures and supplementary figures. Other additional data will be made available on reasonable request.
Abbreviations
- AR:
-
Adrenoceptor
- DEX:
-
Dexmedetomidine
- EPP:
-
End-plate potential
- GIRK:
-
Channel, G Protein-gated inwardly rectifying potassium channel
- MEPP:
-
Miniature end-plate potential
- NMJ:
-
Neuromuscular junction
- nAChR:
-
Nicotinic acetylcholine receptor
- TPQ:
-
Tertiapin-Q
- VGCC:
-
Voltage-gated Ca2+ channel
References
Wang ZM, Messi ML, Rodrigues ACZ, Delbono O (2022) Skeletal muscle sympathetic denervation disrupts the neuromuscular junction postterminal organization: a single-cell quantitative approach. Mol Cell Neurosci 120:103730
Dmitrieva SA, Vologin SG, Tsentsevitsky AN, Arkhipov AY, Khuzakhmetova VF, Sibgatullina GV, Bukharaeva EA (2023) Sympathetic innervation and endogenous catecholamines in neuromuscular preparations of muscles with different functional profiles. Biochem (Mosc) 88:364–373
Delbono O, Rodrigues ACZ, Bonilla HJ, Messi ML (2021) The emerging role of the sympathetic nervous system in skeletal muscle motor innervation and sarcopenia. Ageing Res Rev 67:101305
Khan MM, Lustrino D, Silveira WA, Wild F, Straka T, Issop Y, O’Connor E, Cox D, Reischl M, Marquardt T, Labeit D, Labeit S, Benoit E, Molgo J, Lochmuller H, Witzemann V, Kettelhut IC, Navegantes LC, Pozzan T, Rudolf R (2016) Sympathetic innervation controls homeostasis of neuromuscular junctions in health and Disease. Proc Natl Acad Sci U S A 113:746–750
Petrov AM, Zakirjanova GF, Kovyazina IV, Tsentsevitsky AN, Bukharaeva EA (2022) Adrenergic receptors control frequency-dependent switching of the exocytosis mode between full-collapse and kiss-and-run in murine motor nerve terminal. Life Sci 296:120433
Straka T, Vita V, Prokshi K, Horner SJ, Khan MM, Pirazzini M, Williams MPI, Hafner M, Zaglia T, Rudolf R (2018) Postnatal Development and Distribution of Sympathetic Innervation in mouse skeletal muscle. Int J Mol Sci 19
Bukharaeva E, Khuzakhmetova V, Dmitrieva S, Tsentsevitsky A (2021) Adrenoceptors modulate cholinergic synaptic transmission at the Neuromuscular Junction. Int J Mol Sci 22
Wang ZM, Messi ML, Grinevich V, Budygin E, Delbono O (2020) Postganglionic sympathetic neurons, but not locus coeruleus optostimulation, activates neuromuscular transmission in the adult mouse in vivo. Mol Cell Neurosci 109:103563
Tsentsevitsky A, Nurullin L, Tyapkina O, Bukharaeva E (2020) Sympathomimetics regulate quantal acetylcholine release at neuromuscular junctions through various types of adrenoreceptors. Mol Cell Neurosci 108:103550
Wang ZM, Rodrigues ACZ, Messi ML, Delbono O (2020) Aging blunts sympathetic Neuron Regulation of motoneurons synaptic vesicle release mediated by beta1- and alpha2B-Adrenergic receptors in geriatric mice. J Gerontol A Biol Sci Med Sci 75:1473–1480
Clausen L, Cossins J, Beeson D (2018) Beta-2 adrenergic receptor agonists enhance AChR Clustering in C2C12 myotubes: implications for Therapy of Myasthenic disorders. J Neuromuscul Dis 5:231–240
Li S, Sun B, Nilsson MI, Bird A, Tarnopolsky MA, Thurberg BL, Bali D, Koeberl DD (2013) Adjunctive beta2-agonists reverse neuromuscular involvement in murine pompe Disease. FASEB J 27:34–44
Ghazanfari N, Morsch M, Tse N, Reddel SW, Phillips WD (2014) Effects of the ss2-adrenoceptor agonist, albuterol, in a mouse model of anti-MuSK myasthenia gravis. PLoS ONE 9:e87840
Bartus RT, Betourne A, Basile A, Peterson BL, Glass J, Boulis NM (2016) beta2-Adrenoceptor agonists as novel, safe and potentially effective therapies for Amyotrophic Lateral Sclerosis (ALS). Neurobiol Dis 85:11–24
McMacken GM, Spendiff S, Whittaker RG, O’Connor E, Howarth RM, Boczonadi V, Horvath R, Slater CR, Lochmuller H (2019) Salbutamol modifies the neuromuscular junction in a mouse model of ColQ myasthenic syndrome. Hum Mol Genet 28:2339–2351
Rodriguez Cruz PM, Cossins J, Cheung J, Maxwell S, Jayawant S, Herbst R, Waithe D, Kornev AP, Palace J, Beeson D (2020) Congenital myasthenic syndrome due to mutations in MUSK suggests that the level of MuSK phosphorylation is crucial for governing synaptic structure. Hum Mutat 41:619–631
Breuer T, Bleilevens C, Rossaint R, Marx G, Gehrenkemper J, Dierksen H, Delpierre A, Weis J, Gayan-Ramirez G, Bruells CS (2018) Dexmedetomidine impairs diaphragm function and increases oxidative stress but does not aggravate diaphragmatic atrophy in mechanically ventilated rats. Anesthesiology 128:784–795
Cheng W, Wu Z, Zhang J, Ren W (2023) Effect of dexmedetomidine on tourniquet-induced skeletal muscle injury. Rev Assoc Med Bras (1992) 69:228–232
Liu M, Liu Y, Li X, Pei M, Han M, Qi F (2022) Dexmedetomidine inhibits abnormal muscle hypertrophy of myofascial trigger points via TNF-alpha/ NF-kappaB signaling pathway in rats. Front Pharmacol 13:1031804
Haouzi P, Tubbs N (2022) Effects of fentanyl overdose-induced muscle rigidity and dexmedetomidine on respiratory mechanics and pulmonary gas exchange in sedated rats. J Appl Physiol (1985) 132:1407–1422
Kundra TS, Thimmarayappa A, Dhananjaya M, Manjunatha N (2018) Dexmedetomidine for prevention of skeletal muscle ischaemia-reperfusion injury in patients with chronic limb ischaemia undergoing aortobifemoral bypass Surgery: a prospective double-blind randomized controlled study. Ann Card Anaesth 21:22–25
Cheng M, Gao T, Xi F, Cao C, Chen Y, Zhao C, Li Q, Yu W (2017) Dexmedetomidine ameliorates muscle wasting and attenuates the alteration of hypothalamic neuropeptides and inflammation in endotoxemic rats. PLoS ONE 12:e0174894
Li SP, Zhou XL, Zhao Y (2020) Sedation with midazolam worsens the diaphragm function than dexmedetomidine and propofol during mechanical ventilation in rats. Biomed Pharmacother 121:109405
Weinger MB, Partridge BL, Henry AF (1995) Dexmedetomidine does not modify the neuromuscular blocking action of vecuronium in the anaesthetized rat. Br J Anaesth 74:455–457
Sugita S, Fleming LL, Wood C, Vaughan SK, Gomes MP, Camargo W, Naves LA, Prado VF, Prado MA, Guatimosim C, Valdez G (2016) VAChT overexpression increases acetylcholine at the synaptic cleft and accelerates aging of neuromuscular junctions. Skelet Muscle 6:31
Zakyrjanova GF, Giniatullin AR, Mukhutdinova KA, Kuznetsova EA, Petrov AM (2021) Early differences in membrane properties at the neuromuscular junctions of ALS model mice: effects of 25-hydroxycholesterol. Life Sci 273:119300
Nervo A, Calas AG, Nachon F, Krejci E (2019) Respiratory Failure triggered by cholinesterase inhibitors may involve activation of a reflex sensory pathway by acetylcholine spillover. Toxicology 424:152232
Cisterna BA, Vargas AA, Puebla C, Fernandez P, Escamilla R, Lagos CF, Matus MF, Vilos C, Cea LA, Barnafi E, Gaete H, Escobar DF, Cardozo CP, Saez JC (2020) Active acetylcholine receptors prevent the atrophy of skeletal muscles and favor reinnervation. Nat Commun 11:1073
Kavalali ET (2020) Neuronal ca(2+) signalling at rest and during spontaneous neurotransmission. J Physiol 598:1649–1654
Homan AE, Meriney SD (2018) Active zone structure-function relationships at the neuromuscular junction. Synapse 72:e22057
Tsentsevitsky AN, Gafurova CR, Mukhutdinova KA, Giniatullin AR, Fedorov NS, Malomouzh AI, Petrov AM (2023) Sphingomyelinase modulates synaptic vesicle mobilization at the mice neuromuscular junctions. Life Sci 318:121507
Khuzakhmetova V, Bukharaeva E (2021) Adrenaline facilitates synaptic transmission by Synchronizing Release of Acetylcholine Quanta from Motor nerve endings. Cell Mol Neurobiol 41:395–401
Del Castillo J, Katz B (1954) The effect of magnesium on the activity of motor nerve endings. J Physiol 124:553–559
Bukcharaeva EA, Kim KC, Moravec J, Nikolsky EE, Vyskocil F (1999) Noradrenaline synchronizes evoked quantal release at frog neuromuscular junctions. J Physiol 517(Pt 3):879–888
Tsentsevitsky AN, Petrov AM (2022) L-type ca(2+) channels at Low External Calcium differentially regulate neurotransmitter release in proximal-distal compartments of the Frog Neuromuscular Junction. Cell Mol Neurobiol 42:2833–2847
Mikami M, Zhang Y, Kim B, Worgall TS, Groeben H, Emala CW (2017) Dexmedetomidine’s inhibitory effects on acetylcholine release from cholinergic nerves in guinea pig trachea: a mechanism that accounts for its clinical benefit during airway irritation. BMC Anesthesiol 17:52
Protti DA, Uchitel OD (1993) Transmitter release and presynaptic Ca2 + currents blocked by the spider toxin omega-Aga-IVA. NeuroReport 5:333–336
Katz E, Ferro PA, Weisz G, Uchitel OD (1996) Calcium channels involved in synaptic transmission at the mature and regenerating mouse neuromuscular junction. J Physiol 497(Pt 3):687–697
Protti DA, Szczupak L, Scornik FS, Uchitel OD (1991) Effect of omega-conotoxin GVIA on neurotransmitter release at the mouse neuromuscular junction. Brain Res 557:336–339
Almog M, Korngreen A (2009) Characterization of voltage-gated ca(2+) conductances in layer 5 neocortical pyramidal neurons from rats. PLoS ONE 4:e4841
Kaufmann K, Romaine I, Days E, Pascual C, Malik A, Yang L, Zou B, Du Y, Sliwoski G, Morrison RD, Denton J, Niswender CM, Daniels JS, Sulikowski GA, Xie XS, Lindsley CW, Weaver CD (2013) ML297 (VU0456810), the first potent and selective activator of the GIRK potassium channel, displays antiepileptic properties in mice. ACS Chem Neurosci 4:1278–1286
Tsentsevitsky AN, Khaziev EF, Kovyazina IV, Petrov AM (2022) GIRK channel as a versatile regulator of neurotransmitter release via L-type ca(2+) channel-dependent mechanism in the neuromuscular junction. Neuropharmacology 209:109021
Bukharaeva EA, Skorinkin AI, Samigullin DV, Petrov AM (2022) Presynaptic acetylcholine receptors modulate the Time Course of Action Potential-Evoked Acetylcholine Quanta Secretion at Neuromuscular junctions. Biomedicines 10
Bunemann M, Bucheler MM, Philipp M, Lohse MJ, Hein L (2001) Activation and deactivation kinetics of alpha 2A- and alpha 2 C-adrenergic receptor-activated G protein-activated inwardly rectifying K + channel currents. J Biol Chem 276:47512–47517
Luscher C, Slesinger PA (2010) Emerging roles for G protein-gated inwardly rectifying potassium (GIRK) channels in health and Disease. Nat Rev Neurosci 11:301–315
Luo H, Marron Fernandez de Velasco E, Wickman K (2022) Neuronal G protein-gated K(+) channels. Am J Physiol Cell Physiol 323:C439–C460
Jin W, Lu Z (1998) A novel high-affinity inhibitor for inward-rectifier K + channels. Biochemistry 37:13291–13299
Zurawski Z, Thompson Gray AD, Brady LJ, Page B, Church E, Harris NA, Dohn MR, Yim YY, Hyde K, Mortlock DP, Jones CK, Winder DG, Alford S, Hamm HE (2019) Disabling the Gbetagamma-SNARE interaction disrupts GPCR-mediated presynaptic inhibition, leading to physiological and behavioral phenotypes. Sci Signal 12
Dolphin AC (1998) Mechanisms of modulation of voltage-dependent calcium channels by G proteins. J Physiol 506(Pt 1):3–11
Urbano FJ, Rosato-Siri MD, Uchitel OD (2002) Calcium channels involved in neurotransmitter release at adult, neonatal and P/Q-type deficient neuromuscular junctions (review). Mol Membr Biol 19:293–300
Yang L, Tang J, Dong J, Zheng J (2015) Alpha2-adrenoceptor-independent inhibition of acetylcholine receptor channel and sodium channel by dexmedetomidine in rat superior cervical ganglion neurons. Neuroscience 289:9–18
Cui LN, Sun N, Li BX, Wang LF, Zhang XY, Qiu DL, Chu CP (2020) Noradrenaline inhibits complex spikes activity via the presynaptic PKA signaling pathway in mouse cerebellar slices. Neurosci Lett 729:135008
Dong CJ, Guo Y, Ye Y, Hare WA (2014) Presynaptic inhibition by alpha2 receptor/adenylate cyclase/PDE4 complex at retinal rod bipolar synapse. J Neurosci 34:9432–9440
DeBock F, Kurz J, Azad SC, Parsons CG, Hapfelmeier G, Zieglgansberger W, Rammes G (2003) Alpha2-adrenoreceptor activation inhibits LTP and LTD in the basolateral amygdala: involvement of Gi/o-protein-mediated modulation of Ca2+-channels and inwardly rectifying K+-channels in LTD. Eur J Neurosci 17:1411–1424
Conductier G, Nahon JL, Guyon A (2011) Dopamine depresses melanin concentrating hormone neuronal activity through multiple effects on alpha2-noradrenergic, D1 and D2-like dopaminergic receptors. Neuroscience 178:89–100
Alberto CO, Trask RB, Hirasawa M (2011) Dopamine acts as a partial agonist for alpha2A adrenoceptor in melanin-concentrating hormone neurons. J Neurosci 31:10671–10676
Tsentsevitsky AN, Gafurova CR, Petrov AM (2022) K(ATP) channels as ROS-dependent modulator of neurotransmitter release at the neuromuscular junctions. Life Sci 310:121120
Crawford DC, Kavalali ET (2015) Molecular underpinnings of synaptic vesicle pool heterogeneity. Traffic 16:338–364
Melom JE, Akbergenova Y, Gavornik JP, Littleton JT (2013) Spontaneous and evoked release are independently regulated at individual active zones. J Neurosci 33:17253–17263
Robitaille R (1998) Modulation of synaptic efficacy and synaptic depression by glial cells at the frog neuromuscular junction. Neuron 21:847–855
Ladera C, del Carmen Godino M, Jose Cabanero M, Torres M, Watanabe M, Lujan R, Sanchez-Prieto J (2008) Pre-synaptic GABA receptors inhibit glutamate release through GIRK channels in rat cerebral cortex. J Neurochem 107:1506–1517
Vazquez-Vazquez H, Gonzalez-Sandoval C, Vega AV, Arias-Montano JA, Barral J (2022) Histamine H(3) receptor activation modulates glutamate release in the Corticostriatal Synapse by acting at ca(V)2.1 (P/Q-Type) calcium channels and GIRK (K(IR)3) Potassium channels. Cell Mol Neurobiol 42:817–828
Guo J, Ikeda SR (2004) Endocannabinoids modulate N-type calcium channels and G-protein-coupled inwardly rectifying potassium channels via CB1 cannabinoid receptors heterologously expressed in mammalian neurons. Mol Pharmacol 65:665–674
Ginebaugh SP, Badawi Y, Tarr TB, Meriney SD (2022) Neuromuscular Active Zone Structure and Function in Healthy and Lambert-Eaton Myasthenic Syndrome States. Biomolecules 12
Nishimune H, Badawi Y, Mori S, Shigemoto K (2016) Dual-color STED microscopy reveals a sandwich structure of Bassoon and Piccolo in active zones of adult and aged mice. Sci Rep 6:27935
Giovannini F, Sher E, Webster R, Boot J, Lang B (2002) Calcium channel subtypes contributing to acetylcholine release from normal, 4-aminopyridine-treated and myasthenic syndrome auto-antibodies-affected neuromuscular junctions. Br J Pharmacol 136:1135–1145
Kaja S, Van de Ven RC, Ferrari MD, Frants RR, Van den Maagdenberg AM, Plomp JJ (2006) Compensatory contribution of Cav2.3 channels to acetylcholine release at the neuromuscular junction of tottering mice. J Neurophysiol 95:2698–2704
Pardo NE, Hajela RK, Atchison WD (2006) Acetylcholine release at neuromuscular junctions of adult tottering mice is controlled by N-(cav2.2) and R-type (cav2.3) but not L-type (cav1.2) Ca2 + channels. J Pharmacol Exp Ther 319:1009–1020
Molina-Campos E, Xu Y, Atchison WD (2015) Age-dependent contribution of P/Q- and R-type Ca2 + channels to neuromuscular transmission in lethargic mice. J Pharmacol Exp Ther 352:395–404
Pagani R, Song M, McEnery M, Qin N, Tsien RW, Toro L, Stefani E, Uchitel OD (2004) Differential expression of alpha 1 and beta subunits of voltage dependent Ca2 + channel at the neuromuscular junction of normal and P/Q Ca2 + channel knockout mouse. Neuroscience 123:75–85
Todorov B, van de Ven RC, Kaja S, Broos LA, Verbeek SJ, Plomp JJ, Ferrari MD, Frants RR, van den Maagdenberg AM (2006) Conditional inactivation of the Cacna1a gene in transgenic mice. Genesis 44:589–594
Perissinotti PP, Giugovaz Tropper B, Uchitel OD (2008) L-type calcium channels are involved in fast endocytosis at the mouse neuromuscular junction. Eur J Neurosci 27:1333–1344
Depetris RS, Nudler SI, Uchitel OD, Urbano FJ (2008) Altered synaptic synchrony in motor nerve terminals lacking P/Q-calcium channels. Synapse 62:466–471
Urbano FJ, Piedras-Renteria ES, Jun K, Shin HS, Uchitel OD, Tsien RW (2003) Altered properties of quantal neurotransmitter release at endplates of mice lacking P/Q-type Ca2 + channels. Proc Natl Acad Sci U S A 100:3491–3496
Gilmanov IR, Samigullin DV, Vyskocil F, Nikolsky EE, Bukharaeva EA (2008) Modeling of quantal neurotransmitter release kinetics in the presence of fixed and mobile calcium buffers. J Comput Neurosci 25:296–307
Wang X, Pinter MJ, Rich MM (2010) Ca2 + dependence of the binomial parameters p and n at the mouse neuromuscular junction. J Neurophysiol 103:659–666
Ermolyuk YS, Alder FG, Surges R, Pavlov IY, Timofeeva Y, Kullmann DM, Volynski KE (2013) Differential triggering of spontaneous glutamate release by P/Q-, N- and R-type Ca2 + channels. Nat Neurosci 16:1754–1763
Kailey B, van de Bunt M, Cheley S, Johnson PR, MacDonald PE, Gloyn AL, Rorsman P, Braun M (2012) SSTR2 is the functionally dominant somatostatin receptor in human pancreatic beta- and alpha-cells. Am J Physiol Endocrinol Metab 303:E1107–1116
Kuzhikandathil EV, Oxford GS (1999) Activation of human D3 dopamine receptor inhibits P/Q-type calcium channels and secretory activity in AtT-20 cells. J Neurosci 19:1698–1707
Zamponi GW, Currie KP (2013) Regulation of ca(V)2 calcium channels by G protein coupled receptors. Biochim Biophys Acta 1828:1629–1643
Acknowledgements
ANT, VFK were supported by assignment for Kazan Institute of Biochemistry and Biophysics, FRC Kazan Scientific Center of Russian Academy of Sciences.
Funding
This work was supported by the by Russian Science Foundation, grant number [23-15-00124], https://www.rscf.ru/project/23-15-00124/.
Author information
Authors and Affiliations
Contributions
A.T. performed electrophysiological experiments, visualization, and data analysis. V.K. performed electrophysiological experiments. EB was responsible for conceptualization, methodology, project administration, supervision, writing, review & editing; AMP wrote the main text and was responsible for conceptualization, review & editing. All authors have read and agreed to the published version of the manuscript.
Corresponding author
Ethics declarations
Conflict of Interest
The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper. All authors consent to publish the article.
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Electronic Supplementary Material
Below is the link to the electronic supplementary material.
Rights and permissions
Springer Nature or its licensor (e.g. a society or other partner) holds exclusive rights to this article under a publishing agreement with the author(s) or other rightsholder(s); author self-archiving of the accepted manuscript version of this article is solely governed by the terms of such publishing agreement and applicable law.
About this article

Cite this article
Tsentsevitsky, A.N., Khuzakhmetova, V.F., Bukharaeva, E.A. et al. The Mechanism of α2 adrenoreceptor-dependent Modulation of Neurotransmitter Release at the Neuromuscular Junctions. Neurochem Res 49, 453–465 (2024). https://doi.org/10.1007/s11064-023-04052-1
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s11064-023-04052-1