
Abstract
Alzheimer’s disease (AD) is the most common type of dementia characterized by the deposition of amyloid beta (Aβ) plaques and tau-neurofibrillary tangles in the brain. Visceral obesity (VO) is usually associated with low-grade inflammation due to higher expression of pro-inflammatory cytokines by adipose tissue. The objective of the present review was to evaluate the potential link between VO and the development of AD. Tissue hypoxia in obesity promotes tissue injury, production of adipocytokines, and release of pro-inflammatory cytokines leading to an oxidative-inflammatory loop with induction of insulin resistance. Importantly, brain insulin signaling is involved in the pathogenesis of AD and lower cognitive function. Obesity and enlargement of visceral adipose tissue are associated with the deposition of Aβ. All of this is consonant with VO increasing the risk of AD through the dysregulation of adipocytokines which affect the development of AD. The activated nuclear factor kappa B (NF-κB) pathway in VO might be a potential link in the development of AD. Likewise, the higher concentration of advanced glycation end-products in VO could be implicated in the pathogenesis of AD. Taken together, different inflammatory signaling pathways are activated in VO that all have a negative impact on the cognitive function and progression of AD except hypoxia-inducible factor 1 which has beneficial and neuroprotective effects in mitigating the progression of AD. In addition, VO-mediated hypoadiponectinemia and leptin resistance may promote the progression of Aβ formation and tau phosphorylation with the development of AD. In conclusion, VO-induced AD is mainly mediated through the induction of oxidative stress, inflammatory changes, leptin resistance, and hypoadiponectinemia that collectively trigger Aβ formation and neuroinflammation. Thus, early recognition of VO by visceral adiposity index with appropriate management could be a preventive measure against the development of AD in patients with VO.
Graphical Abstract

Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Alzheimer’s disease (AD) is the most frequent type of dementia. AD was first identified in 1907 by Alois Alzheimer [1]. AD is recognized as a single neurological entity; its frequency is very much increased after the age of 65 [2, 3]. AD should be distinguished from other types of dementia including reversible dementia, Parkinson-associated dementia, vascular dementia, and frontotemporal dementia [1]. AD represents 70% of all dementia types; it upsurges with age, doubling every 10 years. It has been reported that AD prevalence is 3% in individuals aged 65–74, to about 50% in subjects aged 85 years and older [4, 5]. The prevalence of AD was about 5 million in 2007 that is suspected to increase to 13 million in 2050 [6]. Furthermore, head trauma, female sex, low education level, cardio-metabolic disorders, and vascular diseases are considered risk factors for the progress of AD [7]. Nonetheless, till now there is no specific biomarker that differentiates AD from other types of dementia. As well, brain biopsy and brain positron emission tomography were used in the diagnosis of AD but with conflicting findings [4].
The pathologic findings of AD include senile and neuritic plaques with noteworthy dendritic loss, dystrophic neuritis, neuropil threads, and cerebrovascular amyloid [8]. However, synaptic loss, amyloid pathway, and Lewy body deposition are the main pathological feature of AD [8, 9]. Also, AD is characterized by the deposition of amyloid beta (Aβ) plaques and tau-neurofibrillary tangles (TNTs) in the brain. Remarkably, deposition of Aβ occurs earlier than the pathology of cortical tau. Deposition of Aβ-induces tau-mediated neuronal and synaptic loss in AD, though tau neuropathology can progress independently of Aβ accumulation [8,9,10]. The neuropathological changes in AD are related to the deposition of amyloid plaques, neurofibrillary tangles, and progression of neuroinflammation, neuronal mitochondrial dysfunction, autophagy dysfunction, and cholinergic synaptic dysfunction [11, 12] (Fig. 1).

Pathophysiology of Alzheimer’s disease (AD)
The clinical presentation of AD patients is characterized by cognitive dysfunction, speech dysfunction, sensory-motor disorders, memory loss, and personality disorders. However, some AD patients stay stable for several years and may improve with time [13]. Additionally, early-onset AD is more progressive and characterized by less memory loss with higher psychosocial disorders [13, 14]. Authentic AD patients presented with apraxia, judgment dysfunction, speech abnormalities, disorientation, and personality changes [13, 14]. The main clinical findings in AD patients are related to neuropsychiatric disorders (Fig. 2).

Clinical features of Alzheimer’s disease (AD)
Management of AD patients is mostly symptomatic by treatment with cholinesterase inhibitors like donepezil, rivastigmine, tacrine, and galantamine. Also, N-methyl-D-aspartate (NMDA) antagonists like memantine are used in treating cognitive deficits in AD patients [15, 16]. Nonetheless, these agents provide symptomatic relief without affecting the etiopathologic progression of AD [15, 16] (Fig. 3).

Management of Alzheimer’s disease (AD)
It has been shown that different metabolic disorders including obesity, insulin resistance (IR), and related inflammatory disorders are associated with an increased risk for the development of AD [17, 18]. Notably, central obesity is associated with the early-onset development of AD due to increased inflammation and IR [19, 20]. The visceral adiposity index (VAI, please see below for its calculation) reflects adipose tissue dysfunction and central obesity. Thus, this review is aimed to evaluate the potential link between VO and the development of AD.
Obesity and VAI
Obesity is a medical condition due to the excessive accumulation of body fat which adversely affects health [21]. Obesity is defined as body mass index (BMI) ≥ 30 kg/m2. Obesity is correlated with the development of cardiometabolic disorders and other diseases including dyslipidemia, hypertension, IR, type 2 diabetes mellitus (T2DM) and coronary heart diseases as well as malignancies respectively [22, 23]. Different indicators and predictors of obesity are used; one of the most common one is BMI. It has been shown that BMI is incorrectly regarded as a suitable indicator of body fat and obesity as it shows a non-linear correlation with the percentage of body fats in both sexes [24]. Many factors affect the validity of BMI including age, race, gender, hydration status and muscle mass [25]. A retrospective study conducted by Flegal et al. [26] showed that BMI was not a good indicator for assessing the association between obesity and mortality. Furthermore, a meta-analysis and cohort studies illustrated that BMI was not to be used as a predictor of primary and secondary preventions of cardiovascular diseases, as it does not differentiate between body fat and lean mass [27]. Notably, BMI is correlated significantly with indicators of true body fat and does not consider the effect of age, race, and sex [27]. Besides, BMI was not correlated significantly with abdominal obesity and related cardiovascular risk factors as other indicators like waist circumference (WC) and VAI. However, WC cannot differentiate between visceral and subcutaneous fats [28].
Currently, VAI is used to estimate central visceral obesity (VO) which is more correlated with cardiovascular risk factors in the development of cardiovascular diseases [29]. VAI is regarded as an independent indicator associated with 10 years of cardiovascular risk in men with little association in women [29]. Thus, VAI can be routinely used to recognize men obese individuals at risk for cardiovascular complications. A prospective study in Europe found that VAI is an additional indicator to detect cardiovascular risk in men without previous cardiovascular diseases [29]. Amato et al. [30] illustrated that VAI was a reliable indicator of central obesity and visceral fat dysfunction, and was associated with cardiometabolic risk. A retrospective study involved 315 non-obese individuals revealed that VAI was correlated with cardiovascular risk [OR 2.45, 95% CI 1.5–3.95, P < 0.0001]. In addition, VAI was negatively correlated with insulin sensitivity [Rs = − 0.72, P < 0.001] [30]. This observation suggests that VAI is a valuable predictor and indicator of central obesity and IR in patients with cardiometabolic disorders. To date, many studies have shown that VAI is linked with VO-induced inflammation, atherosclerosis, and coronary heart diseases [31, 32]. Li and colleagues [31] in a retrospective study observed that neutrophil-lymphocyte ratio (NLR) and VAI are increased in patients with carotid atherosclerosis which predict 10 years of cardiovascular risk. In Chinese patients with coronary heart disease, VAI was higher.
Of note, VAI can be easily calculated by the following equation according to Nusrianto et al. [33] that depends on many variables including WC, BMI, high-density lipoprotein (HDL), and triglyceride (TG) levels.
VAI is an empirical mathematical method for the assessment function and distribution of body fat [33]. VAI is regarded as a potential indicator of cardiometabolic risk and the development of metabolic syndrome [34]. Furthermore, VAI is considered a better predictor for the development of T2DM and hypertension [35, 36]. VAI is also superior to WC and BMI in predicting the function and distribution of visceral fats in post-menopausal women with low-calorie diets [37]. All this taken together points to VAI being an accurate potential predictor of cardiovascular risk in obese patients. The cut-off value of VAI and associated adipose tissue dysfunction (ATD) according to age is illustrated in Table 1 [20].
Despite this valuable role of VAI, the application of this method is incorrect in small sample size, patients with morbid obesity and obese children less than 16 years. In addition, VAI is affected by fibrate drugs which reduce TG level [20, 38].
Visceral Obesity (VO) and Inflammation
VO is commonly associated with low-grade inflammation due to higher expression of pro-inflammatory cytokines by adipose tissue [39, 40]. An experimental study illustrated that visceral adipose tissue (VAT) affects the reactivity of immune cells within visceral lymph nods. Inflammatory cytokines promote the migration of dendritic cells (DCs) from small intestines to the visceral lymph nodes in mice with a high-fat diet [39]. Thus, visceral lymph nodes seem to be the potential nexus between adipose tissue and intestinal inflammation with exacerbation of systemic inflammation [39].
There is an emerging body of work indicating that genes, epigenetics, and the in-utero environment can impact whether or not a child is obese [41]. While certain genes have been identified that increase one’s risk for becoming obese, other factors such as excess gestational weight gain, gestational diabetes mellitus, and smoking can also influence this risk [41, 42]. However, further research is needed to determine which efforts are effective at decreasing the incidence of obesity and to develop new methods of prevention. Over the past several years, many discoveries have been made regarding the genetic variation that influences complex diseases like cardiovascular disease and obesity [43]. These new discoveries have largely resulted from genome-wide association studies where the application of high-throughput genotyping of millions of genetic markers enables researchers to examine genetic associations on a genome-wide basis. Recent reports indicate that at least 32 genes contribute to common forms of obesity [44]. A number of these have also been confirmed as contributors to pediatric obesity [44]. Many of these genes are thought to be related to the development of obesity through the dysregulation of leptin or other metabolic hormones in the body. A majority of the newly discovered genes are expressed in the brain, emphasizing the role of the central nervous system in obesity risk [43,44,45].
Various studies confirm that VO is connected with abnormal cytokine production and activation of inflammatory signaling pathways [46, 47]. An observational study involved 600 obese patients demonstrated that VAT but not subcutaneous adipose tissue (SAT) is associated with systemic inflammation as evidenced by high levels of c-reactive protein (CRP) and NLR [47]. VAT is regarded as a possible source of inflammation due to the infiltration of macrophages, the main source of pro-inflammatory cytokines. It has been documented that obese patients had a higher level of tumor necrosis factor alpha (TNF-α) as compared to lean subjects [47, 48]. A previous experimental study showed that TNF-α increased exponentially with the development of VAT in obese mice [49]. Obesity and associated metabolic disorders promote macrophage accumulations with the induction of abnormal immune response. Interestingly, TNF-α activates hormone-sensitive lipase with further release of non-esterified fatty acids into the circulation with impairment of insulin sensitivity [50, 51]. Obesity-induced inflammation is linked with the progression of systemic disorders like atherosclerosis, endothelial dysfunction (ED), and hypertension [52]. Of note, obese subjects have high circulating levels of interleukin (IL)-6, TNF-α, CRP and pro-inflammatory adipocytokines like leptin. Pro-inflammatory adipocytokines trigger chronic inflammation [53, 54]. The anti-inflammatory adiponectin plays a crucial role in the mitigation of VO-induced inflammation by inhibiting the expression of nuclear factor kappa B (NF-κB) and TNF-α [55,56,57,58]. Of interest, adiponectin serum level is reduced in obese subjects [59]. A prospective study involving 193 obese subjects showed that adiponectin serum level was low mainly in subjects with high VAI and VAT [59]. Therefore, increasing visceral fat in overweight and obese subjects augments the risk for the development of systemic inflammation due to the reduction in the circulating anti-inflammatory adiponectin level.
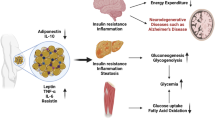
Taken together, the development of VO is associated with the augmentation of pro-inflammatory and proatherogenic mediators leading to ED, IR and atherosclerosis (Fig. 4).

Visceral obesity and development of systemic inflammation
Visceral Obesity (VO) and Insulin Resistance (IR)
IR represents glucose intolerance with a reduction of physiological response to insulin resulting in the development of hyperinsulinemia with euglycemia [60]. Obesity is the major contributor to the progression of IR which interrupts many intracellular signaling [61]. IR induces the over-production of non-esterified fatty acids with the progression of inflammation, oxidative stress, and mitochondrial dysfunction [62, 63]. VAT-induced oxidative stress triggers IR through the modulation of insulin receptors and insulin action [64]. High-fat diet activates the generation of reactive oxygen species (ROS) which impairs insulin sensitivity and triggers inflammatory changes with the development of hyperinflammation [48, 65, 66]. In addition, tissue hypoxia in obesity promotes tissue injury, production of adipocytokines and release of pro-inflammatory cytokines leading to an oxidative-inflammatory loop with induction of IR [63, 66]. Oxidized low density lipoprotein (LDL) and free fatty acids induce the development of IR and associated cardiometabolic changes like atherosclerosis [63, 66]. The precise mechanisms of IR in obesity seem to be multifactorial including glucolipotoxicity, endoplasmic reticulum stress, and oxidative stress [67, 68]. Hardy et al. [69] found that VAT is mainly associated with the development of IR due to inflammatory and oxidative stress interactions. In addition, the accumulation of VAT is thought to affect whole-body insulin sensitivity, unlike peripheral adipose tissue accumulation which does not affect the development of IR [70]. The molecular mechanism of IR in obesity is mainly related to the upregulation of peroxisome proliferator activator receptor gamma (PPARγ) [71, 72]. PPARγ deficient mice are protected from the development of IR following a high-fat diet. Therefore, targeting PPARγ could be a possible therapeutic option for IR [71]. Furthermore, metabolic inflammation could be the main cause of the propagation of IR in obesity [73]. Notoriously, VAT promotes the polarization of immune and inflammatory cells towards skeletal muscle, liver, adipose tissue, brain and pancreatic islets causing metabolic derangement and development of IR [73]. Thus, anti-inflammatory agents by targeting inflammatory cells might attenuate VAT-induced metabolic disorders and related diseases like T2DM and atherosclerosis.
These findings proposed a strong link between VO and IR through the creation of an inflammatory/oxidative loop that disturbs the insulin signaling pathway (Fig. 5).

Visceral obesity and development of insulin resistance
Visceral Obesity (VO) and Brain IR
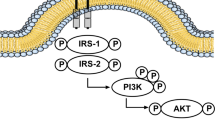
It has been shown that brain insulin signaling is involved in the pathogenesis of AD through AKT phosphorylation which induces AD pathology and lowers cognitive function [74]. A community-based clinic-pathological cohort study involving 150 old patients with and without T2DM reported that insulin signaling was severely disturbed by postmortem analysis [74]. Interestingly, activation of neuronal insulin signaling has been observed to prevent memory loss in an AD model consonant with neuroprotection[75]. Also, the expression of insulin-like growth factor 1 receptor (IGF1R) was increased in postmortem hippocampal tissue from AD patients [75]. Of note, brain IGF1 signaling was severely impaired in AD patients. Thus, boosting brain insulin and IGF1 signaling could be a promising therapy for AD. It has been shown that the administration of intranasal insulin improves memory performance through the enhancement of synaptic plasticity and neurotransmitter function and could be a good treatment for AD [76].
Insulin receptors are widely expressed in certain brain regions involved in the regulation of cognition, appetite, olfaction, and autonomic function [77]. Insulin receptors regulate neuronal plasticity and metabolism and afford a neuroprotective effect. The functional activity of insulin receptors in the brain is distinct from peripheral metabolic effects. Dysfunction of insulin receptors and insulin signaling pathways promote the progression of various brain disorders [77]. Insulin concentration in different brain areas like the brain cortex, hypothalamus, and hippocampus is higher than plasma [78]. Of note, insulin does not affect neuronal glucose transport and expression of glucose transporter 4 (GLUT-4) [78].
Interestingly, insulin in the brain is involved in the regulation of cognitive function rather than glucose uptake as in the peripheral tissues. It exerts neuromodulatory and neuroprotective effects with a positive impact on memory function through the modulation of synaptic activity and neurotransmitter release [79]. Peripheral glucose uptake and cognitive function are reduced with age by an unknown mechanism. Dysregulation of peripheral glucose signaling may affect hippocampal functional activity and stability [79, 80]. Different studies have shown that insulin decreases the risk for the progression of AD. Insulin increases Aβ clearance [74, 81]. In contrast, disturbances in brain insulin levels may lead to detrimental effects through the phosphorylation of tau protein (Fig. 6).

Visceral obesity and brain IR
In this context, VO-induced IR may also promote brain IR with impairment of cognitive function and development of AD [82]. Moreover, high-fat diet (HFD)-induced obesity increases deposition of Aβ with significant impairment of cognitive function in mice via deregulation of the brain insulin signaling pathway [83]. Development of brain IR promotes neurodegeneration and neuroinflammation with progress to AD through activation of neuronal mitogen-activated protein kinase (MAPK) which enhances aggregation of Aβ and formation of neurofibrillary tangles (NFTs) [74, 82]. In turn, high Aβ and NFTs reduce the expression of neuronal insulin receptors with the development of IR and further deterioration of AD pathology [84]. Activated neuronal MAPK triggers neuroinflammation and tau phosphorylation via inhibition of adenosine monophosphate (AMP) and activation of glycogen synthase kinase (GSK) leading to Aβ accumulation and progression of the pathogenesis of AD [85]. Thus, regulation of brain insulin function enhances neuronal activity and prevents the development of AD neuropathology [82].
The possible mechanisms of insulin in the enhancement of cognitive function in AD are related to various regulations of neuronal signaling pathways. Insulin improves catecholamine release and uptake, regulates the expression of NMDA and gamma-Aminobutyric Acid (GABA) receptors, and enhances the cerebral cortex AMP pathway and cortisol circadian rhythm [76, 85] (Fig. 7).

Cognitive enhancing effects of insulin
These findings suggest that insulin plays a critical role in the promotion of cognitive function and the prevention progression of AD. Thus, VO-induced IR could be one of the important pathways in the pathogenesis of AD.
Visceral Obesity (VO) and Risk of AD
Of note, AD is developing due to Aβ accumulation via the amyloidogenic pathway with the formation of Aβ plaques. The processing and production of Aβ plaques affect neuronal activity through the interruption of synaptic activity and induction of cell death [86]. Obesity and enlargement of VAT are associated with the deposition of Aβ [87]. Obesity is regarded as an independent risk factor for the development of AD, and 7.3% of AD patients over the age of 65 years are directly attributed to previous midlife central obesity [87]. Interestingly, apolipoprotein J known as clusterin which is involved in the pathogenesis of AD is increased in various cardiometabolic disorders including obesity [87]. Oh et al. [88] illustrated that the expression of clusterin is involved in the pathogenesis of early-stage AD via enhancement of Aβ-induced neurotoxicity and advancement of cognitive impairments in mice. Besides, adipose tissue-clusterin promotes IR via the induction release of pro-inflammatory cytokines and oxidative stress [89]. Thus, there is a potential link between obesity and the development of AD through the clusterin pathway [89].
On the other hand, high-fat diet-induced obesity promotes brain Aβ pathology. Shie et al. [90] demonstrated that hepatic steatosis and obesity are linked with high circulating Aβ in metabolically stressed mice. Furthermore, increasing circulating levels of Aβ in T2DM and obesity promote the development of ED due to impairment of the nitric oxide (NO) pathway [91]. An experimental study demonstrated that leptin resistance in obese mice promotes the deposition of tau protein [92]. Thus, leptin resistance-induced obesity enhances tauopathy and the development of NFT [92]. Remarkably, VAT triggers the activation of MAPK which is involved in tau pathology and neuroinflammation [93]. Hyperphosphorylation of tau proteins by MAPK promotes the generation of NFT which trigger neuroinflammation and neurodegeneration with subsequent cognitive impairment [93]. Herein, an activated MAPK signaling pathway in obese patients could a possible association between VO and the development of AD.
Moreover, irisin which is a myokine produced from adipose tissue, muscles and hippocampus has a neuroprotective role against the development of AD. Irisin improves neuronal synaptic plasticity with a positive impact on the cognitive function and development of dementia [94,95,96]. Notably, irisin level is reduced in the AD model in both hippocampus and cerebrospinal fluid (CSF). The deficiency of irisin enhances tau pathology and Aβ formation [97]. It has been reported that irisin level is positively correlated with fat mass and BMI and negatively correlated with adiponectin serum level. Thus, the irisin level is reduced following weight loss and bariatric surgery [98]. Leung et al. [99] observed that irisin level was reduced in Chinese adults with obesity due to uncontrolled inflammatory changes. Therefore, the reduction of irisin activity in obese patients may exacerbate neuroinflammation and the development of AD.
Indeed, central obesity with a high waist-height ratio (WHR) is regarded as a high-risk factor for the development of late-onset AD [19]. In addition, high BMI in middle age is correlated with a higher incidence of AD in late life. In contrast, low BMI was also reported to be a risk factor for the development of AD [100]. Loss of olfaction is the initial manifestation of AD that cause poor appetite and weight reduction [101]. Thus, weight loss and reduction of BMI are the consequences of AD, and the reverse is not true. Likewise, the reduction of lean body mass and the increase in body fat percentage may increase the risk of AD [102]. Loss of lean mass is accelerated in AD and is associated with brain atrophy and cognitive performance, perhaps as a direct or indirect consequence of AD pathophysiology or through shared mechanisms common to both AD and sarcopenia [102]. In particular, the percentage of fat was positively associated with cortical thickness and the highest WHR group showed significantly decreased cortical thickness in men compared with the reference group. WHR showed an inverted U-shaped association with total cortical thickness and frontal lobe thickness in men, this association was not detected in women [103]. As well, the increase in body fat percentage is associated with the risk of AD [104]. Therefore, the increase of BMI and body fat percentage may increase the risk of AD development through the induction of Aβ accumulation and release of pro-inflammatory cytokines which promote the development of neuroinflammation.
In this state of affairs, VAI which includes both WC and BMI might be more appropriate in the estimation of VO and its relation with the development of AD. Adiposity indices have a strong relationship with cognitive decline in AD patients [105]. An elegant study revealed that high adiposity indices like the VAT ratio are correlated with abnormal white matter hyperdensity. Thus, higher VAI which reflects VO is regarded as an independent risk factor for cognitive dysfunction in AD patients [105].
These findings are consonant with VO increasing the risk of AD through the dysregulation of adipocytokines which affect the development of AD (Fig. 8).

Visceral obesity and risk of Alzheimer’s disease (AD)
Taking into account all these mechanisms, one is tempted to propose that targeting obesity-induced IR and inflammation reduces the risk of AD through modulation of systemic inflammation, neuroinflammation and brain IR. An experimental study revealed that administration of natural raspberry ketone in obese rats reduced body weight, BMI and improved cholinergic neuron activity [106] suggesting that concomitant supplementation of natural raspberry ketone with calorie restricted regimen effectively modulate the neurodegenerative changes induced by obesity and delay of the progression of AD. In addition, modulation of peripheral and brain IR in obesity by metformin may reduce the risk of neurodegeneration and AD [107]. Therefore, metformin could be a promising therapeutic agent in the management of AD [107]. Furthermore, quercetin (QT) is one of the most abundant polyphenolic flavonoids, which is present in fruits and vegetables and displays many biological, health-promoting effects in a wide range of diseases. In vitro, in vivo, and clinical evidence regarding the anti-Alzheimer, anti-diabetic and anti-obesity effects of QT were approved [108]. Thus, mitigation of T2DM-induced systemic inflammation by metformin and QT may reduce the risk for development of AD.
Signaling Pathways Link Visceral Obesity and AD
Different signaling pathways are involved in the pathogenesis of VO and AD, and they might be a potential link between VO and AD is summarized in Table 2.
MAPK and mTOR
In VO, different inflammatory and metabolic pathways are more active which in direct or indirect ways could affect the development of AD. It has been reported that MAPK is implicated in obesity-mediated complications [109]. Activation of MAPK in obesity triggers systemic inflammation and apoptosis causing peripheral and central effects [109]. Obesity-induced IR and hyperinsulinemia through IGF promote the expression of MAPK and mechanistic targets of rapamycin (mTOR) are involved in the development of systemic complications [109]. Notably, MAPK plays a crucial role in the pathogenesis of AD through the induction of neuroinflammation [110]. MAPK promotes excitotoxicity, synaptic dysfunction and tau phosphorylation. Besides, mTOR is also involved in the pathogenesis of AD and other types of dementia through the modulation of neuronal lysosomal and autophagy processes [110]. The administration of rapamycin in mice after the development of Aβ deposition was effective in the reduction of Aβ plaque accumulation [111]. In addition, the beneficial effects of glucagon-like peptide (GLP-1) in treating AD are mediated through the induction of mTOR [127]. This may explain the double-edge effects of mTOR in the pathogenesis of AD. Therefore, targeting mTOR and MAPK pathways could be effective in the management of AD. Thus, mTOR and MAPK pathways could be a potential link between VO and the development of AD.
NLRP3 Inflammasome
Furthermore, activation of nod-like receptor pyrin 3 (NLRP3) inflammasome is intricate with the progression of the inflammatory process in obesity [112]. Chronic inflammation in obesity may be mediated by activated NLRP3 inflammasome which induces expression cleavage of caspase 1 and release of IL-1β and development of lipotoxicity and IR [112]. Ablation of NLRP3 inflammasome in VAT and liver reduced inflammation and increase insulin sensitivity in mice. As well, the removal of NLRP3 inflammasome reduces VAT interferon-gamma (INFγ) and increases native T cell count in mice [112]. This finding suggests that activation of NLRP3 inflammasome is linked with obesity-induced obesity and the development of IR. A systematic review indicates that excess nutrients trigger activation of NLRP3 inflammasome with subsequent pro-inflammatory response and development of IR [128]. Legrand-Poels revealed that the toxic effects of free and saturated free fatty acids induce the development of ROS and inflammatory reactions which promote the activation of NLRP3 inflammasome [129]. It is of interest that omega-free fatty acids inhibit NLRP3 inflammasome activation and the development of hyperinflammation in obesity.
Different studies implicate NLRP3 inflammasome activation in the pathogenesis of AD [113, 130]. Recently, it has been shown that NLRP3 inflammasome activation results in microglia stimulation, assembly and aggregation of Aβ, and enhancement of tau-associated neurobiology [113]. In an in vitro study it has been found that the deposition of Aβ and NFT was linked with the activation of NLRP3 inflammasome in the microglia [114]. Different experimental studies revealed that NLRP3 inflammasome activation was associated with tau aggregation and exacerbation of AD in mice [115, 131]. Notably, caspase-1 activation and release of pro-inflammatory cytokines precede AD neuropathology implying that activation of NLRP3 inflammasome could be the initial pathogenic event in AD [132]. These findings support that VO-induced NLRP3 inflammasome activation might be a possible cause for the development of AD neuropathology.
NF-κB
NF-κB is an inflammatory signaling pathway implicated in the development of inflammation and IR in obesity [116, 133]. The NF-κB pathway is activated in insulin-sensitive tissue and highly dysregulated in obesity. Inhibition of the NF-κB pathway by aspirin improves insulin sensitivity in VAT [134]. NF-κB maintains inflammation and induces the development of IR with the progression of metabolic derangement in obesity [134]. Targeting of the NF-κB pathway by lipoxin A4 can attenuate obesity-related systemic inflammation and glomerulopathy [135, 136]. In addition, inhibition of the NF-κB pathway by major vault protein inhibits obesity and related atherosclerosis in mice treated with HFD [137]. Therefore, an activated NF-κB pathway in obesity could increase the risk of IR and systemic inflammation hallmarks of AD.
Indeed, the NF-κB pathway is involved in the pathogenesis of AD and has been reported to be a potential link between T2DM and the progression of AD [117]. Certainly, upstream regulators like Aβ, cytokine storm, and ROS activate the NF-κB signaling pathway. As well, many inflammatory signaling pathways like MAPK, AKT, PI3K and GSK3 increase the pathogenesis of AD via induction expression of NF-κB [138]. Lukiw [139] found that the NF-κB pathway was involved in the induction of pro-inflammatory miRNA in AD. An experimental study illustrated that enhancement of NF-κB immunoreactivity is linked with abnormal integrity of the cerebral cortex and hippocampus in AD [140]. Thus, NF-κB expression is higher in the brain pathological area affected by AD. Ju Hwang et al. [141] observed that NF-κB was the main mediator of neuroinflammation in AD. NF-κB involves in the amyloidogenesis process by inducing the release of chemokines, inflammatory cytokines and microglial activation [141, 142]. Thus, inhibition of the NF-κB pathway by aspirin may prevent the progression development of AD. Different studies showed that polyphenols and phytochemicals attenuate the progression of AD through the inhibition of NF-κB-induced neuroinflammation [143, 144]. Of interest, forsythoside B which is a glycoside isolated from Fructus has anti-inflammatory and prevents the pathogenesis of AD [145]. It has been shown that forsythoside B could prevent NF-κB-mediated neuroinflammation in mice with experimental AD [145]. Thus, forsythoside B and other herbal medicine might be effective in preventing the progression of AD.
Therefore, the activated NF-κB pathway in VO might be a potential link in the development of AD, so targeting of NF-κB pathway could be a possible therapeutic value in the management of AD.
Advanced Glycation End-Products
Advanced glycation end-products (AGEs) are toxic compounds produced due to the combination of glucose with protein or lipid molecules in the glycation process [146]. AGEs act on a specific receptor called receptor for AGEs (RAGEs) which is involved in body metabolism, adipose tissue macrophage content, systemic inflammation, and regulation of body mass [147]. The interaction between AGEs and RAGEs triggers vascular inflammation and the development of vascular inflammation [118, 147]. It has been observed that obesity is associated with augmentation of AGEs levels which mediate oxidative stress and inflammation. Indeed, the interaction between AGEs and RAGEs promotes the expression of inflammatory signaling pathways and adhesion molecules leading to inflammatory disorders in obesity and T2DM [118, 147]. Therefore, anti-AGEs medication attenuates hyperinsulinemia and glucose intolerance, promoting weight loss and reduction of body fat [148].
Prolonged use of a diet containing AGEs induces the development of hepatosteatosis and obesity with the increment of leptin and pro-inflammatory cytokines in mice [149]. Activation of RAGEs by AGEs promotes the development of many diseases including IR, T2DM, cancer, and diabetic neuropathy and retinopathy [149]. A study conducted by den-Engelsen et al. [150] illustrated that patients with central obesity have a higher accumulation of AGEs measured by skin autofluorescence. A systematic review revealed that low AGEs diets reduce the risk for the development of central obesity and related complications [151]. AGEs act as pro-inflammatory and pro-oxidants in the progression of AD [119].
Furthermore, AGEs/RAGEs are implicated in the progression of neuroinflammation and AD via glycoxidation of Aβ and the formation of toxic molecules [119]. AGEs /RAGEs complex is involved in the pathogenesis of AD and vascular dementia through activation of tau phosphorylation and GSK3b pathway with subsequent cognitive decline [119]. Analysis of AGEs in both serum and brain of postmortem previously diagnosed with T2DM and AD illustrated that AGEs were augmented and involved in the pathogenesis of AD [119].
Therefore, the higher concentration of AGEs in VO could be implicated in the pathogenesis of AD. Thus, a reduction of the diet containing AGEs or weight loss may reduce the risk of VO-induced AD.
Hypoxia-Inducible Factor 1
The fundamental effects of hypoxia on the progression of AD are related to the types of hypoxia. Mild and intermittent hypoxias are protective while severe and chronic hypoxias are detrimental to AD neuropathology [152]. Chronic hypoxia induces the formation and accumulation of Aβ with aggravation of Aβ neurotoxicity by increasing Ca+ 2 dyshomeostasis and generation of ROS [152]. Taken together, Aβ and chronic hypoxia provoke the progression of neuroinflammation which increase amyloidogenesis and AD pathology. Thus, restoration of normal oxygen tension or the increase of HIF-1 activity in the brain may attenuate the development of AD and other neurodegenerative brain disorders [153]. In addition, chronic hypoxia shifts amyloid precursor protein toward the amyloidogenic pathway with the generation of neuronal Aβ through suppression of alpha-secretase activity and neprilysin which degraded amyloid precursors [154].
HIF-1 is a key molecule that regulates cell response to hypoxia and inflammation as it is essential for cell survival and function [120]. HIF-1 is involved in VAT metabolism and induction of inflammation and IR. Dysregulation of HIF-1 is intricate in the development of obesity and related complications [120]. HIF-1 is regulated by hypoxia, ROS, NO, tricarboxylic acid metabolites, pro-inflammatory cytokines, and hormonal factors [155]. Notably, HIF-1 promotes the expansion of VAT and the increase of body mass with the reduction of basal body metabolic rate [156]. It has been shown that HFD induces the expression of hypothalamic HIF-1 causing hypothalamic inflammation with the development of glucose intolerance and increased body weight [156]. Thus, HIF-1 is regarded as a potential regulator of energy homeostasis and lipid metabolism, herein it is considered a possible target in the management of obesity.
Of note, brain hypometabolism in the aging process due to the gradual reduction of oxygen and glucose supply mainly in certain brain regions including the hippocampus and temporo-frontal cortex trigger AD neuropathology [157]. Brain hypometabolism promotes the expression of Aβ with a reduction of its clearance. In turn, aggregation of brain Aβ induces the progression of oxidative stress, inflammation, and neuronal cell death [157]. Among cellular adaptation mechanisms in response to the effect of Aβ-induced inflammatory changes and neuronal hypoxia, transcription of HIF-1 is augmented as a compensatory mechanism [121]. Thus, maintaining the HIF-1 level by agents like heavy metals like nickel, and iron chelators could be an effective strategy to attenuate AD-induced inflammation and oxidative stress [121, 157]. Remarkably, different studies documented that HIF-1 inhibits the expression of neprilysin and increases the cleavage of amyloid precursor proteins [122, 158]. Also, HIF-1 exerts a neuroprotective effect against the development of AD via suppression of oxidative stress and neuroinflammation induced by Aβ toxicity [123]. Indeed, HIF-1 prevents metabolic derangement and mitochondrial dysfunction triggered by chronic hypoxia in AD. Besides, HIF-1 promotes cerebral circulation by regulating the expression of neuronal erythropoietin receptors [159]. Erythropoietin inhibits the generation and aggregation of neuronal Aβ [159]. Thus, HIF-1 hydroxylase inhibitors could be effective in the management of AD.
These findings revealed that the increase of HIF-1 in patients with VO could be protective against the development of AD.
Taken together, different inflammatory signaling pathways are activated in VO that all have a negative impact on the cognitive function and progression of AD except HIF-1 which has beneficial and neuroprotective effects in mitigating the progression of AD.
Sirtuin1 Pathway
Sirtuin1 (SIRT-1) also known as NAD-dependent deacetylase sirtuin-1, is a protein that in humans encoded by the SIRT1 gene [160]. SIRT-1 is an enzyme located primarily in the cell nucleus that deacetylates transcription factors that contribute to cellular regulation. The SIRT family comprises seven NAD+-dependent deacetylases which control the overall health of organisms through the regulation of pleiotropic metabolic pathways [160, 161]. SIRT-1 is an important modulator of adipose tissue metabolism and its expression is higher in lean than obese subjects [162]. Of note, SIRT-1 inactivation is involved in lipid metabolism and VO [163]. SIRT1 and SIRT2 modulate the differentiation of human adipose stem cells [162]. Reduced expression of SIRT1 and SIRT2 may enhance the differentiation capacity of visceral adipose stem cells in human obesity, fostering VAT expansion [163]. SIRT1levels in adipose-derived stem cells are consistent with protective effects against VO and inflammation and suggest a transcriptional mechanism through which acute hypoxia up-regulates SIRT1in the visceral adipose-derived stem cells of obese patients [124]. SIRT-1 is critical to neuron proliferation and SIRT-1 inactivation is associated with defective Aβ clearance and AD. SIRT-1 is involved with the prevention of oxidative stress, inflammatory changes, leptin resistance and adiponectin levels [45, 125]. SIRT-1 is responsible for the transcriptional regulation of various transcription factors and signaling pathways including MAPK, mTOR, HIF-1, NF-κB and NLRP3 [164]. These findings suggest that the downregulation of SIRT-1 in VO may induce the development of AD. Therefore, SIRT-1 could be a potential link between VO and AD, and targeting this pathway may mitigate VO-induced AD. It has been observed that SIRT1 protects against memory loss through mechanisms that involve oxidative stress, Aβ toxicity, neurofibrillary degeneration, vascular injury, mitochondrial dysfunction, and neuronal loss. In addition, SIRT1 relies upon other avenues that can include trophic factors, such as erythropoietin, and signaling pathways, such as Wnt1 inducible signaling pathway protein 1 (WISP1/CCN4). Yet, SIRT1 can have detrimental effects as well that involve tumorigenesis and blockade of stem cell differentiation and maturation that can limit reparative processes for cognitive loss. Further investigations with SIRT1should be able to capitalize upon these novel targets for dementia and cognitive loss [165, 166]. Poor et al. [107] showed that metformin improves cognitive function in AD through the activation of the SIRT1 pathway. As well, resveratrol has antioxidant, anti-inflammatory, and neuroprotective properties and can decrease the toxicity and aggregation of Aβ peptides in the hippocampus of AD patients, promote neurogenesis, and prevent hippocampal damage [167]. In addition, the antioxidant activity of resveratrol plays an important role in neuronal differentiation through the activation of the SIRT1 pathway. SIRT1 plays a vital role in the growth and differentiation of neurons and prevents the apoptotic death of these neurons by deacetylating and repressing p53 activity; however, the exact mechanisms remain unclear. Resveratrol also has anti-inflammatory effects as it suppresses M1 microglia activation, which is involved in the initiation of neurodegeneration, and promotes T helper 2 (Th2) response by increasing anti-inflammatory cytokines and SIRT1 expression [167]. A recent study conducted by Sousa et al. [126] illustrated that monoterpenes as SIRT1 activators could be effective potential therapeutic treatments against aging and aging-related disorders including AD. SIRT1 expression and activity reduce with aging, and increasing its activity extends the life span in various mammals, and improves many age-related diseases. Consequently, many natural and synthetic SIRT1 activators and inhibitors have been developed. Known SIRT1 activators of natural origin are mainly polyphenols. Nonetheless, various classes of non-polyphenolic monoterpenoids have been identified as inducers of SIRT1 expression and/or activity [126]. Besides, SIRT1 activators attenuate ED and systemic complications induced by obesity [168]. Therefore, SIRT1 activators could be effective against obesity-mediated complications and the development of AD.
These findings indicate that the SIRT1 pathway is highly dysregulated in obesity and this dysregulation contributes to the pathogenesis of AD. Therefore, SIRT1 activators could be effective in the mitigation of VO-induced AD.
Adipocytokines in Obesity and AD
Different types of adipocytokines are dysregulated in VO which affect the metabolic profile and brain functions. Interestingly, obese subjects have high circulating levels of IL-6, TNF-α, CRP and pro-inflammatory adipocytokines like leptin. Pro-inflammatory adipocytokines trigger chronic inflammation [53]. In contrast, the anti-inflammatory adiponectin plays a crucial role in the mitigation of VO-induced inflammation by inhibiting the expression of NF-κB and TNF-α [55]. In consonance, adiponectin serum level is reduced in obese subjects [59]. A prospective study involved 193 obese subjects showed that adiponectin serum level was low mainly in subjects with high VAI and VAT [59]. Therefore, the expression of leptin and adiponectin are dysregulated in VO leading to systemic inflammation and oxidative disorders. Bonda et al. [169] showed that dysregulation of leptin signaling is involved in AD. Notably, leptin receptors and concentration are increased in specific brain areas and CSF in AD patients. Leptin resistance in the hippocampus and dysregulation of leptin signaling are associated with the development of AD. There is a significant correlation between leptin levels in both CSF and the brain with the progression of AD [170]. It has been shown that leptin promotes the pathogenesis of AD. A prospective study involving 785 subjects showed that high circulating leptin was associated with a low risk for the development of AD [170]. Since leptin level highly fluctuates, and most of the measured leptin was performed in experimental and preclinical studies, a potentially positive correlation between high leptin and cognitive function in AD patients needs to be elucidated in clinical studies. It was suggested that high leptin in presymptomatic AD could be protective and attenuate the progression of AD. Notably, brain leptin level is correlated with the volume of gray matter and hippocampus, and can reverse neurocognitive deficits in humans with congenital leptin deficiency [171]. Different experimental studies reported that the administration of leptin can reduce the formation and aggregation of Aβ [172, 173].
Recent studies confirmed that leptin has neuroprotective against the progression of AD [174, 175]. Leptin improves neurogenesis and antioxidant potential with reduction in the expression of Aβ and tau phosphorylation in mice with experimental AD [174, 176]. Therefore, VO-induced leptin resistance may increase the risk for AD neuropathology through augmentation of neurotoxicity [175]. Thus, disruption of brain leptin signaling may induce the development of AD in mice [175]. These findings indicated that VO-induced leptin resistance could be a possible mechanism for the development of AD via disruption of brain leptin singling (Fig. 9).

Role of leptin and adiponectin in the pathogenesis of Alzheimer’s disease (AD)
Notably, adiponectin plays a crucial role in promoting cognitive function and amelioration the pathogenesis of AD. Adiponectin has a neuroprotective effect against the development of AD through anti-inflammatory and antioxidant effects [177]. A prospective study showed that adiponectin level was reduced in AD patients [178]. Decreasing brain adiponectin favors Aβ accumulation and associated neurodegenerative changes in mice [179]. Therefore, brain hypoadiponectinemia is implicated in the progression of neurodegeneration and AD via induction of brain IR and tau phosphorylation with subsequent propagation of neuroinflammation [178, 179]. Thus, oxidative and inflammatory disorders induced by peripheral and central metabolic disorders may trigger a reduction of circulating adiponectin leading to the development of AD (Fig. 9). On the other hand, the anti-inflammatory adiponectin is highly reduced in VO leading to the development of IR and metabolic derangements [59]. Notably, increasing VAT in obese and overweight individuals is linked with a significant reduction of circulating adiponectin. A prospective study comprising 206 obese subjects who received a structured diet for weight control illustrated that weight loss is associated with increasing total adiponectin levels [59]. There is an inverse relationship between VAT and adiponectin expression. Besides, adiponectin is positively correlated with HDL and negatively correlated with pro-inflammatory cytokines [180, 181]. Thus, VO-induced reduction in circulating adiponectin promotes systemic inflammatory and metabolic changes which in turn reduce the expression of adiponectin and propagation of systemic complications. Reduction of adiponectin as in T2DM and VO increases free fatty acid levels, impairment of insulin signaling, and development of IR and hyperinsulinemia (Fig. 10).

Adiponectin and development of hyperinsulinemia
Therefore, VO-mediated hypoadiponectinemia and leptin resistance may promote the progression of Aβ formation and tau phosphorylation with the development of AD.
Conclusion
AD is a common type of dementia, characterized by the deposition of Aβ plaques and TNTs in the brain. Remarkably, deposition of Aβ occurs prior to the pathology of cortical tau. Deposition of Aβ-induces tau-mediated neuronal and synaptic loss in AD, though tau neuropathology can progress independently of Aβ accumulation. The neuropathological changes in AD are related to the deposition of amyloid plaques and TNTs, the progression of neuroinflammation, neuronal mitochondrial dysfunction, autophagy dysfunction, and cholinergic synaptic dysfunction.
Obesity is a medical condition due to excessive accumulation of body fat which adversely affects health. VO is commonly associated with low-grade inflammation due to higher expression of pro-inflammatory cytokines by adipose tissue. Development of VO is associated with augmentation of pro-inflammatory and proatherogenic mediators leading to ED, IR and atherosclerosis. VAT-induced oxidative stress triggers IR through the modulation of insulin receptors and insulin action. In addition, tissue hypoxia in obesity promotes tissue injury, production of adipocytokines and release of pro-inflammatory cytokines leading to an oxidative-inflammatory loop with induction of IR. Also, brain insulin signaling is involved in the pathogenesis of AD through AKT phosphorylation which induces AD pathology and lowers cognitive function. Of note, AD is developing due to Aβ accumulation via an amyloidogenic pathway with the formation of Aβ plaques. The processing and production of Aβ plaques affect neuronal activity through the interruption of synaptic activity and induction of cell death. Obesity and enlargement of VAT are associated with the deposition of Aβ. Obesity is regarded as an independent risk factor for the development of AD. These findings are consonant with VO increasing the risk of AD through the dysregulation of adipocytokines which affect the development of AD.
Activated NF-κB pathway in VO might be a potential link in the development of AD, so targeting of NF-κB pathway could be a possible therapeutic value in the management of AD. Likewise, the higher concentration of AGEs in VO could be implicated in the pathogenesis of AD. Thus, the reduction of the diet containing AGEs or weight loss may reduce the risk of VO-induced AD.
Different inflammatory signaling pathways are activated in VO that all have a negative impact on the cognitive function and progression of AD except HIF-1 which has beneficial and neuroprotective effects in mitigating the progression of AD. In addition, VO-mediated hypoadiponectinemia and leptin resistance may promote the progression of Aβ formation and tau phosphorylation with the development of AD.
Taken together, VO-induced AD is mainly mediated through the induction of oxidative stress, inflammatory changes, leptin resistance, and hypoadiponectinemia that collectively trigger Aβ formation and neuroinflammation. Thus, early recognition of VO by VAI with appropriate management could be a preventive measure against the development of AD in patients with VO. Large-scale prospective studies are warranted in this regard.
Data Availability
All data are available in the manuscript.
References
Alsubaie N, Al-kuraishy HM, Al-Gareeb AI, Alharbi B, De Waard M, Sabatier J-M, Saad HM, Batiha GE-S (2022) Statins use in Alzheimer disease: bane or boon from frantic search and narrative review. Brain Sci 12:1290
Reitz C, Rogaeva E, Beecham GW (2020) Late-onset vs nonmendelian early-onset Alzheimer disease: a distinction without a difference? Neurol Genet. https://doi.org/10.1212/NXG.0000000000000512
Knopman DS, Petersen RC, Jack CR (2019) A brief history of “Alzheimer disease”: multiple meanings separated by a common name. Neurology 92:1053–1059
Jack CR, Bennett DA, Blennow K, Carrillo MC, Feldman HH, Frisoni GB, Hampel H, Jagust WJ, Johnson KA, Knopman DS (2016) A/T/N: an unbiased descriptive classification scheme for Alzheimer disease biomarkers. Neurology 87:539–547
Kern S, Zetterberg H, Kern J, Zettergren A, Waern M, Höglund K, Andreasson U, Wetterberg H, Börjesson-Hanson A, Blennow K (2018) Prevalence of preclinical Alzheimer disease: comparison of current classification systems. Neurology 90:e1682–e1691
Zhu CW, Sano M (2006) Economic considerations in the management of Alzheimer’s disease. Clin Interv Aging 1:143
Reitz C, Mayeux R (2014) Alzheimer disease: epidemiology, diagnostic criteria, risk factors and biomarkers. Biochem Pharmacol 88:640–651
van der Kant R, Goldstein LS, Ossenkoppele R (2020) Amyloid-β-independent regulators of tau pathology in Alzheimer disease. Nat Rev Neurosci 21:21–35
Long JM, Holtzman DM (2019) Alzheimer disease: an update on pathobiology and treatment strategies. Cell 179:312–339
Al-Kuraishy HM, Abdulhadi MH, Hussien NR, Al-Niemi MS, Rasheed HA, Al-Gareeb AI (2020) Involvement of orexinergic system in psychiatric and neurodegenerative disorders: a scoping review. Brain Circ 6:70
Al-Kuraishy HM (2016) Central additive effect of Ginkgo biloba and Rhodiola rosea on psychomotor vigilance task and short-term working memory accuracy. J Intercult Ethnopharmacol 5:7
Al-Kuraishy HM, Al-Gareeb AI (2020) Citicoline improves human vigilance and visual working memory: the role of neuronal activation and oxidative stress. Basic Clin Neurosci 11:423
Petersen C, Nolan AL, de Paula França Resende E, Miller Z, Ehrenberg AJ, Gorno-Tempini ML, Rosen HJ, Kramer JH, Spina S, Rabinovici GD (2019) Alzheimer’s disease clinical variants show distinct regional patterns of neurofibrillary tangle accumulation. Acta Neuropathol 138:597–612
Mendez MF (2019) Early-onset Alzheimer disease and its variants. Continuum 25:34
Sharma A, Pachauri V, Flora S (2018) Advances in multi-functional ligands and the need for metal-related pharmacology for the management of Alzheimer disease. Front Pharmacol 9:1247
Yiannopoulou KG, Papageorgiou SG (2020) Current and future treatments in Alzheimer disease: an update. J Cent Nerv Syst Dis 12:1179573520907397
Hildreth KL, Van Pelt RE, Schwartz RS (2012) Obesity, insulin resistance, and Alzheimer’s disease. Obesity 20:1549
Verdile G, Keane KN, Cruzat VF, Medic S, Sabale M, Rowles J, Wijesekara N, Martins RN, Fraser PE, Newsholme P (2015) Inflammation and oxidative stress: the molecular connectivity between insulin resistance, obesity, and Alzheimer’s disease. Mediat Inflamm 2015:105828. https://doi.org/10.1155/2015/105828
Luchsinger JA, Cheng D, Tang MX, Schupf N, Mayeux R (2012) Central obesity in the elderly is related to late onset Alzheimer’s disease. Alzheimer Dis Assoc Disord 26:101
Amato MC, Giordano C (2014) Visceral adiposity index: an indicator of adipose tissue dysfunction. Int J Endocrinol 2014:730827. https://doi.org/10.1155/2014/730827
Al-Kuraishy HM, Al-Gareeb AI, Al-Buhadilly AK (2018) Rosuvastatin improves vaspin serum levels in obese patients with acute coronary syndrome. Diseases 6:9
Al-Kuraishy HM, Al-Gareeb AI (2016) Effect of orlistat alone or in combination with Garcinia cambogia on visceral adiposity index in obese patients. J Intercult Ethnopharmacol 5:408
Alomair BM, Al-kuraishy HM, Al-Gareeb AI, Al-Hamash SM, De Waard M, Sabatier J-M, Saad HM, El-Saber Batiha G (2022) Montelukast and acute coronary syndrome: the endowed drug. Pharmaceuticals 15:1147
Gallagher D, Heymsfield SB, Heo M, Jebb SA, Murgatroyd PR, Sakamoto Y (2000) Healthy percentage body fat ranges: an approach for developing guidelines based on body mass index. Am J Clin Nutr 72:694–701
Freedman DS, Sherry B (2009) The validity of BMI as an indicator of body fatness and risk among children. Pediatrics 124:S23–S34
Flegal KM, Graubard BI, Williamson DF, Gail MH (2005) Excess deaths associated with underweight, overweight, and obesity. JAMA 293:1861–1867
Romero-Corral A, Montori VM, Somers VK, Korinek J, Thomas RJ, Allison TG, Mookadam F, Lopez-Jimenez F (2006) Association of bodyweight with total mortality and with cardiovascular events in coronary artery disease: a systematic review of cohort studies. Lancet 368:666–678
Pinho CPS, Diniz AdS, Arruda IKGd, Leite APDL, Petribu MdMV, Rodrigues IG (2018) Waist circumference measurement sites and their association with visceral and subcutaneous fat and cardiometabolic abnormalities. Arch Endocrinol Metab 62:416–423
Kouli G-M, Panagiotakos DB, Kyrou I, Georgousopoulou EN, Chrysohoou C, Tsigos C, Tousoulis D, Pitsavos C (2017) Visceral adiposity index and 10-year cardiovascular disease incidence: the ATTICA study. Nutr Metab Cardiovasc Dis 27:881–889
Amato MC, Giordano C, Galia M, Criscimanna A, Vitabile S, Midiri M, Galluzzo A, Group AS (2010) Visceral Adiposity Index: a reliable indicator of visceral fat function associated with cardiometabolic risk. Diabetes Care 33:920–922
Li B, Lai X, Yan C, Jia X, Li Y (2020) The associations between neutrophil-to-lymphocyte ratio and the Chinese visceral adiposity index, and carotid atherosclerosis and atherosclerotic cardiovascular disease risk. Exp Gerontol 139:111019
Xie Y, Zhang Y, Qin P, Ping Z, Wang C, Peng X, Chen H, Zhao D, Xu S, Wang L (2022) The association between Chinese Visceral Adipose Index and coronary heart disease: a cohort study in China. Nutr Metab Cardiovasc Dis 32:550–559
Nusrianto R, Tahapary DL, Soewondo P (2019) Visceral adiposity index as a predictor for type 2 diabetes mellitus in Asian population: a systematic review. Diabetes Metab Syndr: Clin Res Rev 13:1231–1235
Pekgor S, Duran C, Berberoglu U, Eryilmaz MA (2019) The role of visceral adiposity index levels in predicting the presence of metabolic syndrome and insulin resistance in overweight and obese patients. Metab Syndr Relat Disord 17:296–302
Nusrianto R, Ayundini G, Kristanti M, Astrella C, Amalina N, Riyadina W, Tahapary DL, Soewondo P (2019) Visceral adiposity index and lipid accumulation product as a predictor of type 2 diabetes mellitus: the Bogor cohort study of non-communicable diseases risk factors. Diabetes Res Clin Pract 155:107798
Fiorentino TV (2018) Visceral adiposity index (VAI), a powerful predictor of incident hypertension in prehypertensives. Intern Emerg Med 13:471–473
Elisha B, Messier V, Karelis A, Coderre L, Bernard S, Prud’homme D, Rabasa-Lhoret R (2013) The visceral adiposity index: relationship with cardiometabolic risk factors in obese and overweight postmenopausal women—a MONET group study. Appl Physiol Nutr Metab 38:892–899
Alkazmi L, Al-kuraishy HM, Batiha GE-S, Mostafa-Hedeab G, De Waard M, Sabatier J-M, Kabrah SM, Saad HM, Al-Gareeb AI, Simal-Gandara J (2022) Roxadustat for SARS-CoV-2 infection: old signaling raised new hopes. Drugs R&D 22(3):183–186
Magnuson AM, Fouts JK, Regan DP, Booth AD, Dow SW, Foster MT (2018) Adipose tissue extrinsic factor: obesity-induced inflammation and the role of the visceral lymph node. Physiol Behav 190:71–81
Al-kuraishy HM, Batiha GE-S, Faidah H, Al-Gareeb AI, Saad HM, Simal-Gandara J (2022) Pirfenidone and post-Covid-19 pulmonary fibrosis: invoked again for realistic goals. Inflammopharmacology. https://doi.org/10.1007/s10787-022-01027-6
Rhee KE, Phelan S, McCaffery J (2012) Early determinants of obesity: genetic, epigenetic, and in utero influences. Int J Pediatr 2012:463850. https://doi.org/10.1155/2012/463850
Batiha GE-S, Al-kuraishy HM, Al-Maiahy TJ, Al-Buhadily AK, Saad HM, Al-Gareeb AI, Simal-Gandara J (2022) Plasminogen activator inhibitor 1 and gestational diabetes: the causal relationship. Diabetol Metab Syndr 14:127
Nevalainen T, Kananen L, Marttila S, Jylhävä J, Mononen N, Kähönen M, Raitakari OT, Hervonen A, Jylhä M, Lehtimäki T (2017) Obesity accelerates epigenetic aging in middle-aged but not in elderly individuals. Clin Epigenetics 9:1–9
Ou XH, Zhu CC, Sun SC (2019) Effects of obesity and diabetes on the epigenetic modification of mammalian gametes. J Cell Physiol 234:7847–7855
Martins IJ (2016) The role of clinical proteomics, lipidomics, and genomics in the diagnosis of Alzheimer’s disease. Proteomes 4:14
Khan S, Chan YT, Revelo XS, Winer DA (2020) The immune landscape of visceral adipose tissue during obesity and aging. Front Endocrinol 11:267
Yu J-Y, Choi W-J, Lee H-S, Lee J-W (2019) Relationship between inflammatory markers and visceral obesity in obese and overweight Korean adults: an observational study. Medicine 98:e14740
El-Saber Batiha G, Al-Gareeb AI, Saad HM, Al-Kuraishy HM (2022) COVID-19 and corticosteroids: a narrative review. Inflammopharmacology. https://doi.org/10.1007/s10787-022-00987-z
Hotamisligil GS, Shargill NS, Spiegelman BM (1993) Adipose expression of tumor necrosis factor-α: direct role in obesity-linked insulin resistance. Science 259:87–91
Ghigliotti G, Barisione C, Garibaldi S, Fabbi P, Brunelli C, Spallarossa P, Altieri P, Rosa G, Spinella G, Palombo D (2014) Adipose tissue immune response: novel triggers and consequences for chronic inflammatory conditions. Inflammation 37:1337–1353
Mostafa-Hedeab G, Al-Kuraishy HM, Al-Gareeb AI, Jeandet P, Saad HM, Batiha GE-S (2022) A raising dawn of pentoxifylline in management of inflammatory disorders in Covid-19. Inflammopharmacology. https://doi.org/10.1007/s10787-022-00993-1
Henning RJ (2021) Obesity and obesity-induced inflammatory disease contribute to atherosclerosis: a review of the pathophysiology and treatment of obesity. Am J Cardiovasc Dis 11:504
Singh M, Benencia F (2019) Inflammatory processes in obesity: focus on endothelial dysfunction and the role of adipokines as inflammatory mediators: we reviewed obesity-induced metabolic and immunological changes at the level of vasculature and emphasize on the importance of adipokines. Int Rev Immunol 38:157–171
Alorabi M, Cavalu S, Al-Kuraishy HM, Al-Gareeb AI, Mostafa-Hedeab G, Negm WA, Youssef A, El-Kadem AH, Saad HM, Batiha GE-S (2022) Pentoxifylline and berberine mitigate diclofenac-induced acute nephrotoxicity in male rats via modulation of inflammation and oxidative stress. Biomed Pharmacother 152:113225
Battineni G, Sagaro GG, Chintalapudi N, Amenta F, Tomassoni D, Tayebati SK (2021) Impact of obesity-induced inflammation on cardiovascular diseases (CVD). Int J Mol Sci 22:4798
Babalghith AO, Al-kuraishy HM, Al-Gareeb AI, De Waard M, Sabatier J-M, Saad HM, Batiha GE-S (2022) The potential role of growth differentiation factor 15 in COVID-19: a corollary subjective effect or not? Diagnostics 12:2051
Al-Kuraishy HM, Al-Gareeb AI, Bungau SG, Radu A-F, Batiha GE-S (2022) The potential molecular implications of adiponectin in the evolution of SARS-CoV-2: inbuilt tendency. J King Saud Univ Sci. https://doi.org/10.1016/j.jksus.2022.102347
Abdulhadi MH, Al-Kuraishy HM, Al-Gareeb AI (2021) Beneficial effects of levothyroxine replacement therapy on leptin adiponectin ratio in patients with idiopathic primary hypothyroidism. J Pak Med Assoc 71:S17–21
Gariballa S, Alkaabi J, Yasin J, Al Essa A (2019) Total adiponectin in overweight and obese subjects and its response to visceral fat loss. BMC Endocr Disord 19:1–6
Hurtado-Roca Y, Bueno H, Fernandez-Ortiz A, Ordovas JM, Ibañez B, Fuster V, Rodriguez-Artalejo F, Laclaustra M (2017) Oxidized LDL is associated with metabolic syndrome traits independently of central obesity and insulin resistance. Diabetes 66:474–482
Al-Kuraishy HM, Hussian NR, Al-Naimi MS, Al-Gareeb AI, Al-Mamorri F, Al-Buhadily AK (2021) The potential role of pancreatic γ-aminobutyric acid (GABA) in diabetes mellitus: a critical reappraisal. Int J Prev Med 12:19
Al-Kuraishy HM, Al-Gareeb AI, Waheed HJ, Al-Maiahy TJ (2018) Differential effect of metformin and/or glyburide on apelin serum levels in patients with type 2 diabetes mellitus: concepts and clinical practice. J Adv Pharm Technol Res 9:80
Healy L, Ryan A, Carroll P, Ennis D, Crowley V, Boyle T, Kennedy M, Connolly E, Reynolds J (2010) Metabolic syndrome, central obesity and insulin resistance are associated with adverse pathological features in postmenopausal breast cancer. Clin Oncol 22:281–288
Al-Kuraishy HM, Al-Gareeb AI, Shams HA, Al-Mamorri F (2019) Endothelial dysfunction and inflammatory biomarkers as a response factor of concurrent coenzyme Q10 add-on metformin in patients with type 2 diabetes mellitus. J Lab Physicians 11:317–322
Rasheed HA, Al-Kuraishy HM, Al-Gareeb AI, Hussien NR, Al-Nami MS (2019) Effects of diabetic pharmacotherapy on prolactin hormone in patients with type 2 diabetes mellitus: bane or boon. J Adv Pharm Technol Res 10:163
Maciejczyk M, Żebrowska E, Chabowski A (2019) Insulin resistance and oxidative stress in the brain: what’s new? Int J Mol Sci 20:874
Al-Naimi MS, Rasheed HA, Al-Kuraishy HM, Al-Gareeb AI (2019) Berberine attenuates olanzapine induced-metabolic syndrome. J Pak Med Assoc 69(Suppl 3):S88–s92
Ye J (2013) Mechanisms of insulin resistance in obesity. Front Med 7:14–24
Hardy OT, Czech MP, Corvera S (2012) What causes the insulin resistance underlying obesity? Curr Opin Endocrinol Diabetes Obes 19:81
Greenfield JR, Campbell LV (2004) Insulin resistance and obesity. Clin Dermatol 22:289–295
Kadowaki T, Hara K, Yamauchi T, Terauchi Y, Tobe K, Nagai R (2003) Molecular mechanism of insulin resistance and obesity. Exp Biol Med 228:1111–1117
Al-Nami MS, Al-Kuraishy HM, Al-Gareeb AI (2020) Impact of thioctic acid on glycemic indices and associated inflammatory-induced endothelial dysfunction in patients with type 2 diabetes mellitus: a case control study. Int J Crit Illn Inj Sci 10:21
Wu H, Ballantyne CM (2020) Metabolic inflammation and insulin resistance in obesity. Circ Res 126:1549–1564
Arvanitakis Z, Wang HY, Capuano AW, Khan A, Taïb B, Anokye-Danso F, Schneider JA, Bennett DA, Ahima RS, Arnold SE (2020) Brain insulin signaling, Alzheimer disease pathology, and cognitive function. Ann Neurol 88:513–525
Selles MC, Fortuna JT, Zappa-Villar MF, de Faria YP, Souza AS, Suemoto CK, Leite RE, Rodriguez RD, Grinberg LT, Reggiani PC (2020) Adenovirus-mediated transduction of insulin-like growth factor 1 protects hippocampal neurons from the toxicity of Aβ oligomers and prevents memory loss in an Alzheimer mouse model. Mol Neurobiol 57:1473–1483
Hallschmid M (2021) Intranasal insulin for Alzheimer’s disease. CNS Drugs 35:21–37
Pomytkin I, Costa-Nunes JP, Kasatkin V, Veniaminova E, Demchenko A, Lyundup A, Lesch KP, Ponomarev ED, Strekalova T (2018) Insulin receptor in the brain: mechanisms of activation and the role in the CNS pathology and treatment. CNS Neurosci Ther 24:763–774
Kuwabara T, Kagalwala MN, Onuma Y, Ito Y, Warashina M, Terashima K, Sanosaka T, Nakashima K, Gage FH, Asashima M (2011) Insulin biosynthesis in neuronal progenitors derived from adult hippocampus and the olfactory bulb. EMBO Mol Med 3:742–754
Convit A (2005) Links between cognitive impairment in insulin resistance: an explanatory model. Neurobiol Aging 26:31–35
Gerozissis K (2003) Brain insulin: regulation, mechanisms of action and functions. Cell Mol Neurobiol 23:1–25
Chapman CD, Schiöth HB, Grillo CA, Benedict C (2018) Intranasal insulin in Alzheimer’s disease: food for thought. Neuropharmacology 136:196–201
Kellar D, Craft S (2020) Brain insulin resistance in Alzheimer’s disease and related disorders: mechanisms and therapeutic approaches. Lancet Neurol 19:758–766
Kothari V, Luo Y, Tornabene T, O’Neill AM, Greene MW, Geetha T, Babu JR (2017) High fat diet induces brain insulin resistance and cognitive impairment in mice. Biochim Biophys Acta 1863:499–508
De Felice FG, Vieira MN, Bomfim TR, Decker H, Velasco PT, Lambert MP, Viola KL, Zhao W-Q, Ferreira ST, Klein WL (2009) Protection of synapses against Alzheimer’s-linked toxins: insulin signaling prevents the pathogenic binding of Aβ oligomers. Proc Natl Acad Sci USA 106:1971–1976
Gratuze M, Julien J, Petry FR, Morin F, Planel E (2017) Insulin deprivation induces PP2A inhibition and tau hyperphosphorylation in hTau mice, a model of Alzheimer’s disease-like tau pathology. Sci Rep 7:1–13
Breijyeh Z, Karaman R (2020) Comprehensive review on Alzheimer’s disease: causes and treatment. Molecules 25:5789
Bradley D (2020) Clusterin as a potential biomarker of obesity-related Alzheimer’s disease risk. Biomark Insights 15:1177271920964108
Oh SB, Kim MS, Park S, Son H, Kim SY, Kim MS, Jo DG, Tak E, Lee JY (2019) Clusterin contributes to early stage of Alzheimer’s disease pathogenesis. Brain Pathol 29:217–231
Wittwer J, Bradley D (2021) Clusterin and its role in insulin resistance and the cardiometabolic syndrome. Front Immunol 12:612496
Shie F-S, Shiao Y-J, Yeh C-W, Lin C-H, Tzeng T-T, Hsu H-C, Huang F-L, Tsay H-J, Liu H-K (2015) Obesity and hepatic steatosis are associated with elevated serum amyloid beta in metabolically stressed APPswe/PS1dE9 mice. PLoS ONE 10:e0134531
Meakin PJ, Coull BM, Tuharska Z, McCaffery C, Akoumianakis I, Antoniades C, Brown J, Griffin KJ, Platt F, Ozber CH (2020) Elevated circulating amyloid concentrations in obesity and diabetes promote vascular dysfunction. J Clin Investig 130:4104–4117
Platt TL, Beckett TL, Kohler K, Niedowicz DM, Murphy MP (2016) Obesity, diabetes, and leptin resistance promote tau pathology in a mouse model of disease. Neuroscience 315:162–174
Kelleher I, Garwood C, Hanger DP, Anderton BH, Noble W (2007) Kinase activities increase during the development of tauopathy in htau mice. J Neurochem 103:2256–2267
Kim OY, Song J (2018) The role of irisin in Alzheimer’s disease. J Clin Med 7:407
Jin Y, Sumsuzzman DM, Choi J, Kang H, Lee S-R, Hong Y (2018) Molecular and functional interaction of the myokine irisin with physical exercise and Alzheimer’s disease. Molecules 23:3229
Rotimi DE, Ben-Goru GM, Evbuomwan IO, Elebiyo TC, Alorabi M, Farasani A, Batiha GE-S, Adeyemi OS (2022) Zingiber officinale and Vernonia amygdalina infusions improve redox status in rat brain. Evid Based Complement Alternat Med. https://doi.org/10.1155/2022/9470178
de Freitas GB, Lourenco MV, De Felice FG (2020) Protective actions of exercise-related FNDC5/Irisin in memory and Alzheimer’s disease. J Neurochem 155:602–611
Arhire LI, Mihalache L, Covasa M (2019) Irisin: a hope in understanding and managing obesity and metabolic syndrome. Front Endocrinol 10:524
Leung WK, Yu AP, Lai CW, Siu PM (2018) Association of markers of proinflammatory phenotype and beige adipogenesis with metabolic syndrome in chinese centrally obese adults. J Diabetes Research. https://doi.org/10.1155/2018/8956509
Atti AR, Palmer K, Volpato S, Winblad B, De Ronchi D, Fratiglioni L (2008) Late-life body mass index and dementia incidence: nine‐year follow‐up data from the Kungsholmen Project. J Am Geriatr Soc 56:111–116
Gustafson D (2008) A life course of adiposity and dementia. Eur J Pharmacol 585:163–175
Burns JM, Johnson DK, Watts A, Swerdlow RH, Brooks WM (2010) Reduced lean mass in early Alzheimer disease and its association with brain atrophy. Arch Neurol 67:428–433
Kim HJ, Kim C, Jeon S, Kang M, Kim YJ, Lee J-M, Shin H-Y, Cho H, Ye BS, Kim J-H (2015) Association of body fat percentage and waist-hip ratio with brain cortical thickness. Alzheimer Dis Assoc Disord 29:279–286
Bates KA, Sohrabi HR, Rodrigues M, Beilby J, Dhaliwal SS, Taddei K, Criddle A, Wraith M, Howard M, Martins G (2009) Association of cardiovascular factors and Alzheimer’s disease plasma amyloid-β protein in subjective memory complainers. J Alzheimers Dis 17:305–318
Pasha EP, Birdsill A, Parker P, Elmenshawy A, Tanaka H, Haley AP (2017) Visceral adiposity predicts subclinical white matter hyperintensities in middle-aged adults. Obes Res Clin Pract 11:177–187
Mohamed HE, Abo-ELmatty DM, Mesbah NM, Saleh SM, Ali A-MA, Sakr AT (2018) Raspberry ketone preserved cholinergic activity and antioxidant defense in obesity induced Alzheimer disease in rats. Biomed Pharmacother 107:1166–1174
Poor SR, Ettcheto M, Cano A, Sanchez-Lopez E, Manzine PR, Olloquequi J, Camins A, Javan M (2021) Metformin a potential pharmacological strategy in late onset Alzheimer’s disease treatment. Pharmaceuticals 14:890
Ebrahimpour S, Zakeri M, Esmaeili A (2020) Crosstalk between obesity, diabetes, and alzheimer’s disease: Introducing quercetin as an effective triple herbal medicine. Ageing Res Rev 62:101095
Donohoe F, Wilkinson M, Baxter E, Brennan DJ (2020) Mitogen-activated protein kinase (MAPK) and obesity-related cancer. Int J Mol Sci 21:1241
Munoz L, Ammit AJ (2010) Targeting p38 MAPK pathway for the treatment of Alzheimer’s disease. Neuropharmacology 58:561–568
Lin A-L, Zheng W, Halloran JJ, Burbank RR, Hussong SA, Hart MJ, Javors M, Shih Y-YI, Muir E, Fonseca RS (2013) Chronic rapamycin restores brain vascular integrity and function through NO synthase activation and improves memory in symptomatic mice modeling Alzheimer’s disease. J Cereb Blood Flow Metab 33:1412–1421
Vandanmagsar B, Youm Y-H, Ravussin A, Galgani JE, Stadler K, Mynatt RL, Ravussin E, Stephens JM, Dixit VD (2011) The NLRP3 inflammasome instigates obesity-induced inflammation and insulin resistance. Nat Med 17:179–188
Hanslik KL, Ulland TK (2020) The role of microglia and the Nlrp3 inflammasome in Alzheimer’s disease. Front Neurol 11:570711
Halle A, Hornung V, Petzold GC, Stewart CR, Monks BG, Reinheckel T, Fitzgerald KA, Latz E, Moore KJ, Golenbock DT (2008) The NALP3 inflammasome is involved in the innate immune response to amyloid-β. Nat Immunol 9:857–865
Ising C, Venegas C, Zhang S, Scheiblich H, Schmidt SV, Vieira-Saecker A, Schwartz S, Albasset S, McManus RM, Tejera D (2019) NLRP3 inflammasome activation drives tau pathology. Nature 575:669–673
Shoelson S, Lee J, Yuan M (2003) Inflammation and the IKKβ/IκB/NF-κB axis in obesity-and diet-induced insulin resistance. Int J Obes 27:S49–S52
Granic I, Dolga AM, Nijholt IM, van Dijk G, Eisel UL (2009) Inflammation and NF-kappaB in Alzheimer’s disease and diabetes. J Alzheimers Dis 16:809–821
Egaña-Gorroño L, López-Díez R, Yepuri G, Ramirez LS, Reverdatto S, Gugger PF, Shekhtman A, Ramasamy R, Schmidt AM (2020) Receptor for advanced glycation end products (RAGE) and mechanisms and therapeutic opportunities in diabetes and cardiovascular disease: insights from human subjects and animal models. Front Cardiovasc Med 7:37
Beeri MS, Uribarri J, Cai W, Buchman AS, Haroutunian V (2020) Human Brain and serum advanced glycation end products are highly correlated: preliminary results of their role in Alzheimer disease and type 2 diabetes. Endocr Pract 26:576–577
Gaspar JM, Velloso LA (2018) Hypoxia inducible factor as a central regulator of metabolism— implications for the development of obesity. Front Neurosci 12:813
Hassan H, Chen R (2021) Hypoxia in Alzheimer’s disease: effects of hypoxia inducible factors. Neural Regener Res 16:310–311
Zhang F, Zhong R, Qi H, Li S, Cheng C, Liu X, Liu Y, Le W (2018) Impacts of acute hypoxia on Alzheimer’s disease-like pathologies in APPswe/PS1dE9 mice and their wild type littermates. Front Neurosci 12:314
Lall R, Mohammed R, Ojha U (2019) What are the links between hypoxia and Alzheimer’s disease? Neuropsychiatr Dis Treat 15:1343
Mariani S, Di Giorgio MR, Rossi E, Tozzi R, Contini S, Bauleo L, Cipriani F, Toscano R, Basciani S, Barbaro G (2020) Blood SIRT1 shows a coherent association with leptin and adiponectin in relation to the degree and distribution of adiposity: a study in obesity, normal weight and anorexia nervosa. Nutrients 12:3506
Kuang H, Tan CY, Tian HZ, Liu LH, Yang MW, Hong FF, Yang SL (2020) Exploring the bi-directional relationship between autophagy and Alzheimer’s disease. CNS Neurosci Ther 26:155–166
Sousa C, Mendes AF (2022) Monoterpenes as sirtuin-1 activators: therapeutic potential in aging and related diseases. Biomolecules 12:921
Li L (2017) The molecular mechanism of glucagon-like peptide-1 therapy in Alzheimer’s disease, based on a mechanistic target of rapamycin pathway. CNS Drugs 31:535–549
Rheinheimer J, de Souza BM, Cardoso NS, Bauer AC, Crispim D (2017) Current role of the NLRP3 inflammasome on obesity and insulin resistance: a systematic review. Metabolism 74:1–9
Legrand-Poels S, Esser N, L’homme L, Scheen A, Paquot N, Piette J (2014) Free fatty acids as modulators of the NLRP3 inflammasome in obesity/type 2 diabetes. Biochem Pharmacol 92:131–141
Tan M-S, Yu J-T, Jiang T, Zhu X-C, Tan L (2013) The NLRP3 inflammasome in Alzheimer’s disease. Mol Neurobiol 48:875–882
Stancu I-C, Cremers N, Vanrusselt H, Couturier J, Vanoosthuyse A, Kessels S, Lodder C, Brône B, Huaux F, Octave J-N (2019) Aggregated Tau activates NLRP3–ASC inflammasome exacerbating exogenously seeded and non-exogenously seeded Tau pathology in vivo. Acta Neuropathol 137:599–617
Heneka MT, McManus RM, Latz E (2018) Inflammasome signalling in brain function and neurodegenerative disease. Nat Rev Neurosci 19:610–621
Al-Kuraishy HM, Al-Gareeb AI, Al-Maiahy TJ, Alexiou A, Mukerjee N, Batiha GE-S (2022) Prostaglandins and non-steroidal anti-inflammatory drugs in Covid-19. Biotechnol Genet Eng Rev:1–21
Tornatore L, Thotakura AK, Bennett J, Moretti M, Franzoso G (2012) The nuclear factor kappa B signaling pathway: integrating metabolism with inflammation. Trends Cell Biol 22:557–566
Guo YP, Jiang HK, Jiang H, Tian HY, Li L (2018) Lipoxin A4 may attenuate the progression of obesity-related glomerulopathy by inhibiting NF-κB and ERK/p38 MAPK-dependent inflammation. Life Sci 198:112–118
Batiha GE-S, Al-Gareeb AI, Elekhnawy E, Al-Kuraishy HM (2022) Potential role of lipoxin in the management of COVID-19: a narrative review. Inflammopharmacology. https://doi.org/10.1007/s10787-022-01070-3
Ben J, Jiang B, Wang D, Liu Q, Zhang Y, Qi Y, Tong X, Chen L, Liu X, Zhang Y, Zhu X, Li X, Zhang H, Bai H, Yang Q, Ma J, Wiemer EAC, Xu Y, Chen Q (2019) Major vault protein suppresses obesity and atherosclerosis through inhibiting IKK-NF-κB signaling mediated inflammation. Nat Commun 10:1801
Shi ZM, Han YW, Han XH, Zhang K, Chang YN, Hu ZM, Qi HX, Ting C, Zhen Z, Hong W (2016) Upstream regulators and downstream effectors of NF-κB in Alzheimer’s disease. J Neurol Sci 366:127–134
Lukiw WJ (2012) NF-κB-regulated, proinflammatory miRNAs in Alzheimer’s disease. Alzheimers Res Ther 4:47
Terai K, Matsuo A, McGeer PL (1996) Enhancement of immunoreactivity for NF-kappa B in the hippocampal formation and cerebral cortex of Alzheimer’s disease. Brain Res 735:159–168
Ju Hwang C, Choi DY, Park MH, Hong JT (2019) NF-κB as a key mediator of brain inflammation in Alzheimer’s disease. CNS Neurol Disord Drug Targets 18:3–10
Al-Thomali AW, Al-Kuraishy HM, Al-Gareeb AI, De AKA-BM, Sabatier JM, Khan Khalil AA, Saad HM, Batiha GE (2022) Role of neuropilin 1 in COVID-19 patients with acute ischemic stroke. Biomedicines 10:2032
Uddin MS, Hasana S, Ahmad J, Hossain MF, Rahman MM, Behl T, Rauf A, Ahmad A, Hafeez A, Perveen A, Ashraf GM (2021) Anti-neuroinflammatory potential of polyphenols by inhibiting NF-κB to halt Alzheimer’s disease. Curr Pharm Des 27:402–414
Seo EJ, Fischer N, Efferth T (2018) Phytochemicals as inhibitors of NF-κB for treatment of Alzheimer’s disease. Pharmacol Res 129:262–273
Kong F, Jiang X, Wang R, Zhai S, Zhang Y, Wang D (2020) Forsythoside B attenuates memory impairment and neuroinflammation via inhibition on NF-κB signaling in Alzheimer’s disease. J Neuroinflamm 17:305
Al-Kuraishy HM, Al-Gareeb AI, Faidah H, Al-Maiahy TJ, Cruz-Martins N, Batiha GE (2021) The looming effects of estrogen in Covid-19: a rocky rollout. Front Nutr 8:649128
Asadipooya K, Uy EM (2019) Advanced glycation end products (AGEs), receptor for AGEs, diabetes, and bone: review of the literature. J Endocr Soc 3:1799–1818
Tsoukas MA, Farr OM, Mantzoros CS (2015) Leptin in congenital and HIV-associated lipodystrophy. Metabolism 64:47–59
Sayej WN, Knight Iii PR, Guo WA, Mullan B, Ohtake PJ, Davidson BA, Khan A, Baker RD, Baker SS (2016) Advanced glycation end products induce obesity and hepatosteatosis in CD-1 wild-type mice. Biomed Res Int 2016:7867852
den Engelsen C, van den Donk M, Gorter KJ, Salomé PL, Rutten GE (2012) Advanced glycation end products measured by skin autofluorescence in a population with central obesity. Dermatoendocrinol 4:33–38
Sohouli MH, Sharifi-Zahabi E, Lari A, Fatahi S, Shidfar F (2020) The impact of low advanced glycation end products diet on obesity and related hormones: a systematic review and meta-analysis. Sci Rep 10:22194
Zhang F, Niu L, Li S, Le W (2019) Pathological impacts of chronic hypoxia on Alzheimer’s disease. ACS Chem Neurosci 10:902–909
Cunnane SC, Trushina E, Morland C, Prigione A, Casadesus G, Andrews ZB, Beal MF, Bergersen LH, Brinton RD, de la Monte S, Eckert A, Harvey J, Jeggo R, Jhamandas JH, Kann O, la Cour CM, Martin WF, Mithieux G, Moreira PI, Murphy MP, Nave KA, Nuriel T, Oliet SHR, Saudou F, Mattson MP, Swerdlow RH, Millan MJ (2020) Brain energy rescue: an emerging therapeutic concept for neurodegenerative disorders of ageing. Nat Rev Drug Discov 19:609–633
Babusikova E, Dobrota D, Turner AJ, Nalivaeva NN (2021) Effect of global brain ischemia on amyloid precursor protein metabolism and expression of amyloid-degrading enzymes in rat cortex: role in pathogenesis of Alzheimer’s disease. Biochemistry 86:680–692
Wu Y, Li Z, McDonough MA, Schofield CJ, Zhang X (2021) Inhibition of the oxygen-sensing asparaginyl hydroxylase factor inhibiting hypoxia-inducible factor: a potential hypoxia response modulating strategy. J Med Chem 64:7189–7209
Gaspar JM, Mendes NF, Corrêa-da-Silva F, Lima-Junior JC, Gaspar RC, Ropelle ER, Araujo EP, Carvalho HM, Velloso LA (2018) Downregulation of HIF complex in the hypothalamus exacerbates diet-induced obesity. Brain Behav Immun 73:550–561
Ashok BS, Ajith TA, Sivanesan S (2017) Hypoxia-inducible factors as neuroprotective agent in Alzheimer’s disease. Clin Exp Pharmacol Physiol 44:327–334
Lall R, Mohammed R, Ojha U (2019) What are the links between hypoxia and Alzheimer’s disease? Neuropsychiatr Dis Treat 15:1343–1354
Li J, Tao T, Xu J, Liu Z, Zou Z, Jin M (2020) HIF–1α attenuates neuronal apoptosis by upregulating EPO expression following cerebral ischemia–reperfusion injury in a rat MCAO model. Int J Mol Med 45:1027–1036
Martins IJ (2016) Anti-aging genes improve appetite regulation and reverse cell senescence and apoptosis in global populations. Adv Aging Res 5:9–26. https://doi.org/10.4236/aar.2016.51002
Martins IJ (2017) Single gene inactivation with implications to diabetes and multiple organ dysfunction syndrome. J Clin Epigenet 3:24. https://doi.org/10.21767/2472-1158.100058
Martins I (2019) Appetite regulation and the peripheral sink amyloid beta clearance pathway in diabetes and Alzheimer’s disease. In: Top 10 commentaries in Alzheimer’s disease. Avid Sci, pp 2–11
Song YS, Lee SK, Jang YJ, Park HS, Kim J-H, Lee YJ, Heo Y-S (2013) Association between low SIRT1 expression in visceral and subcutaneous adipose tissues and metabolic abnormalities in women with obesity and type 2 diabetes. Diabetes Res Clin Pract 101:341–348
Sasaki T (2015) Age-associated weight gain, leptin, and SIRT1: a possible role for hypothalamic SIRT1 in the prevention of weight gain and aging through modulation of leptin sensitivity. Front Endocrinol 6:109
Maiese K (2018) Sirtuins: developing innovative treatments for aged-related memory loss and Alzheimer’s disease. Curr Neurovasc Res 15:367–371
Martins IJ (2013) Increased risk for obesity and diabetes with neurodegeneration in developing countries. J Mol Genet Med S1:001. https://doi.org/10.4172/1747-0862.S1-001
Gomes BAQ, Silva JPB, Romeiro CFR, Dos Santos SM, Rodrigues CA, Gonçalves PR, Sakai JT, Mendes PFS, Varela ELP, Monteiro MC (2018) Neuroprotective mechanisms of resveratrol in Alzheimer’s disease: role of SIRT1. Oxid Med Cell Longev 2018:8152373. https://doi.org/10.1155/2018/8152373
San Cheang W, Wong WT, Wang L, Cheng CK, Lau CW, Ma RCW, Xu A, Wang N, Huang Y, Tian XY (2019) Resveratrol ameliorates endothelial dysfunction in diabetic and obese mice through sirtuin 1 and peroxisome proliferator-activated receptor δ. Pharmacol Res 139:384–394
Bonda DJ, Stone JG, Torres SL, Siedlak SL, Perry G, Kryscio R, Jicha G, Casadesus G, Smith MA, Zhu X (2014) Dysregulation of leptin signaling in Alzheimer disease: evidence for neuronal leptin resistance. J Neurochem 128:162–172
Lieb W, Beiser AS, Vasan RS, Tan ZS, Au R, Harris TB, Roubenoff R, Auerbach S, DeCarli C, Wolf PA (2009) Association of plasma leptin levels with incident Alzheimer disease and MRI measures of brain aging. JAMA 302:2565–2572
Paz-Filho GJ, Babikian T, Asarnow R, Esposito K, Erol HK, Wong M-L, Licinio J (2008) Leptin replacement improves cognitive development. PLoS ONE 3:e3098
Greco SJ, Sarkar S, Johnston JM, Tezapsidis N (2009) Leptin regulates tau phosphorylation and amyloid through AMPK in neuronal cells. Biochem Biophys Res Commun 380:98–104
Marwarha G, Dasari B, Prasanthi JR, Schommer J, Ghribi O (2010) Leptin reduces the accumulation of Aβ and phosphorylated tau induced by 27-hydroxycholesterol in rabbit organotypic slices. J Alzheimers Dis 19:1007–1019
Calió ML, Mosini AC, Marinho DS, Salles GN, Massinhani FH, Ko GM, Porcionatto MA (2021) Leptin enhances adult neurogenesis and reduces pathological features in a transgenic mouse model of Alzheimer’s disease. Neurobiol Dis 148:105219
King A, Brain A, Hanson K, Dittmann J, Vickers J, Fernandez-Martos C (2018) Disruption of leptin signalling in a mouse model of Alzheimer’s disease. Metab Brain Dis 33:1097–1110
Shafiq S, Zahan R, Yesmin S, Khan A, Mahmud MS, Reza MA, Albogami SM, Alorabi M, De Waard M, Saad HM, Sabatier J-M, Naz T, Batiha GE-S (2022) Phytochemical analysis and understanding the antioxidant and anticancer properties of methanol extract from Litsea glutinosa: in vitro and in vivo studies. Molecules 27:6964
Ng RC-L, Chan K-H (2017) Potential neuroprotective effects of adiponectin in Alzheimer’s disease. Int J Mol Sci 18:592
Teixeira AL, Diniz BS, Campos AC, Miranda AS, Rocha NP, Talib LL, Gattaz WF, Forlenza OV (2013) Decreased levels of circulating adiponectin in mild cognitive impairment and Alzheimer’s disease. Neuromol Med 15:115–121
Chan K-H, Lam KS-L, Cheng O-Y, Kwan JS-C, Ho PW-L, Cheng KK-Y, Chung SK, Ho JW-M, Guo VY, Xu A (2012) Adiponectin is protective against oxidative stress induced cytotoxicity in amyloid-beta neurotoxicity. PLoS ONE 7:e52354
Nova E, San Mauro-Martín I, Díaz-Prieto LE, Marcos A (2019) Wine and beer within a moderate alcohol intake is associated with higher levels of HDL-c and adiponectin. Nutr Res 63:42–50
Tong HV, Luu NK, Son HA, Hoan NV, Hung TT, Velavan TP, Toan NL (2017) Adiponectin and pro-inflammatory cytokines are modulated in Vietnamese patients with type 2 diabetes mellitus. J Diabetes Investig 8:295–305
Funding
Not applicable.
Author information
Authors and Affiliations
Contributions
HMA, AIA, AAA and ZHH conceptualized the manuscript, wrote, edited and reviewed the main text and approved the final edition of the manuscript. NAK, HMS, GE-SB and MDW prepared the figures, wrote, corrected, amended and approved the final edition of the manuscript.
Corresponding authors
Ethics declarations
Competing Interests
The authors declare no conflict of interest.
Ethical Approval
Not applicable.
Consent to Participate
Not applicable.
Consent for Publication
Not applicable.
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Springer Nature or its licensor (e.g. a society or other partner) holds exclusive rights to this article under a publishing agreement with the author(s) or other rightsholder(s); author self-archiving of the accepted manuscript version of this article is solely governed by the terms of such publishing agreement and applicable law.
About this article

Cite this article
Al-Kuraishy, H.M., Al-Gareeb, A.I., Alsayegh, A.A. et al. A Potential Link Between Visceral Obesity and Risk of Alzheimer’s Disease. Neurochem Res 48, 745–766 (2023). https://doi.org/10.1007/s11064-022-03817-4
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s11064-022-03817-4