
Abstract
Post-traumatic epilepsy (PTE) is a serious complication that can occur following traumatic brain injury (TBI). Sustained secondary changes after TBI promote the process of PTE. Here, we aim to evaluate changes in behavior, electrocorticogram, and histomorphology in rats following chronic TBI models. We observed intensive 7–8 Hz spike-wave-discharges (SWDs) at frontal recording sites and quantified them in SD rats with different degrees of TBI and compared them with age-matched sham rats to evaluate the association between SWDs and injury severity. Notably, although SWDs were even presented in the sham group, the number and duration of events were much lower than those in the TBI groups. SWDs have numerous similarities to absence seizures, such as abrupt onset, termination, and lack of postictal suppression, which may be the nonconvulsive characteristics of PTE. Retigabine, a novel antiepileptic drug, is ineffective in reducing SWDs. In addition, we examined chronic histopathological changes in TBI rats. Rats subjected to moderate and severe TBI exhibited significantly impaired neurological function, which was accompanied by marked cortical injury, hippocampus deformation, reactive gliosis, and mossy fiber sprouting. Long-term progressive structural changes in the brain are one of the characteristics of epileptogenesis after TBI. Our study provided the potential value of epileptiform SWDs in reflecting the nonconvulsive characteristic of PTE and highlighted the vital role of chronic pathological changes, such as reactive gliosis, in promoting the epileptogenesis following TBI.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Traumatic brain injury (TBI) is a primary cause of acquired epilepsy [1], which is characterized by spontaneous repetitive seizures that are usually focal and nonconvulsive but can be generalized as convulsive seizures. Post-traumatic epilepsy (PTE) is often refractory to anti-seizure medications or surgery. Therefore, it is crucial to use appropriate preclinical models to explore the mechanisms underlying the development of PTE. One of the most common animal models for TBI is the controlled cortical impact (CCI) model, which mimics penetrating brain trauma and is significantly more likely to produce PTE than other models [2,3,4]. To date, spontaneous convulsive (infrequent and delayed) and nonconvulsive seizures have been reported in CCI rats [5, 6], and the increase in the risk of seizures depends on injury severity [5,6,7]. The CCI model is well characterized histopathologically and has been used successfully in rats to demonstrate the development of PTE. Brain structural changes, such as neuronal injury and reactive gliosis, have been observed in the ipsilateral cortex, hippocampus (CA1–CA3 and dentate gyrus [DG] regions), and thalamus [6].
Absence seizures (ASs) are a form of nonconvulsive seizure characterized by impaired consciousness, sudden arrest of ongoing behavior, and occurrence of bilaterally synchronous spike-wave-discharges (SWDs) in the electrocorticogram (ECoG) [8, 9]. The SWDs of ASs are typically episodes of sudden onset, termination, and lack of postictal suppression that last from seconds to minutes and mainly occur during light sleep and passive wakefulness [10]. These events are often accompanied by behavioral arrest (interruption of ongoing movement) with or without rhythmic facial twitching. Inherent 7–12 Hz SWDs are commonly recorded in both inbred (Genetic Absence Epilepsy Rats from Strasbourg [GAERS] and Wistar Albino Glaxo Rats from Rijswijk [WAG/Rij]) and outbred (Sprague–Dawley and Long-Evans) rat strains. Moreover, SWDs are recorded in both sham and TBI rats and are identical to those recorded in photothrombotic brain infarction models of post-stroke epilepsy [11, 12]. However, the association between SWDs and TBI still remains unknown. TBI can lead to acute cell death, BBB damage, and excitatory toxicity shortly after injury. Nevertheless, chronic neuropathology, such as neuronal injury, hippocampal cell loss, mossy fiber sprouting (MFS), and synaptic reorganization, may be primarily involved in the process of post-traumatic epileptogenesis. Therefore, in this study, we visually examined continuous video-ECoG recordings for possible seizure attacks and quantified the frequency and duration of SWDs to describe the electrographic signatures of SWDs and examine the relation between SWDs and TBI. In addition, we tested the effect of a novel drug, retigabine, on reducing SWDs. At last, we also discussed the corresponding neuropathological changes associated with chronic TBI to establish relationship between structure and function.
Materials and Methods
Animals
A total of 24 adult male Sprague–Dawley rats weighing 250 ± 10 g (Beijing Vital River Laboratory Animal Technology Co., Ltd., Beijing, China) were housed in an animal facility in standard laboratory conditions (temperature 22 °C ± 1 °C; humidity 50–60%; 12-h light–dark cycle) with food and water ad libitum. All procedures were performed in accordance with the guidelines of the Capital Medical University Animal Care and Use Committee. Rats were randomly divided into the following groups: sham-operated (n = 6), CCI-mild (n = 7), CCI-moderate (n = 7), and CCI-severe (n = 4).
Controlled Cortical Impact Injury
We used the CCI model of TBI. Rats were anesthetized with 3–5% isoflurane, fixed in a stereotaxic frame, and maintained with 1.5% isoflurane via a nose cone throughout the surgery. A 6-mm diameter craniotomy was centered at anteroposterior (AP) − 3.0 mm and mediolateral (ML) + 3.0 mm, with the dura mater kept intact. Cortical contusion was produced using CCI equipment (Leica 9969S) with a 5 mm-diameter tip. The impact velocity was 4.0 m/s, and the contact duration was 100 ms. The impact depth was 1.0, 2.0, or 3.0 mm (mild, moderate, or severe injury, respectively). The rats were placed in a heating pad for recovery post-TBI. Sham-injured rats received identical surgical preparation but were not subjected to CCI injury.
Behavioral Test
The modified Neurological Severity Score (mNSS) test [13] was performed in all rats before TBI and at 1, 3, 7, 10, and 14 days following TBI by two investigators who were blinded to experimental group. The mNSS comprehensively evaluates neurological functions, such as motor, sensory, reflex, and balance functions. Scores range from 0 to 18 (minimum 0, maximum 18) and higher scores indicate more serious injury.
Chronic Video-ECoG Recording
Ten months after injury, rats were implanted with intracranial screw electrodes (1 × 4 mm, Plexon). Two recording electrodes were placed over the ipsilateral frontal (AP 3.0 mm, ML 2.5 mm) and contralateral frontal (AP 3.0 mm, ML − 2.5 mm) cortices. A reference screw and a ground screw were implanted in the bilateral occipital bones, respectively. Screws were wrapped with silver wire, which was welded to a multi-pin connector and secured using dental acrylic. After a 1-week recovery period, video-ECoGs were recorded for 7 days using the Omniplex Server System (Plexon, USA) at a sampling rate of 2000 Hz. On the final day of video-ECoG monitoring, rats received a single intraperitoneal [i.p.] injection of retigabine (10 mg/kg; R823985, Macklin, Shanghai, China), followed by video-ECoG monitoring for 4 h to observe changes in SWDs and behavior.
Quantification of SWD Events
At the end of video-ECoG monitoring, 3-day (6 h/day) long records were extracted for each rat and manually reviewed for subsequent SWD-event and related behavior analyses. An SWD event was defined as abnormal rhythmic discharge with high-amplitude spikes that are distinct from baseline activity and have a minimum duration of 10 s. The asynchronicity and synchronicity of SWDs were assessed by comparing SWD initiation between the two electrodes. If there was no detectable SWD delay between the two recording electrodes, onsets were considered synchronous, whereas an SWD delay exceeding 100 ms was considered an asynchronous onset. The behavior of rats during SWDs was also observed using time-locked video.
Histological Assessment
TBI-moderate rats were deeply anesthetized with 10% chloral hydrate (0.04 ml/kg, i.p.) and transcardially perfused with 0.9% NaCl and 4% paraformaldehyde (PFA). The brains were removed and placed in 4% PFA at 4 ℃ to be post-fixed overnight. Brains were cryosectioned (CM1950, Leica, Bensheim, Germany) into serial coronal slices (25 µm). The sections were placed onto gelatin-covered microscope slides and stained with Cresyl Violet for morphological analysis. The damage volume was calculated by the formula: [(VC − VL)/VC] × 100% (VC: volume of the contralateral hemisphere; VL: volume of the ipsilateral hemisphere). Numbers of neurons in the hippocampal CA1 region were counted with ImageJ and analyzed with Prism (GraphPad Software, San Diego, CA, USA).
Floating slices were washed three times with PBSTT (phosphate-buffered saline [PBS] with 0.3% Triton X-100 and 0.1% Tween). Slices were blocked with 1% goat serum to avoid binding of non-specific antibodies and incubated at 4 ℃ overnight in a solution containing the primary antibody for ionized calcium binding adaptor molecule 1 (Iba1; CST, 17,198, 1:300), glial fibrillary acidic protein (GFAP; Invitrogen, 13–0300, 1:300) and zinc transporter 3 (ZnT3; SySy, 197,002, 1:300). After thoroughly washing in PBSTT, the slices were incubated with secondary antibodies (Alexa Fluor 488 donkey anti-rat or goat anti-rabbit immunoglobulin G) for 1.5 h and counterstained with 4’,6-diamidino-2-phenylindole. Images were captured using a confocal microscope (LSM710, Zeiss).
Statistical Analysis
Statistical analysis was performed using SPSS version 22 and GraphPad Prism 8. Data were analyzed using ANOVA, and t test. Two-tailed Fisher’s exact tests were used to evaluate the incidence of SWDs in each group. Results are expressed as Means ± SEM, and p < 0.05 was considered statistically significant.
Results
CCI Injury and Neurological Dysfunction
The design of the study is illustrated in Fig. 1A. The rats were subjected to different degrees (sham, mild, moderate, and severe) of unilateral cortical contusion using the CCI model, and body weight and neurological function were evaluated before injury and 1, 3, 7, 10, and 14 days after injury. The body weight of the rats decreased on the first day following TBI and gradually increased during subsequent days, with no significant difference between groups (Fig. 1B). The mNSS test was used to evaluate the behavior of rats at different injury time points. Severe brain injury rats showed obvious neurological dysfunction, which did not recover at 14 days after TBI, which resulted in obvious sequelae. Rats subjected to moderate TBI exhibited significantly impaired neurological function on days 1 and 3 post-TBI and largely recovered after 7 days. There was no significant difference in neurological function between the mild injury and sham groups (Fig. 1C). To effectively measure parameters of the chronic CCI models, we should take the mortality into consideration. Due to a mortality rate of up to 25%, rats in the severe injury group were not appropriate for long-term observation. Therefore, we recruited rats in mild and moderate groups for the subsequent procedure.

Controlled cortical impact (CCI) injury and neurological dysfunction. A Study design. The rats were subjected to different degrees (sham, mild, moderate and severe) of CCI injury. Behavioral test was performed before CCI and 1, 3, 7, 10, and 14 days after CCI. Skull electrodes were implanted at 10 months post-traumatic brain injury (TBI). Video-electrocorticogram(v-ECoG) was monitored for 1 weeks, then followed by retigabine injection(10 mg/kg). B The body weight of the rats decreased on the first day after TBI and then gradually increased, with no significant differences among groups. C Behavioral test, indicated by modified Neurological Severity Score (mNSS), was assessed at the time points mentioned above. (*p < 0.05, **p < 0.01, ***p < 0.001 vs. the sham group)
Appearance and Characteristics of SWD Events
At 10 months after TBI, rats received video-ECoG monitoring to assess spontaneous seizures. Two rats (one in the sham group and one in the moderate TBI group) were excluded from analysis because of neocortical damage that occurred during electrode implantation surgery. In one case, the headstage detached from the rat 3 days after the electrodes were implanted. The other rats all showed spontaneous SWD bursts in the continuously recorded video-ECoG, which typically had large amplitudes, high-frequency activity, abrupt onset, and termination. Figure 2A and B shows a 200-s ECoG recording and corresponding power spectra of sham and moderately injured rats 10 months following TBI. SWDs were characterized by abrupt initiations and terminations and no suppression post-event and had spectral power in the theta (4–8 Hz) and beta bands (12–18 Hz). Waveforms were selected from the sham and TBI groups and expanded at different timescales, as shown in grey box of Fig. 2. The spike frequency was approximately 7 Hz (6–8 Hz), and morphology did not appear significantly different, which is consistent with previous reports [14]. Another feature of these abnormal discharges was that they could be interrupted by warning stimuli, such as sudden changes in noise or light (Fig. 3A). The corresponding videos showed stereotyped behaviors of rats, such as exhibiting a freezing-like posture (interruption of ongoing movement), vibrissa extension, and slight vibrational tremor, which is similar to non-convulsive seizures (Fig. 3B), which have been described in many other studies [15, 16].

Characteristics of spike-wave-discharges (SWD) events. A–B A sample 200-s ECoG recording and corresponding power spectra from sham and TBI moderately injured rats. Grey boxes showed the extended waveforms from the sham and TBI groups at different time scales

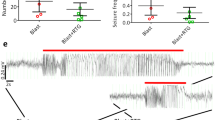
Interruption of ongoing SWD events with noise. A SWDs interrupted by warning stimuli were recorded using ECoG and spectrogram. A noise occurred (red line) and interrupted the SWD episode. B Corresponding videos showed stereotyped behaviors in rats, such as exhibiting a freezing-like posture (interruption of ongoing movement), vibrissa extension, and slight vibrational tremor
TBI Increased the Frequency and Duration of SWD Events
The SWD incidence rate was no significant difference between TBI rats (80%) and sham rats (75%) (Fisher’s Exact Test, p = 1.0). Most SWD events began synchronously, and few initiated asynchronously (Fig. 4A and B). To compare SWD events among the sham, mild, and moderate injury groups, we measured the number and duration of SWD events in each group that were > 10 s. The comparison of the number of SWD events between the different groups showed that the TBI moderate injury group had significantly more SWD events than did the sham (p = 0.0042) and mild injury groups (p = 0.0216) (Fig. 4C). The total SWD duration per day of the TBI moderate injury rats differed significantly from that of the sham (p = 0.0087) and mildly injured rats (p = 0.0212) (Fig. 4D). In addition, the longest SWD event recorded in the sham group lasted 38.4 s, whereas that in the moderate injury group lasted 154.9 s.

TBI injury increased the frequency and duration of SWD events. A Spontaneous SWD bursts occurred in ipsilateral and contralateral hemispheres of TBI rat brains. B Expansion of approximately 9 s of samples showed that SWD events appeared to start synchronously. C Comparisons of the frequency of SWD events among different groups showed that moderate injury rats exhibited significantly more SWD events. D SWD duration was longer in moderate group than that in sham and mild groups. (**p < 0.01 vs. the sham group; #p < 0.05 vs. the mild group)
Retigabine Increases the Frequency of SWD Events
Retigabine, a novel anti-seizure medication, has been reported to be effective in controlling convulsive seizures in animal models [17, 18]. However, the efficacy of retigabine in the treatment of nonconvulsive seizures remains unclear. Having observed SWDs with characteristics similar to ASs, we tested whether retigabine can reduce SWDs in these TBI rats. Therefore, we administered an anticonvulsant, retigabine (10 mg/kg; i.p.) to rats at 40 ± 8 weeks post-TBI and monitored rats via video-ECoG. After 2 h of baseline recording, retigabine was injected, followed by 4 h of observation. We found that the frequency of SWD events was significantly increased after retigabine intervention. Figure 5A and B shows the ECoG and the corresponding power spectra before and after retigabine intervention. However, SWD morphology did not differ significantly between before and after the intervention. Overall, the data indicated that administering retigabine post-TBI aggravates spontaneous SWDs; however, further studies are necessary to clarify the possible mechanisms.

Retigabine aggravated SWDs in TBI rats. A Spontaneous SWD bursts were recorded using ECoG, the frequency of SWD events significantly increased following retigabine intervention (10 mg/kg; i.p.). B The corresponding power spectra are shown before and after retigabine intervention
Chronic Neuropathology after TBI
Nissl staining performed on the brain slices of chronic moderate TBI and age-matched sham rats. The analysis of cortical loss and hippocampal damage, which is considered to be associated with epileptogenesis following TBI, indicated that compared with the sham group, there was a significantly greater volumetric loss of focal cortex and more pronounced hippocampal deformation in the TBI group, particularly in the ipsilateral brain tissue (Fig. 6A and C). The results of quantitative analysis show that compared with the sham group, the TBI group showed significantly reduced cortical tissue (Fig. 6B). In the TBI group, the number of neurons in the ipsilateral CA1 was significantly lower than that in the contralateral CA1, which was calculated using the stereological strategy. No significant differences between the contralateral and ipsilateral sides were observed in the sham group (Fig. 6D). Immunofluorescence for Iba1 and GFAP in the damaged cortex was elevated in the TBI group compared with that in the sham group (Fig. 7A and B). Hippocampal astrocyte proliferation based on GFAP were evident in TBI rats, particularly in the CA3 region (Fig. 8A). MFS was detected by staining hippocampal sections with ZnT3, a member of mammalian zinc transporters, which is expressed in reactive zinc-enriched brain regions such as entorhinal cortex, hippocampus and amygdala (as observed previously with Timm staining) [19, 20]. ZnT3 is enriched in mossy fibers that project from hippocampal dentate granule cells to hilar interneurons and CA3 pyramidal neurons [20]. MFS in the CA3 was detected in TBI rats (Fig. 8B), which may create a functional excitatory feedback loop and contribute to epileptogenesis.

Chronic structural abnormalities in TBI rats. A Tissue loss were investigated using Cresyl Violet staining at 10 months after TBI in the sham and TBI groups. B Compared with the sham group, the cortical tissue of TBI group decreased significantly. C Hippocampal neuronal loss was significant in the TBI group, particularly in the ipsilateral brain tissue. D The number of neurons in CA1 was quantified using the stereological strategy and was significantly lower in the ipsilateral than in the contralateral hemisphere in the TBI group. (*p < 0.05, **p < 0.01)

Reactive gliosis in peripheral cortex after TBI. A-B Microglia and astrocytes all increased in damaged cortex, the fluorescence intensity of ionized calcium binding adaptor molecule 1 (Iba1) and glial fibrillary acidic protein (GFAP) in the ipsilateral cortex of the TBI group were significantly higher than that of the sham group

Astrocyte proliferation and mossy fiber sprouting (MFS) in the ipsilateral hippocampus after TBI. A Compared with the sham group, the immunofluorescence of GFAP increased significantly more in the TBI group. B Region-matched hippocampal sections were stained for Zinc transporter 3 (ZnT3), a zinc transporter that is abundant in synaptic vesicles of mossy fibers, to examine MFS. Compared with sham rats, MFS was evident in TBI rats, especially in the CA3 region (white arrow)
Discussion
PTE is a chronic recurrent spontaneous brain dysfunction caused by cellular and molecular alterations of the brain and increased neuronal excitability following brain trauma. In this study, we evaluated changes in behavior, ECoG, and histomorphology in chronic TBI models. High amplitude and frequent SWDs of 7–8 Hz were observed at frontal recording sites, and they were correlated with the degree of injury. SWDs were characterized by abrupt onset and termination, and were often accompanied by behavioral arrest, vibrissa extension, and slight facial twitching. In addition, Food and Drug Administration-approved anticonvulsant drug, retigabine, was ineffective in reducing these SWDs. Histopathological results showed significant cortical damage, hippocampal deformation, glial cell proliferation and MFS in chronic moderate TBI model.
SWD after TBI May Reflect ASs
In one study, two distinct types of spontaneous seizures were observed in adult rats months (rather than weeks) after TBI. These types of seizures included convulsive seizures, which are infrequent but last for several minutes, and nonconvulsive seizures, which are frequent but only last for tens of seconds. Nonconvulsive seizures were four times more frequent and two times longer in the TBI rats than in the uninjured rats [21]. D’Ambrosio and Miller [22] observed transient rhythmic electrical activity accompanied by behavioral arrest in rats following lateral fluid percussion injury (FPI), which they considered similar to SWDs and ASs. Our study findings are consistent with SWDs previously observed in Sprague–Dawley rats [12]. The observed SWD characteristics can be summarized as follows: (1) high-voltage rhythmic and frequent SWDs of 7–8 Hz were observed at frontal recording sites, which correlated with the degree of TBI; (2) SWDs were equally prominent in age-matched sham rats, which had a shorter duration and lower frequency, and had similar morphological and behavioral features to the SWDs of the TBI group; (3) most SWD events in both the sham and TBI groups started synchronously; those that were not if not synchronous differed by only several hundreds milliseconds; (4) SWDs were often accompanied by stereotyped behaviors of rats, such as behavioral arrest (interruption of ongoing movement), vibrissa extension, and slight facial twitching, which are similar to the behaviors of ASs; (5) sensory stimuli, such as a click, can interrupt the development of SWDs.
ASs, a common form of genetic epilepsy, can result in impaired attention and emotional and social deficits [23]. ASs are characterized by a short duration (a few seconds) and are usually accompanied by behavioral interruptions and impairment in consciousness [24, 25]. One of the most striking electrographic features of ASs is synchronous 3 Hz SWDs across the cerebral cortex [26]. There has been controversy over whether SWDs at 6–8 Hz in rodents is a pathological or normal rhythm [27]. Some researchers suspect that these events are not a pathological characteristic (lack of postictal suppression) of seizures that are observed clinically in posttraumatic epilepsy or other forms of acquired epilepsy [28, 29]. In addition, these high-voltage rhythmic spikes, which are described as SWDs, are present in uninjured rats, including Sprague–Dawley rats used in previous FPI experiments. Two rat strains, GAERS and WAG/Rij, have been used as animal models for genetic absence epilepsy, which also exhibit spontaneous SWDs at 7–12 Hz, accompanied by behavioral arrest. However, a counter-argument is that rodent SWDs resemble human ASs: (1) SWDs recorded in TBI rats occur numerous times per day and last for several seconds with rhythmic spikes in the range of 6–8 Hz, which are similar to ASs; (2) SWDs with an abrupt onset, termination, and lack of postictal suppression are properties that are identical to classical ASs; (3) SWDs can be blocked by anti-absence medications, such as ethosuximide, which strengthen their association with ASs [10]. Although focal onsets may be a key characteristic to distinguish between subconvulsive events after injury and ASs, studies have shown that ASs in rodents start asynchronously; moreover, the ‘cortical focus’ theory suggests that cortical focus drives the widespread corticothalamic network during ASs [29, 30]. Furthermore, signal analytical techniques have demonstrated that a local cortical onset zone of SWD exists in both rodents and humans, albeit in different locations, and SWDs occur through repetitive cyclic activity of the cortico–thalamo–cortical circuits [31].
Retigabine Increases the Frequency and Duration of SWD Events
Although the mechanisms that give rise to the paroxysmal oscillations of ASs are not fully understood, evidence suggests that synchronous bidirectional cortico–thalamo–cortical circuits are crucial [32]. ASs constitute a specific type of epilepsy that is likely associated with excess gamma-aminobutyric acid (GABA)ergic transmission in the thalamus. Studies have shown that the inhibition and disinhibition mediated by extrasynaptic GABAA receptors (eGABAAR) in thalamocortical (TC) neurons play a bidirectional role in the generation of ASs. The anticonvulsant retigabine is a novel antiseizure drug that is used for partial-onset seizures in adults with epilepsy. Its anticonvulsive effects involve Kv7 channels, which are encoded by KCNQ genes, as well as eGABAAR [33]. Retigabine is effective for nearly all types of seizure models except ASs, even though Kv7 channels in the thalamus are highly expressed [17]. Retigabine is a subtype-selective regulator of GABAAR, with a preference for extrasynaptic δ-containing receptors [33]. Although enhanced eGABAAR activity in TC neurons is a typical pathophysiological mechanism in several absence epilepsy models, drugs that activate δ-containing GABAAR can aggravate ASs [34, 35]. This explains our results where retigabine exacerbated seizure expression (reflected by increases in both the duration and number of SWDs). Consistent with these findings, we also found that the frequency and duration of SWD events were significantly increased following retigabine intervention, although SWD morphology did not change. This indicates that using retigabine for patients with absence-like seizures after TBI is not recommended.
Histopathology of the Post-TBI Epileptic Rat Brain
The reason for the high frequency of SWDs in TBI rats is not well understood. However, it is hypothesized that it is related to the severity of brain injury and the degree of midline shift. Although damage following CCI is largely unilateral, moderate or severe injury may cause relative widespread damage of the ipsilateral brain, which includes the medulla, thalamus, and hippocampus. Before spontaneous seizures occur, progressive brain damage occurs over months to years. Magnetic resonance images from 3 h to 60 days after FPI shows that lesions involve the damaged cortex, hippocampus, and thalamus (with hemi-atrophy occurring over time) [36]. Our study found the chronic structural abnormalities in TBI rats, including the cortical tissue loss, hippocampal deformation, and reactive gliosis in the injured cerebral cortex after brain trauma. These findings suggest that persistent progressive structural change is one of the characteristics of epileptic seizures after brain injury.
At the cellular level, TBI leads to neuronal injury [37] and reactive gliosis [38], which are characteristics of the human PTE brain. Variable subpial and subcortical gliosis were observed in ten epileptic patients who underwent focal cortical resection surgery [39]. One study showed that astrocytes begin to increase at approximately 7–10 days after injury, and in the subsequent weeks, months, and even years, astrocytes increase in number and fibrillary appearance, which causes glial scarring in and around the damaged area and eventually promotes seizures [40]. Conversely, long-term chronic epileptic seizures can also result in astrocyte proliferation involved in hippocampal sclerosis, which affects the normal physiological regulation of the brain and aggravates seizures. Moreover, in addition to gliogenesis and neurogenesis, MFS plays a vital role in promoting epileptogenesis. Mossy fibers are axons emitted by granule cells in the hippocampal DG. Typically, few synapses are established between granule cells in the DG; however, sprouted mossy fibers form an abnormal positive feedback circuit among granule cells, which significantly promotes the spread of seizure-like activity [19, 41]. Consistent with previous studies, we observed intense glial GFAP + immunoreactivity as well as significant MFS in the hippocampus during the chronic period following injury.
Controversial Role of SWDs in TBI Model of SD Rats and Limitations
The spontaneous occurrence of SWDs in normal non-pathological rats complicates the interpretation of nonconvulsive seizures after TBI. Although the current studies of SWDs cannot confirm that TBI causes de novo nonconvulsive seizures, as Sick’s study suggests, TBI may deteriorate pre-existing epileptic or hyperexcitable states [42]. For example, it has not been established from previous studies whether TBI causes changes in brain excitability, or leads to conversion of normal brain to epileptic brain that increase SWDs in the brain already expressing these events. However, our data provided the indirect evidence that cortical injury significantly increased the incidence of SWDs, which was worsened by increased injury severity. In addition, the age-dependent expression of SWDs in rats also makes the occurrence of acquired epileptogenesis after TBI difficult to explain, since the time course of this acquired epilepsy is very similar to normal aging. Thus, caution is required when using this model to study acquired epilepsy after TBI.
Astrocytes play a crucial role in neuronal growth, restruction and homeostatic balance. It is well known that TBI causes reactive gliosis, which is primarily involved in boundary formation around areas of tissue damage or inflammation [43]. The upregulation of GFAP in reactive astrocytes is perhaps the best-known hallmark of reactive gliosis after brain trauma [44]. In this study, we described the chronic histopathological alterations in TBI rats, such as reactive gliosis in peripheral cortex. However, one specific limitation was that we were unable to explain the possible causal relationship between SWDs expression and these histopathological changes after TBI. Therefore, given the complicated interaction between astrocytes and neurons, dissecting the cellular and molecular mechanisms and exploring the role of reactive glial cells in the acquired nonconvulsive seizures represents key goals of future research.
Conclusion
The behavioral, electrographic, and structural abnormalities observed in this study may reflect the dynamic functional reorganization of the brain post-TBI that leads to epileptogenesis. High-voltage rhythmic and frequent SWDs of 7–8 Hz were recorded in both sham and TBI rats and were characterized by abrupt onset and termination, variable duration, and lack of postictal suppression. These were accompanied by behavioral arrest and slight facial twitching, which are properties that are identical to classical ASs. In addition, the frequency and duration of SWDs increased with the increasing severity of TBI. These measures may serve as predictive markers of trauma severity, advanced post-traumatic pathology, or other disorders. Consistent with previous studies, brain structural changes, such as glial proliferation and MFS, were observed in the cortex and hippocampus during the chronic period following injury, which are characteristics of human PTE. The generation of SWDs depends on the impairment of the corticothalamic circuit, which may be related to excessive GABAergic transmission in the thalamus. The novel antiepileptic drug, retigabine, was not only ineffective for SWDs but also exacerbated SWDs expression (duration and number); thus, its administration requires clinical vigilance.
Data Availability
The datasets used and/or analyzed during the current study are available from the corresponding author on reasonable request.
Abbreviations
- TBI:
-
Traumatic brain injury
- PTE:
-
Post-traumatic epilepsy
- CCI:
-
Controlled cortical impact
- DG:
-
Dentate gyrus
- ASs:
-
Absence seizures
- SWDs:
-
Spike-wave-discharges
- ECoG:
-
Electrocorticogram
- GAERS:
-
Genetic Absence Epilepsy Rats from Strasbourg
- WAG/Rij:
-
Wistar Albino Glaxo Rats from Rijswijk
- AP:
-
Anteroposterior
- ML:
-
Mediolateral
- mNSS:
-
Modified Neurological Severity Score
- PFA:
-
Paraformaldehyde
- Iba1:
-
Ionized calcium binding adaptor molecule 1
- GFAP:
-
Glial fibrillary acidic protein
- MFS:
-
Mossy fiber sprouting
- ZnT3:
-
Zinc transporter 3
- FPI:
-
Fluid percussion injury
- GABA:
-
Gamma-aminobutyric acid
- eGABAAR:
-
Extrasynaptic GABAA receptors
References
Lucke-Wold BP, Nguyen L, Turner RC, Logsdon AF, Chen YW, Smith KE, Huber JD, Matsumoto R, Rosen CL, Tucker ES, Richter E (2015) Traumatic brain injury and epilepsy: underlying mechanisms leading to seizure. Seizure 33:13–23. https://doi.org/10.1016/j.seizure.2015.10.002
Bolkvadze T, Pitkanen A (2012) Development of post-traumatic epilepsy after controlled cortical impact and lateral fluid-percussion-induced brain injury in the mouse. J Neurotrauma 29:789–812. https://doi.org/10.1089/neu.2011.1954
Hunt RF, Scheff SW, Smith BN (2009) Posttraumatic epilepsy after controlled cortical impact injury in mice. Exp Neurol 215:243–252. https://doi.org/10.1016/j.expneurol.2008.10.005
Statler KD, Scheerlinck P, Pouliot W, Hamilton M, White HS, Dudek FE (2009) A potential model of pediatric posttraumatic epilepsy. Epilepsy Res 86:221–223. https://doi.org/10.1016/j.eplepsyres.2009.05.006
Allan J, Hartman PG, Crane-Robinson C, Aviles FX (1980) The structure of histone H1 and its location in chromatin. Nature 288:675–679
Kelly KM, Miller ER, Lepsveridze E, Kharlamov EA, McHedlishvili Z (2015) Posttraumatic seizures and epilepsy in adult rats after controlled cortical impact. Epilepsy Res 117:104–116. https://doi.org/10.1016/j.eplepsyres.2015.09.009
Pitkänen A, McIntosh TK (2006) Animal models of post-traumatic epilepsy. J Neurotrauma 23:241–261. https://doi.org/10.1089/neu.2006.23.241
Crunelli V, Leresche N (2002) Childhood absence epilepsy: genes, channels, neurons and networks. Nat Rev Neurosci 3:371–382. https://doi.org/10.1038/nrn811
Blumenfeld H (2005) Cellular and network mechanisms of spike-wave seizures. Epilepsia 46(Suppl 9):21–33. https://doi.org/10.1111/j.1528-1167.2005.00311.x
Coenen AM, Drinkenburg WH, Inoue M, van Luijtelaar EL (1992) Genetic models of absence epilepsy, with emphasis on the WAG/Rij strain of rats. Epilepsy Res 12:75–86. https://doi.org/10.1016/0920-1211(92)90029-s
Kharlamov EA, Jukkola PI, Schmitt KL, Kelly KM (2003) Electrobehavioral characteristics of epileptic rats following photothrombotic brain infarction. Epilepsy Res 56:185–203. https://doi.org/10.1016/j.eplepsyres.2003.09.005
Kelly KM, Kharlamov A, Hentosz TM, Kharlamova EA, Williamson JM, Bertram EH 3rd, Kapur J, Armstrong DM (2001) Photothrombotic brain infarction results in seizure activity in aging Fischer 344 and Sprague Dawley rats. Epilepsy Res 47:189–203. https://doi.org/10.1016/s0920-1211(01)00294-7
Chen J, Sanberg PR, Li Y, Wang L, Lu M, Willing AE, Sanchez-Ramos J, Chopp M (2001) Intravenous administration of human umbilical cord blood reduces behavioral deficits after stroke in rats. Stroke 32:2682–2688. https://doi.org/10.1161/hs1101.098367
Rodgers KM, Dudek FE, Barth DS (2015) Progressive, seizure-like, spike-wave discharges are common in both injured and uninjured Sprague-Dawley rats: implications for the fluid percussion injury model of post-traumatic epilepsy. J Neurosci 35:9194–9204. https://doi.org/10.1523/jneurosci.0919-15.2015
Komoltsev IG, Frankevich SO, Shirobokova NI, Volkova AA, Levshina IP, Novikova MR, Manolova AO, Gulyaeva NV (2021) Differential early effects of traumatic brain injury on spike-wave discharges in Sprague-Dawley rats. Neurosci Res 166:42–54. https://doi.org/10.1016/j.neures.2020.05.005
Taylor JA, Reuter JD (2019) Spontaneous recurrent absence seizure-like events in wild-caught rats. J Neurosci 39:4829–4841. https://doi.org/10.1523/jneurosci.1167-18.2019
Large CH, Sokal DM, Nehlig A, Gunthorpe MJ, Sankar R, Crean CS, Vanlandingham KE, White HS (2012) The spectrum of anticonvulsant efficacy of retigabine (ezogabine) in animal models: implications for clinical use. Epilepsia 53:425–436. https://doi.org/10.1111/j.1528-1167.2011.03364.x
Mazarati A, Wu J, Shin D, Kwon YS, Sankar R (2008) Antiepileptogenic and antiictogenic effects of retigabine under conditions of rapid kindling: an ontogenic study. Epilepsia 49:1777–1786. https://doi.org/10.1111/j.1528-1167.2008.01674.x
Buckmaster PS, Lew FH (2011) Rapamycin suppresses mossy fiber sprouting but not seizure frequency in a mouse model of temporal lobe epilepsy. J Neurosci 31:2337–2347. https://doi.org/10.1523/jneurosci.4852-10.2011
Wenzel HJ, Cole TB, Born DE, Schwartzkroin PA, Palmiter RD (1997) Ultrastructural localization of zinc transporter-3 (ZnT-3) to synaptic vesicle membranes within mossy fiber boutons in the hippocampus of mouse and monkey. Proc Natl Acad Sci USA 94:12676–12681. https://doi.org/10.1073/pnas.94.23.12676
Campbell JN, Gandhi A, Singh B, Churn SB (2014) Traumatic brain injury causes a tacrolimus-sensitive increase in non-convulsive seizures in a rat model of post-traumatic epilepsy. Int. J. Neurol. Brain Disord. 2:1–11
D’Ambrosio R, Miller JW (2010) What is an epileptic seizure? Unifying definitions in clinical practice and animal research to develop novel treatments. Epilepsy currents 10:61–66. https://doi.org/10.1111/j.1535-7511.2010.01358.x
Tenney JR, Glauser TA (2013) The current state of absence epilepsy: can we have your attention? Epilepsy currents 13:135–140. https://doi.org/10.5698/1535-7511-13.3.135
Blumenfeld H (2005) Consciousness and epilepsy: why are patients with absence seizures absent? Prog Brain Res 150:271–286. https://doi.org/10.1016/s0079-6123(05)50020-7
Blumenfeld H (2012) Impaired consciousness in epilepsy. Lancet Neurol 11:814–826. https://doi.org/10.1016/s1474-4422(12)70188-6
Smith S (2005) EEG in the diagnosis, classification, and management of patients with epilepsy. J Neurol Neurosurg Psych 76(Suppl 2):ii2–ii7
Wiest MC, Nicolelis MA (2003) Behavioral detection of tactile stimuli during 7–12 Hz cortical oscillations in awake rats. Nat Neurosci 6:913–914. https://doi.org/10.1038/nn1107
D’Ambrosio R, Fairbanks JP, Fender JS, Born DE, Doyle DL, Miller JW (2004) Post-traumatic epilepsy following fluid percussion injury in the rat. Brain 127:304–314. https://doi.org/10.1093/brain/awh038
D’Ambrosio R, Fender JS, Fairbanks JP, Simon EA, Born DE, Doyle DL, Miller JW (2005) Progression from frontal-parietal to mesial-temporal epilepsy after fluid percussion injury in the rat. Brain 128:174–188. https://doi.org/10.1093/brain/awh337
Meeren HK, Pijn JP, Van Luijtelaar EL, Coenen AM, Lopes da Silva FH (2002) Cortical focus drives widespread corticothalamic networks during spontaneous absence seizures in rats. J Neurosci 22:1480–1495. https://doi.org/10.1523/jneurosci.22-04-01480.2002
Lüttjohann A, van Luijtelaar G (2015) Dynamics of networks during absence seizure’s on- and offset in rodents and man. Front Physiol 6:16. https://doi.org/10.3389/fphys.2015.00016
McCafferty C, David F, Venzi M, Lőrincz ML, Delicata F, Atherton Z, Recchia G, Orban G, Lambert RC, Di Giovanni G, Leresche N, Crunelli V (2018) Cortical drive and thalamic feed-forward inhibition control thalamic output synchrony during absence seizures. Nat Neurosci 21:744–756. https://doi.org/10.1038/s41593-018-0130-4
Treven M, Koenig X, Assadpour E, Gantumur E, Meyer C, Hilber K, Boehm S, Kubista H (2015) The anticonvulsant retigabine is a subtype selective modulator of GABAA receptors. Epilepsia 56:647–657. https://doi.org/10.1111/epi.12950
Walker MC, Kullmann DM (2012) Tonic GABA(A) receptor-mediated signaling in epilepsy. In: Noebels JL, Avoli M, Rogawski MA, Olsen RW, Delgado-Escueta AV (eds) Jasper’s basic mechanisms of the epilepsies. National Center for Biotechnology Information (US), Bethesda, MD
Cope DW, Di Giovanni G, Fyson SJ, Orbán G, Errington AC, Lorincz ML, Gould TM, Carter DA, Crunelli V (2009) Enhanced tonic GABAA inhibition in typical absence epilepsy. Nat Med 15:1392–1398. https://doi.org/10.1038/nm.2058
Pitkänen A, Kharatishvili I, Karhunen H, Lukasiuk K, Immonen R, Nairismägi J, Gröhn O, Nissinen J (2007) Epileptogenesis in experimental models. Epilepsia 48(Suppl 2):13–20. https://doi.org/10.1111/j.1528-1167.2007.01063.x
Hicks R, Soares H, Smith D, McIntosh T (1996) Temporal and spatial characterization of neuronal injury following lateral fluid-percussion brain injury in the rat. Acta Neuropathol 91:236–246. https://doi.org/10.1007/s004010050421
Hill-Felberg SJ, McIntosh TK, Oliver DL, Raghupathi R, Barbarese E (1999) Concurrent loss and proliferation of astrocytes following lateral fluid percussion brain injury in the adult rat. J Neurosci Res 57:271–279. https://doi.org/10.1002/(sici)1097-4547(19990715)57:2%3c271::aid-jnr13%3e3.0.co;2-z
Jones AL, Britton JW, Blessing MM, Parisi JE, Cascino GD (2018) Chronic traumatic encephalopathy in an epilepsy surgery cohort: Clinical and pathologic findings. Neurology 90:e474–e478. https://doi.org/10.1212/wnl.0000000000004927
Cervós-Navarro J, Lafuente JV (1991) Traumatic brain injuries: structural changes. J Neurol Sci 103(Suppl):S3-14. https://doi.org/10.1016/0022-510x(91)90002-o
Santhakumar V, Aradi I, Soltesz I (2005) Role of mossy fiber sprouting and mossy cell loss in hyperexcitability: a network model of the dentate gyrus incorporating cell types and axonal topography. J Neurophysiol 93:437–453. https://doi.org/10.1152/jn.00777.2004
Sick T, Wasserman J, Bregy A, Sick J, Dietrich WD, Bramlett HM (2017) Increased expression of epileptiform spike/wave discharges one year after mild, moderate, or severe fluid percussion brain injury in rats. J Neurotrauma 34:2467–2474. https://doi.org/10.1089/neu.2016.4826
Sofroniew MV (2020) Astrocyte reactivity: subtypes, states, and functions in CNS innate immunity. Trends Immunol 41:758–770. https://doi.org/10.1016/j.it.2020.07.004
Pekny M, Nilsson M (2005) Astrocyte activation and reactive gliosis. Glia 50:427–434. https://doi.org/10.1002/glia.20207
Acknowledgements
We thank Sarina Iwabuchi, PhD, from Liwen Bianji (Edanz) (www.liwenbianji.cn) for editing a draft of this manuscript.
Funding
This work was mainly supported by the startup fund from Capital Medical University Advanced Innovation Center for Human Brain Protection (20181101, JW), and partially by grants from National Key R&D Program of China (2017YFC1307501, QW), Beijing-Tianjin-Hebei Cooperative Basic Research Program (H2018206435, QW), the National Natural Science Foundation of China (81870935, JW).
Author information
Authors and Affiliations
Contributions
LS and RL conducted literature review and wrote the initial draft of the manuscript. HY and TYmade preliminary revision. JW and QW made critical revision. All authors contributed to manuscript revision and approved the submitted version.
Corresponding authors
Ethics declarations
Conflict of Interest
The authors declare no conflict of interest.
Ethical Approval
All procedures were approved by the Animal Care and Use Committee of Capital Medical University (Beijing, China).
Consent to participate
Not applicable.
Consent to publish
Not applicable.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
About this article

Cite this article
Sun, L., Liu, R., Yang, H. et al. Characteristics of Epileptiform Spike-wave Discharges and Chronic Histopathology in Controlled Cortical Impact Model of Sprague–Dawley Rats. Neurochem Res 47, 3615–3626 (2022). https://doi.org/10.1007/s11064-022-03542-y
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s11064-022-03542-y