
Abstract
Postoperative cognitive dysfunction (POCD) induced by anesthesia or surgery has become a common complication in the aged population. Sevoflurane, a clinical inhalation anesthetic, could stimulate calcium overload and necroptosis to POCD. In addition, necroptosis inhibitor necrostatin-1 (Nec-1) alleviated cognitive impairment caused by multiple causes, including postoperative cognitive impairment. However, whether Nec-1 exerts a neuroprotective effect on POCD via calcium and necroptosis remains unclear. We anesthetized Sprague–Dawley rats with sevoflurane to construct the POCD model and to explore the mechanism underlying neuroprotective effects of Nec-1 in POCD. Rats were treated with Nec-1 (6.25 mg/kg) 1 h prior to anesthesia. Open field test and Morris water maze were employed to detect the cognitive function. In this study, rats exposed to sevoflurane displayed cognitive dysfunction without changes in spontaneous activity; however, the sevoflurane-induced POCD could be relieved by Nec-1 pretreatment. Nec-1 decreased sevoflurane-induced calcium overload and calpain activity in the hippocampus. In addition, Nec-1 alleviated the expression of p-RIPK1, RIPK1, p-RIPK3, RIPK3, p-MLKL and MLKL. Furthermore, Nec-1 remarkably increased BDNF and p-TrkB/TrkB expression in the hippocampus of aged rats. Ultimately, our research manifests evidence that Nec-1 may play a neuroprotective role against sevoflurane-induced cognitive impairment via the increase of BDNF/TrkB and suppression of necroptosis-related pathway.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Postoperative cognitive dysfunction (POCD) is the constellation of cognitive symptoms involved in anesthesia and surgery, associated with impairments on the patients’ memory, attention, executive function and personality changes [1, 2]. These symptoms can last anywhere from days to months, many POCD cases are unabiding, but importantly, the persistent form of POCD is likely to evolve into AD and are thus, more devastating [3, 4]. Advanced age is known to be the important risk factor for this persistent form of POCD [5]. Individuals with POCD experience poor quality of life, higher death rates and elevated dependency on social security when compared to those without POCD [6]. With the increase of population aging, this age-associated perioperative complication has become one of the public health problems to be solved urgently. Therefore, it is interesting to understand the underlying mechanisms of POCD.
Although many pathophysiological mechanisms have been found related to the occurrence of POCD, the exact cascade remains elusive. Accumulating evidence indicates that inhalation anesthesia elicits neurotoxicity and neurocognitive decline in elderly patients experiencing POCD [7, 8]. Several lines of evidence have indicated that sevoflurane could impair cognitive function, and the cellular calcium homeostasis imbalance may be one of the pathogenic mechanisms. For example, sevoflurane could increase intracellular Ca2+ to attract mitochondrial dysfunction and mitochondria-mediated neuro-apoptosis in neurons [9]. Our previous work also suggested that receiving inhalation of sevoflurane causes learning and memory deficits in rats by inducing neuronal apoptosis through increasing [Ca2+]c and calcium pathway proteins like calcineurin and calpain, we further showed that inhibition of calcium overload effectively alleviated the cognitive impairment induced by sevoflurane [10, 11].
Recent research has identified that a reduction of brain-derived neurotrophic factor (BDNF) as a crucial factor affects the risk of POCD in aging mice [12]. In addition, down-regulated expression of the BDNF/TrkB pathway mediated by overactivation of calpain might bring about postoperative cognitive dysfunction in aged mice [13].
At present, the pathological mechanisms of POCD have not been fully clarified, and available methods of preventive strategy or treatment for POCD are still limited. Necroptosis is a new pathway of non-apoptotic cell death, it is tightly operated and regulated by genetic controls, which emphasizes the notion that it is a modulated process [14]. The pathway relies on the assembly of the apical protein kinases, RIPK1 and RIPK3 termed the necrosome, further phosphorylates the downstream molecule MLKL, which eventually causes cell death [15]. Recent studies have shown that Ca2+ and its downstream signaling molecules are involved in the occurrence of necroptosis [16]. Necroptosis has been implicated in several major CNS diseases and its inhibitor necrostatin-1 (Nec-1) has an ameliorative effect. RIPK1/RIPK3 mediated necroptosis is involved in sevoflurane‑induced neonatal neurotoxicity in the rat hippocampus [17]. Nec-1 rescues from LPS-induced RIP3-mediated necroptosis [18] and alleviates postoperative cognitive impairments in d-galactose-induced aged mice [19], meanwhile, it also directly mitigated cognitive dysfunction in prediabetic rats [20]. Moreover, Nec-1 alleviated SHA-induced synaptic damage in hippocampus by inhibiting necroptosis associated with the BDNF pathway [21]. A TrkB agonistic antibody which mimicked BDNF functionally inhibited both apoptosis and necroptosis in rat model of ischemia/reperfusion [22]. In SH-SY5Y cells, the aggregated Aβ and low levels of BDNF-induced neuronal necroptosis [23]. These studies suggested that BNDF downregulation was associated with necroptosis.
The effect of Nec-1 on cognition and brain pathologies in sevoflurane-induced cognitive impairment in aged rats has not been investigated. Taking all of these theories and research results from previous studies into consideration, we hypothesized that Nec-1 attenuated cognitive decline and brain pathologies via BDNF-TrkB signaling in POCD rats. In the current study, the effect of Nec-1 on cognitive impairment induced by sevoflurane was investigated in aged rats and the potential mechanisms were explored.
Materials and Methods
Animals
A total of 120 male Sprague–Dawley rats (18 month-old) weighing 550–750 g were purchased from the Experimental Animal Center of Hebei Medical University [Permit No. SCXK 2013-2-001]. The rats were accessed food and water ad libitum under controlled temperature 22–25 °C, with a 12-h light/dark cycle (7:00–19:00). All experiments were conducted according to the protocol approved by the Ethics Committee for Animal of the Third Hospital of Hebei Medical University (Shijiazhuang, China). Efforts were made to minimize pain, suffering or discomfort, and to reduce the number of animals used.
Animal Treatment
Rats were randomly assigned into the control (Con) group, control plus necrostatin-1 (Con+Nec-1) group, sevoflurane (Sev) group and sevoflurane plus necrostatin-1 (Sev+Nec-1) group. According to the previous study [24], rats in Sev and Sev+Nec-1 groups were exposed to 2% sevoflurane (Maruishi Pharmaceutical Co., Ltd. Japan) delivered via humidified 30% O2 carrier gas for 5 h, Con and Con+Nec-1 groups were exposed to the carrier gas without sevoflurane for the same period. Besides this, the rats in Con+Nec-1 and Sev+Nec-1 groups were pretreated with Nec-1 at 1 h prior to anesthesia 6.25 mg/kg intraperitoneally, while the rats in Con and Sev groups received an equal volume and concentration of DMSO in the same way [19]. Nec-1 (Abcam, ab141053) was dissolved in DMSO to a concentration of 25 µg/µL, and then dilute 40 times with PBS before use. In order to make sure that the final concentration of Nec-1 was 0.625 µg/µL and DMSO was 2.5%. The experimental program was shown in Fig. 1.

The experimental schematic diagram. Morris water maze (MWM) training was performed 1 to 5 days before anesthesia. Aged rats were pretreated with Nec-1 (6.25 mg/kg, i.p.) 1 h prior to sevoflurane anesthesia. After open field test (OFT) and Morris maze test, rats were sacrificed. Western blot and flow cytometry were performed at 1 day after anesthesia, meanwhile, HE staining and immunofluorescence were performed at 3 days after anesthesia
Open Field Test (OFT)
Open field test was carried out to assess anxiety-like behavior and locomotive activity at 1 day after sevoflurane exposure. An open-field chamber (100 cm × 100 cm) was divided into 16 equally spaced squares bordered with opaque and 50 cm high walls painted black. Before use, the urine and feces left by the previous rat were removed, then 75% ethanol was used to wipe the chamber to eliminate the scent remaining. Rats need to acclimate to the environment for at least 30 min before testing, each animal was placed in the center of the setup and allowed uninterrupted and free movement for 5 min. The traveling distance, traveling speed and duration in the central area were recorded by the EthoVision™ 7.0 automatic camera system (Noldus, Netherlands).
Morris Water Maze Test
Twenty-four hours after anesthesia exposure, the spatial learning and memory ability was tested with the Morris water maze (MWM). The MWM was performed in a circular pool (180 cm diameter, 50 cm deep, filled to a depth of 30 cm with warm water at 24 ± 1 °C), which was divided into four quadrants. An invisible platform (round, 12 cm diameter, 2 cm below surface) was hidden in a designated target quadrant of the pool. Rats were trained for five consecutive days before anesthesia, each rat was tested four times every day. They were taught the location of the platform with a different starting position in a quasi-randomized manner, facing the pool wall. Each test continued until the rat discovered the platform and the rats who failed to locate the escape platform within the allotted time (90 s) were gently put onto it by the experimenter for 15 s. Testing trials were conducted once a day at 1, 3 and 7 days after sevoflurane exposure, and the latency time to locate the hidden platform was recorded. On the 7th day, the platform was removed from the pool and the animals were released from the quadrant at the entry site where the last test was performed and allowed to swim for 60 s, meanwhile, the frequency of crossing the original platform was recorded. MWM test was conducted in JLBehv-MWM (Shanghai Ji’liang Software Technology Co., Ltd. China).
HE Staining
At the end of MWM 3 days after anesthesia exposure, animals (n = 6) were deeply anesthetized with sodium pentobarbital (60 mg/kg, i.p.), the brain tissues were collected and rinsed with phosphate buffer solution. After separation, the tissues were soaked in 4% paraformaldehyde for 24 h. Subsequently, the samples were dehydrated and embedded in paraffin wax. The paraffin-embedded brain sections (5 μm) of hippocampal in the coronal plane were cut and stained with hematoxylin and eosin. Then the pathological changes in the hippocampus were examined by optical microscopy. The necroptosis cell number of CA1 and CA3 was counted by a pathologist.
Western Blot
At 1 day after sevoflurane exposure, after MWM (n = 6), the hippocampus separated from the brain were dissected with lysis buffer and centrifuged at 12,000×g at 4 °C for 10 min. Then the supernatant was collected. The protein concentration was measured with BCA Protein Assay Kit (Beyotime, Shanghai, China). Protein samples were added with loading buffer and heated for 5 min at 100 °C. 30 μg protein from the whole cell lysates was separated by 10% PAGE gels with 5% stacking gels and transferred onto polyvinylidene fluoride (PVDF) membrane (IPVH00010, Millipore, USA). The membranes were soaked in TBST buffer containing 5% skim milk for 1 h at room temperature and then incubated with primary antibodies overnight at 4 °C. The primary antibodies were as follows: rabbit anti-RIPK1 antibody (ABclonal, 1:500, A7414), rabbit anti-RIPK3 antibody (BioSS, 1:500, bs-3551R), rabbit anti-MLKL antibody (Proteintech, 1:1000, 66675-1-Ig), rabbit anti-phospho-RIPK1 (Ser384) (Affinity, 1:600, AF7088), rabbit anti-phospho-RIPK3 (Ser232) (Affinity, 1:600, AF7443), rabbit anti-phospho-MLKL (Ser358) (Affinity, 1:600, AF7420), rabbit anti-calpain antibody (1:1000, Abcam, ab124631), mouse anti-αII spectrin (1:800, Santa Cruz, sc-46696), rabbit anti-BDNF antibody (Proteintech, 1:1000, 28205-1-AP), rabbit anti-p-TrkB antibody (Beyotime, 1:600, AF1963), rabbit anti-TrkB antibody (Affinity, 1:600, AF6461) and rabbit anti-β-actin antibody (Proteintech, 1:2000, 20536-1-AP) at 4 °C reaction of the night. After incubation, membranes were washed three times in TBST and incubated with an HRP-conjugated IgG antibody (Proteintech, 1:2000, HRP-20758) in TBST buffer for 1 h. After washing with TBST buffer repeatedly, the membranes were developed via chemiluminescence reagents from an ECL kit (Abbkine, K22030, China) and were digitally scanned (EPSONV 300, China). To evaluate changes in estimated proteins quantitatively, the appropriate bands were analyzed by densitometry and quantified by Image J software.
Immunofluorescence Assay
At 3 days after sevoflurane exposure, after MWM (n = 6), the hippocampus tissues were studied employing the double fluorescein-labeled method in rats. For immunofluorescence staining, the hippocampus of rats was cut into 20 μm thick slices. And then, the slices were washed with PBS, infiltrated with 0.3% Triton X, and blocked with 10% normal goat serum (NGS). Subsequently, the slices were incubated with primary antibodies overnight, mouse anti-Neun antibody (Millipore, 1:200, MAB377), RIPK1 (ABclonal, 1:200, A7414), RIPK3 (BioSS, 1:100, bs-3551R), MLKL (Proteintech, 1:200, 66,675–1-Ig). After washing with PBS three times, sections were incubated with goat anti-rabbit IgG-FITC secondary antibody (Beyotime, 1:100, P0186) at room temperature for 1 h. Finally, the 4′,6-diamidino-2-phenylindole (DAPI; Beyotime, P0131) was used to stain sections for 10 min. Subsequently, images were captured by a Nikon Eclipse CI fluorescence microscope and the percentages of RIPK1/Neun/DAPI, RIPK3/Neun/DAPI and MLKL/Neun/DAPI were counted.
Measurements of Cytosolic Calcium Concentration ([Ca2+]c) and the Ratio of Necroptosis in the Hippocampus
The hippocampi were dissected at 1 day after anaesthesia and the cell suspension was prepared as previously described [10, 11]. Subsequently, the cell suspension was washed twice with PBS and incubated with 5 μmol/l Fura-3 AM (Solarbio, IF0150) at 37 °C for 30 min. After washing twice, cells were re-suspended by PBS at 37 °C for 15 min. Flow cytometry was performed to measure fluorescence intensity, at the excitation wavelength of 480 nm and emission wavelength of 525 nm. The fluorescence intensity reveals the concentration of intracellular calcium ([Ca2+]c).
The cell suspension was prepared by the same method as above, resuspending the cells in 500 μl of 1 × binding buffer after centrifugation. Subsequently, added 5 μl of 20 μg/ml Annexin V-FITC and 10 μl of 50 mg/ml PI (Invitrogen, 282932-000), incubated at room temperature for 10 min in the dark. The cells were washed and analyzed by FACS Calibur (Becton, Dickinson Company, USA). The percentages of cells in each quadrant were analyzed by FACS Calibur (FC500; Beckman Coulter Inc.) using ModFit software (EXPO32 ADC v1.2; Beckman Coulter Inc.). The ratio of necroptosis in groups was detected by calculating the PI + cells numbers.
Statistical Analysis
All Data were analyzed using the SPSS software (version 21.0). The homogeneity of variance test was used for detecting whether the data were normally distributed, and the data meet normal distribution were expressed as mean ± SD. One-way ANOVA was carried out to determine significant differences followed by the Tukey test to compare means of different groups. Statistical significance was determined as P < 0.05.
Results
Sevoflurane Anesthesia Had No Effect on the Spontaneous Activity of the Aged Rats
The open field test was employed to evaluate whether anesthesia-induced behavioral changes were the result of a decrease in spontaneous activity. There was no significant differences in traveling speed (Fig. 2B, F(3,36) = 0.284, P = 0.837), total moving distance (Fig. 2C, F(3,36) = 0.404, P = 0.751), time spent in the center (Fig. 2D, F(3,36) = 1.240, P = 0.309) among the groups. The above results indicated that the behavioral changes in the anesthetized rats were not due to a decrease in motor ability.

Sevoflurane anesthesia had no effect on the spontaneous activity of the aged rats. The trajectory of spontaneous activity in rats (A). The average traveling speed (B), total moving distance (C), and time spent in the center (D) of the OFT at 1 day after anesthesia. Data are shown as mean ± SD (n = 10 animals per group)
Nec-1 Alleviated Sevoflurane-Induced POCD in Aged Rats
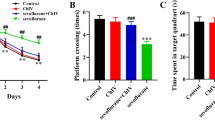
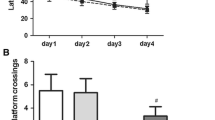
To further investigate the ability of learning and memory, the MWM test was conducted in aged rats. As shown in Fig. 3A, the escape latency in all groups manifested shorter latency as training progressed, and no difference was discovered between any two groups during the same time, suggesting that all experimental rats were able to learn the platform location from practice. However, there was no difference in the average swimming speed on 7th day (Fig. 3B, F(3,36) = 0.442, P = 0.725). After anesthetic, compared to the Con group, the escape latency was significantly higher in the Sev (P < 0.05) and Sev+Nec-1 (P < 0.05) groups, but no difference was found in the Con+Nec-1 group (P > 0.05). In addition, the escape latency was lower in the Sev+Nec-1 group (P < 0.05) on 1, 3 and 7 days compared to the Sev group (Fig. 3C). Meanwhile, times of crossing the platform in the Sev and Sev+Nec-1 groups were decreased compared with Con group and Sev+Nec-1 group was increased compared with Sev group in the 7th day (Fig. 3D, F(3,36) = 42.903, P < 0.05), suggesting sevoflurane-induced cognitive dysfunction was ameliorated by Nec-1.

Nec-1 alleviated sevoflurane-induced POCD in aged rats. The escape latency (time to find the hidden platform) in training phase in the MWM test, day-(5–1) means 1–5 days before anesthesia (A). The average swimming velocity on 7 days after anesthesia (B). The escape latency at 1, 3 and 7 days after rats were anesthetized (C). The times of crossing the hidden platform removed on 7 days after anesthesia (D). Representative swimming traces of rat in MWM test with hidden platform removed on 7 days after anesthesia (E). Data are shown as mean ± SD (n = 10 animals per group). *P < 0.05 vs. Con group, #P < 0.05 vs. Sev group
Nec-1 Improved the Sevoflurane-Induced Pathological Changes of the Hippocampus
HE staining was used for histopathological examination of the hippocampus, representative images showed the CA1 area (Fig. 4, red-lined box) and CA3 area (Fig. 4, black-lined box). In the Con and Con+Nec-1 groups, there were almost no abnormalities in the hippocampus which showed normal hippocampal pyramidal cells with regular morphology, round and pale stained nuclei and evenly stained cytoplasm (Fig. 4A, B). In the Sev group, the hippocampus exhibited significant disordered neuronal arrangement with acidophilic changes (Fig. 4C, red arrow), and the above pathological changes were alleviated in the Sev+Nec-1 group (Fig. 4D). The number of cells that suffered necroptosis was counted in the CA1 and CA3 regions. It was obviously higher after exposure to 2% sevoflurane for 5 h compared with the Con group (p < 0.05), however, Nec-1 inhibited the number of cells that underwent necroptosis (p < 0.05) (Fig. 4E, F(3,20) = 261.947, P < 0.05).

Nec-1 improved the sevoflurane-induced pathological changes of the hippocampus. Histopathological evaluation of the hippocampus of animals 3 days after anesthesia by H&E staining. The representative images of histopathological changes in the hippocampus CA1 (red-lined box) and CA3 (black-lined box) of aged rats (A–D), the red arrow indicates the occurrence of eosinophilic necrosis. Scale bars = 100 μm. The number of cells suffered necroptosis in CA1 and CA3 regions (E). The emblematic plots of necroptosis detection by flow cytometry in the hippocampus of aged rats 1 day after anesthesia in each group (F–I), necrosis cells (B1, Annexin V−/PI+). The statistical analysis of hippocampus cells necrosis (J). Data are presented as mean ± SD (n = 6 animals per group). *P < 0.05 vs. Con group, #P < 0.05 vs. Sev group
In addition, to study the occurrence of necroptosis, we measured the PI-positive cells to estimate necroptosis. With PI/Annexin V double staining by using flow cytometry [25], cells were stained with Annexin FITC and PI, and analyzed by FACS using FL1 (Annexin) and FL3 (PI) channels. As shown in Fig. 4F, the ratio of necroptosis in Sev (p < 0.05) and Sev+Nec-1 (p < 0.05) groups was significantly increased compared with Con group and it was decreased in Sev+Nec-1 group (p < 0.05) when compared to Sev group (F(3,20) = 60.507, P < 0.05). These results illustrated that necroptosis occurred after sevoflurane exposure, and Nec-1 pretreatment inhibited necroptosis.
Nec-1 Decreased the Expression of Marker Proteins of Necroptosis Induced by Sevoflurane in Aged Rats
To certify the presence of necroptosis and detect the role of necroptosis in POCD, expressions of the necroptosis-related proteins like p-RIPK1, RIPK1, p-RIPK3, RIPK3, p-MLKL and MLKL were evaluated in hippocampal tissues using western blot. When compared with Con group, expression of p-RIPK1 and RIPK1 was increased markedly in the Sev (p < 0.05) and Sev+Nec-1 (p < 0.05) groups, but not in the Con+Nec-1 group (P > 0.05), and it was down-regulated in Sev+Nec-1 group (p < 0.05) compared with Sev group (Fig. 5C, p-RIPK1: F(3,20) = 90.351, P < 0.05; RIPK1: F(3,20) = 67.739, P < 0.05). Compared to the Con group, expression of RIPK3 was significantly up-regulated in Sev and Sev+Nec-1 groups, there was no statistical difference in group Con+Nec-1, however, Nec-1 decreased the expression of p-RIPK3 and RIPK3 (Fig. 5D, p-RIPK3: F(3,20) = 35.337, P < 0.05; RIPK3: F(3,20) = 41.171, P < 0.05). The expression trend of p-MLKL and MLKL was coincident with above (Fig. 5E, p-MLKL: F(3,20) = 60.896, P < 0.05; MLKL: F(3,20) = 25.096, P < 0.05).

Nec-1 decreased the expression of marker proteins of necroptosis induced by sevoflurane in aged rats. The representative western blot images of RIPK1, RIPK3 and MLKL in the hippocampi of aged rats on 1 day after anesthesia in each group (A). The quantity analysis of RIPK1 (B), RIPK3 (C) and MLKL (D) expression in the hippocampus of each group. Data are presented as mean ± SD (n = 6 animals per group). *P < 0.05 vs. Con group, #P < 0.05 vs. Sev group
Nec-1 Ameliorates the Expression of RIPK1, RIPK3 and MLKL in Hippocampal Neuronal Cells
To further explore RIPK1/RIPK3/MLKL mediated necroptosis in the hippocampal neurons of rats, immunofluorescence was used to reveal the expression of the proteins in the neuron. As shown by the results of immunofluorescence (Fig. 6A–C), there was little RIPK1, RIPK3 and MLKL positive neuron in the Con and Con+Nec-1 groups 3 days after anesthesia. It indicated that RIPK1-positive, RIPK3-positive and MLKL-positive neurons were notably increased in aged rats exposed to sevoflurane compared with rats exposed to the carrier gas. However, Nec-1 reduced RIPK1, RIPK3 and MLKL expression in the Sev+Nec-1 group prominently compared with Sev group (RIPK1: Fig. 6D, F(3,20) = 73.715, P < 0.05; RIPK3: Fig. 6E, F(3,20) = 96.415, P < 0.05; MLKL: Fig. 6F, F(3,20) = 95.858, P < 0.05). These changing trends were consistent with that of western blot.

Nec-1 ameliorates the expression of RIPK1, RIPK3 and MLKL in hippocampal neuronal cells. Representative photomicrographs from RIPK1/Neun/DAPI staining (RIPK1, green; Neun, red; DAPI, blue) showing hippocampal CA1 neurons 3 days after exposed to sevoflurane (A). Representative photomicrographs from RIPK3/Neun/DAPI staining (RIPK3, green; Neun, red; DAPI, blue) showing hippocampal neurons 3 days after exposed to sevoflurane (B). Representative photomicrographs from MLKL/Neun/DAPI staining (MLKL, green; Neun, red; DAPI, blue) showing hippocampal neurons 3 days after exposed to sevoflurane (C). Scale bars = 50 μm. Percentages of RIPK1/Neun/DAPI, RIPK3/Neun/DAPI and MLKL/Neun/DAPI caused by the indicated stimuli (D–F). Data are presented as mean ± SD (n = 6 animals per group). *P < 0.05 vs. Con group, #P < 0.05 vs. Sev group
Nec-1 Decreased [Ca2+]c and Calpain Expression Level and Activity in the Rat Hippocampus
Our previous work had been suggested that sevoflurane can induce neurotoxicity through increasing [Ca2+]c and calpain expression [10]. To detect the effect of Nec-1 on calcium homeostasis, the [Ca2+]c and calpain activity in the hippocampus were measured after sevoflurane exposure in the aged rats. Spectrin is a well-known calpain substrate and, therefore, cleavage of the protein giving rise to two breakdown products, SBDP145 and SBDP150, can be used to investigate putative alterations in calpain activity. As shown in Fig. 7B, [Ca2+]c was significantly higher in Sev and Sev+Nec-1 group when compared with the Con group, and Nec-1 reduced the [Ca2+]c (F(3,20) = 25.096, P < 0.05). Compared with the Con group, the expression of calpain was markedly higher in the Sev group and Sev+Nec-1 group; compared with the Sev group, the expression of calpain was significantly lower in Sev+Nec-1 group (Fig. 7D, F(3,20) = 25.096, P < 0.05). The expression trend of SBDP150 and SBDP145 was coincident with above (Fig. 7E, SBDP150: F(3,20) = 62.805, P < 0.05; SBDP145: F(3,20) = 37.531, P < 0.05).

Nec-1 decreased [Ca2+]c and calpain expression level and activity in the rat hippocampus. Intracellular [Ca2+]c in hippocampus cells 1 day after sevoflurane exposure in each group, the ‘B’ and ‘C’ gates in the figure represent negative and positive cells respectively (A). Representative histograms of [Ca2+]c by flow cytometry in the hippocampus of aged rats in each group (B). Representative western blot images of calpain, α II Spectrin band as well as the 150 kDa SBDP (SBDP150) and 145 kDa SBDP (SBDP145) in the hippocampus of aged rats at day 1 after anesthesia in each group (C). The quantity analysis of calpain expression in the hippocampus of each group (D). The quantity analysis of SBDP150 and SBDP145 expression in the hippocampus of each group (E). Data are presented as mean ± SD (n = 6 animals per group). *P < 0.05 vs. Con group, #P < 0.05 vs. Sev group
Nec-1 Attenuated the Sevoflurane-Induced Down-Regulation of BDNF and p-TrkB in the Hippocampus
To search for the mechanism by which Nec-1 ameliorates cognitive dysfunction induced by sevoflurane, we detected the expression of BDNF and p-TrkB/TrkB in the hippocampus. As shown in Fig. 8, exposure to sevoflurane significantly decreased BDNF vs the control group (F(3,20) = 28.039, P < 0.05). Furthermore, the down-regulation of BDNF restrained the downstream signaling pathway. As expected, the expression of p-TrkB/TrkB was decreased on 1 day after anesthesia (F(3,20) = 25.168, P < 0.05). Nevertheless, Nec-1 upregulated BDNF(P < 0.05) and p-TrkB/TrkB (P < 0.05) expression in the hippocampus effectively.

Nec-1 attenuated the sevoflurane-induced down-regulation of BDNF and p-TrkB in the hippocampus. The representative western blot images of BDNF and p-TrkB in the hippocampus of aged rats on 1 day after anesthesia in each group (A). The quantity analysis of BDNF (B) and p-TrkB/ TrkB (B) expression in the hippocampus of each group. Data are presented as mean ± SD (n = 6 animals per group). *P < 0.05 vs. Con group, #P < 0.05 vs. Sev group
Discussion
In the present study, our findings suggest that Nec-1 improved the hippocampus-dependent cognitive behavior of rats after anesthesia, consist of spatial learning and memory abilities. Our pathological results showed that sevoflurane could lead to necroptosis in specific sub-regions of the hippocampus. The necroptosis was distributed in neurons of CA1 and CA3, while an anti-necroptotic molecule Nec-1 treatment alleviated neuronal necroptosis in CA1 and CA3 (Fig. 4). This effect of Nec-1 was conducive to the recovery of spatial learning and memory functions eventually. Furthermore, sevoflurane-induced necroptosis owing to the subsequent increase in the expression of P-RIPK1, RIPK1, p-RIPK3, RIPK3, p-MLKL and MLKL. Moreover, the [Ca2+]c and calpain activity were increased, as well as the downregulation of BDNF/TrkB signaling pathway after anesthesia, but Nec-1 mitigated the above changes and eventually contributed to the recovery of spatial learning and memory functions. In spite of the mechanisms that still need to be explored, our research demonstrated that Nec-1 could relieve the sevoflurane-induced cognitive impairment via protecting the hippocampal neurons from necroptosis and possibly by increasing BDNF-TrkB signalling.
The mechanism of POCD is intricate, which is on the basis of central nervous system degeneration in elderly patients caused by surgery/anesthesia. According to the published study [24], animal models of POCD were established by inhalation of 2% sevoflurane for 5 h, and this concentration of sevoflurane was within the range of clinical application [26]. In addition, several studies have shown that a number of elderly patients exposed to sevoflurane anesthesia exhibited different degrees of cognitive dysfunction [27, 28]. Coincidentally, in animal models of sevoflurane anesthesia, the analogical phenomena have also been observed [29, 30]. Studies have shown that the hippocampus played an important role in the formation of spatial memory, in addition, its structure and function were suffered negative effects from external surgical/anesthetic pressure stimulation [31]. Cell death in sevoflurane-induced neurotoxicity can occur in hippocampal neuronal via several means, such as apoptosis, autophagy and necroptosis [10, 17, 32]. The mechanism of cognitive decline induced by sevoflurane has been reported to be closely related to multiple factors, including calcium overload [10], upregulation of inflammatory chemokines (TNF-α, IL-6, toll-like receptors 4) [33], oxidative stress [34], etc. Notably, researches have indicated that these indicators are involved in the occurrence of necroptosis [35]. Whether these factors are involved in sevoflurane-induced necroptosis is unclear. In this study, we investigated the possible mechanism of calcium overload in sevoflurane induced necroptosis.
Calpain, a downstream calcium signaling molecule, is located in the cytoplasm and nucleus of neurons and is widely distributed in the CNS. A reported study indicated that an increase in GluN2B and excessive activation of calpain-2 participated in isoflurane-induced neurotoxicity and long-term cognitive deficiency, inhibiting GluN2B level and calpain-2 activity obviously alleviated these responses [36]. Furthermore, one recent study suggests that deletion of Sarm1 prevents isoflurane induced cognitive dysfunction through suppression of Calpain-TrkB and MAPK signaling pathways in aged mice [37]. Importantly, as a calcium-dependent papain-like enzyme, calpain is involved in neuronal necrosis [38]. The increase of [Ca2+]c causes the up-regulation of calpain expression, which participates in the necroptosis induced by ischemia–reperfusion in a lung transplant setting [39]. Wang et al. [40] found that inhibition of extracellular signal-regulated kinase/calpain-2 pathway reduces necroptosis induced by cerebral ischemia–reperfusion injury. Here, we found that sevoflurane-induced intracellular calcium elevation and subsequent calpain overactivation participate in necroptosis (Fig. 7), which means calcium may function as an initiator of sevoflurane-induced RIPK1 activation. Nevertheless, the relationship between calcium and necroptosis is complex. For instance, evidence suggests that influx of calcium may act as a downstream factor of MLKL during necroptosis [41]. Nec-1 suppressed calcium elevation and calpain activity changes maybe due to the reducing MLKL-associated calcium influx. Future study should address how MLKL regulates Ca2+ influx in plasma membrane.
Thus far, a series of neuroprotective effects of Nec-1 has been reported [18,19,20]. Previous studies have indicated that Nec-1 can alleviate brain cell death and ameliorate cognitive dysfunction in the AD model [42], SAH rats [21], ischemia/reperfusion rat [43] and CCI model [44]. Notably, Duan et al. [19] showed that Nec-1 treatment ameliorates spatial learning and memory dysfunction in D-galactose-induced aged mice. The inhibition of necroptosis may provide a new target on POCD treatment. However, the neuroprotective effect of Nec-1 has not been clearly clarified. In our study, after cognitive dysfunction occurred, necroptosis was activated following the up-regulation of RIPK1, RIPK3 and MLKL in the hippocampus, consistent with the results of other studies [18, 19]. RIPK1 and RIPK3 are the upstream proteins in necroptosis, after being activated, phosphorylated RIPK3 interacts with RIPK1 to form an amyloid-like necrosome [45], then the necrosome initiates downstream signaling pathways triggering necroptosis eventually [46]. Nec-1 treatment reduces the expression levels of these proteins (Figs. 5, 6).
To determine the effect of Nec-1 on cognitive dysfunction after sevoflurane anesthesia, the MWM test [47], a generally accepted model in rodents, was chosen to detect the learning and memory of rats in the present study. Our results showed that there were no significant differences among groups during the training of rats, however, animals exposed to 2% sevoflurane 5 h had obvious learning and memory disorder as manifested by the prolonged escape latency and the lower number of original platform crossings, which were in accordance with other studies [24, 48, 49]. Recently, we also observed that Nec-1 pretreatment (6.25 mg/kg, i.p.) significantly attenuated the sevoflurane-induced memory deficits. Open field test was conducted to assess spontaneous activity and there were no differences in traveling speed, moving distance and time spent in the center (Fig. 2), which coincided with the results of Duan et al. [19]. These results suggested that anesthesia does not affect the animals' autonomous activities, excluding the influence of abnormal autonomous movement on the MWM test. Collectively, the MWM test suggests that Nec-1 obviously attenuated cognitive impairment induced by sevoflurane in aged rats.
BDNF, a member of the neurotrophin family, is widely expressed in the central nervous system [50]. Recent studies have indicated that deficits in BDNF signaling were contributed to the development of several nervous system diseases, such as Alzheimer’s disease, Huntington’s disease, depression and anxiety disorders [51]. BDNF is a neuroprotective protein critical for neuronal plasticity, learning, and memory [52]. Studies have previously reported that BDNF plays a role in neuronal protective functions might be in the activation of the TrkB receptor [53]. Blocking the phosphorylation of TrkB was reported to be necessary for learning and memory dysfunction induced by sevoflurane [54]. Baek et al. [55] found that upregulation of the BDNF/TrkB pathway was involved in the protection of neuronal cells from cell death in the brain. Before us, the function of the BDNF/TrkB signaling pathway on neuroprotective effects of Nec-1 on sevoflurane-induced cognitive decline was unclear. In this study, we observed that sevoflurane notably down-regulated BDNF levels and subsequently decreased the expression of p-TrkB in the hippocampus (Fig. 8). The suppression of the BDNF/TrkB signaling pathway was possibly related to the function of spatial learning and memory in aged rats. Whereas, pre-treatment with Nec-1 could significantly increase the expressions of BDNF and p-TrkB in the hippocampus, which appears to be associated with improvement in memory and cognitive ability. The underlying mechanism for the decrease in BDNF level might be that sevoflurane-induced calcium overload and calpain overactivation modulate the transcriptional activity of BDNF, which reduces the dephosphorylation of TrkB [13].
There were still limitations in our study. The relationship between calcium and necroptosis is complex, the [Ca2+]c and calpain overactivation not only participates in the occurrence of necroptosis, but also regulated by necroptosis related proteins, the potential regulation mechanism was not further explored. In addition, in the present study, we mainly focused on the effect of Nec-1 on necroptosis and BDNF-TrkB signaling pathway in sevoflurane-induced cognitive impairment rats. Calcium overload and calpain activation are only preliminarily discussed as possible mechanisms, and the specific regulation mechanism of calpain involvement in necroptosis of POCD will be carried out in future studies.
Conclusions
In summary, our data indicate that prolonged exposure to sevoflurane lead to significant cognitive dysfunction and pre-treatment with Nec-1 could reverse the cognition impairment. The underlying mechanism of this protective function may be associated with its inhibition of calcium overload and calpain overactivation, regulation of BDNF/TrkB signaling dysfunction, as well as the downregulation of necroptosis markers, including p-RIPK1, RIPK1, p-RIPK3, RIPK3, p-MLKL and MLKL in hippocampal neurons. Hence, these findings may shed light on a new target point of Nec-1 against POCD caused by sevoflurane.
Data Availability
The data that support the findings of this study are available on request from corresponding author. The data are not publicly available due to privacy or ethical restrictions.
Code Availability
SPSS software (version 21.0).
References
Muscat SM, Deems NP, D’Angelo H, Kitt MM, Grace PM, Andersen ND, Silverman SN, Rice KC, Watkins LR, Maier SF, Barrientos RM (2021) Postoperative cognitive dysfunction is made persistent with morphine treatment in aged rats. Neurobiol Aging 98:214–224
Lv G, Li C, Wang W, Li N, Wang K (2020) Silencing SP1 alleviated sevoflurane-induced POCD development via cholinergic anti-inflammatory pathway. Neurochem Res 45:2082–2090
O’Gara BP, Mueller A, Gasangwa DVI, Patxot M, Shaefi S, Khabbaz K, Banner-Goodspeed V, Pascal-Leone A, Marcantonio ER, Subramaniam B (2020) Prevention of early postoperative decline: a randomized, controlled feasibility trial of perioperative cognitive training. Anesth Analg 130:586–595
Bickel H, Gradinger R, Kochs E, Förstl H (2008) High risk of cognitive and functional decline after postoperative delirium. A three-year prospective study. Dement Geriatr Cogn Disord 26:26–31
Kotekar N, Shenkar A, Nagaraj R (2018) Postoperative cognitive dysfunction - current preventive strategies. Clin Interv Aging 13:2267–2273
Skvarc DR, Berk M, Byrne LK, Dean OM, Dodd S, Lewis M, Marriott A, Moore EM, Morris G, Page RS, Gray L (2018) Post-operative cognitive dysfunction: an exploration of the inflammatory hypothesis and novel therapies. Neurosci Biobehav Rev 84:116–133
Miller D, Lewis SR, Pritchard MW, Schofield-Robinson OJ, Shelton CL, Alderson P, Smith AF (2018) Intravenous versus inhalational maintenance of anaesthesia for postoperative cognitive outcomes in elderly people undergoing non-cardiac surgery. The Cochrane database of systematic reviews 8:Cd012317
Wang Z, Meng S, Cao L, Chen Y, Zuo Z, Peng S (2018) Critical role of NLRP3-caspase-1 pathway in age-dependent isoflurane-induced microglial inflammatory response and cognitive impairment. J Neuroinflammation 15:109
Zhu X, Yao Y, Guo M, Li J, Yang P, Xu H, Lin D (2021) Sevoflurane increases intracellular calcium to induce mitochondrial injury and neuroapoptosis. Toxicol Lett 336:11–20
Liu X, Song X, Yuan T, He J, Wang X, Wang Q (2016) Effects of calpain on sevoflurane-induced aged rats hippocampal neuronal apoptosis. Aging Clin Exp Res 28:633–639
Zhang Q, Li Y, Bao Y, Yin C, Xin X, Guo Y, Gao F, Huo S, Wang X, Wang Q (2018) Pretreatment with nimodipine reduces incidence of POCD by decreasing calcineurin mediated hippocampal neuroapoptosis in aged rats. BMC Anesthesiol 18:42
Luo F, Min J, Wu J, Zuo Z (2020) Histone deacetylases may mediate surgery-induced impairment of learning, memory, and dendritic development. Mol Neurobiol 57:3702–3711
Qiu LL, Pan W, Luo D, Zhang GF, Zhou ZQ, Sun XY, Yang JJ, Ji MH (2020) Dysregulation of BDNF/TrkB signaling mediated by NMDAR/Ca(2+)/calpain might contribute to postoperative cognitive dysfunction in aging mice. J Neuroinflamm 17:23
de Almagro MC, Vucic D (2015) Necroptosis: Pathway diversity and characteristics. Semin Cell Dev Biol 39:56–62
Cao L, Mu W (2021) Necrostatin-1 and necroptosis inhibition: Pathophysiology and therapeutic implications. Pharmacol Res 163:105297
Li C, Ma Q, Toan S, Wang J, Zhou H, Liang J (2020) SERCA overexpression reduces reperfusion-mediated cardiac microvascular damage through inhibition of the calcium/MCU/mPTP/necroptosis signaling pathways. Redox Biol 36:101659
Xu R, Zhu Y, Jia J, Li WX, Lu Y (2021) RIPK1/RIPK3-mediated necroptosis is involved in sevoflurane-induced neonatal neurotoxicity in the rat hippocampus. Cell Mol Neurobiol. https://doi.org/10.1007/s10571-021-01098-z
Nikseresht S, Khodagholi F, Nategh M, Dargahi L (2015) RIP1 inhibition rescues from LPS-induced RIP3-mediated programmed cell death, distributed energy metabolism and spatial memory impairment. J Mol Neurosci 57:219–230
Duan S, Wang X, Chen G, Quan C, Qu S, Tong J (2018) Inhibiting RIPK1 limits neuroinflammation and alleviates postoperative cognitive impairments in d-galactose-induced aged mice. Front Behav Neurosci 12:138
Jinawong K, Apaijai N, Wongsuchai S, Pratchayasakul W, Chattipakorn N, Chattipakorn SC (2020) Necrostatin-1 mitigates cognitive dysfunction in prediabetic rats with no alteration in insulin sensitivity. Diabetes 69:1411–1423
Yang C, Li T, Xue H, Wang L, Deng L, Xie Y, Bai X, Xin D, Yuan H, Qiu J, Wang Z, Li G (2018) Inhibition of necroptosis rescues SAH-induced synaptic impairments in hippocampus via CREB-BDNF pathway. Front Neurosci 12:990
Han F, Guan X, Guo W, Lu B (2019) Therapeutic potential of a TrkB agonistic antibody for ischemic brain injury. Neurobiol Dis 127:570–581
Tagai N, Tanaka A, Sato A, Uchiumi F, Tanuma SI (2020) Low levels of brain-derived neurotrophic factor trigger self-aggregated amyloid β-induced neuronal cell death in an Alzheimer’s cell model. Biol Pharm Bull 43:1073–1080
Chen Y, Zhang P, Lin X, Zhang H, Miao J, Zhou Y, Chen G (2020) Mitophagy impairment is involved in sevoflurane-induced cognitive dysfunction in aged rats. Aging 12:17235–17256
Shang L, Huang JF, Ding W, Chen S, Xue LX, Ma RF, Xiong K (2014) Calpain: a molecule to induce AIF-mediated necroptosis in RGC-5 following elevated hydrostatic pressure. BMC Neurosci 15:63
Mei X, Zheng HL, Li C, Ma X, Zheng H, Marcantonio E, Xie Z, Shen Y (2020) The effects of propofol and sevoflurane on postoperative delirium in older patients: a randomized clinical trial study. Journal of Alzheimer’s disease : JAD 76:1627–1636
Geng YJ, Wu QH, Zhang RQ (2017) Effect of propofol, sevoflurane, and isoflurane on postoperative cognitive dysfunction following laparoscopic cholecystectomy in elderly patients: a randomized controlled trial. J Clin Anesth 38:165–171
Qiao Y, Feng H, Zhao T, Yan H, Zhang H, Zhao X (2015) Postoperative cognitive dysfunction after inhalational anesthesia in elderly patients undergoing major surgery: the influence of anesthetic technique, cerebral injury and systemic inflammation. BMC Anesthesiol 15:154
Peng S, Zhang Y, Sun DP, Zhang DX, Fang Q, Li GJ (2011) The effect of sevoflurane anesthesia on cognitive function and the expression of Insulin-like Growth Factor-1 in CA1 region of hippocampus in old rats. Mol Biol Rep 38:1195–1199
Shan L, Ma D, Zhang C, Xiong W, Zhang Y (2017) miRNAs may regulate GABAergic transmission associated genes in aged rats with anesthetics-induced recognition and working memory dysfunction. Brain Res 1670:191–200
Chen L, Dong R, Lu Y, Zhou Y, Li K, Zhang Z, Peng M (2019) MicroRNA-146a protects against cognitive decline induced by surgical trauma by suppressing hippocampal neuroinflammation in mice. Brain Behav Immun 78:188–201
Xu L, Shen J, Yu L, Sun J, McQuillan PM, Hu Z, Yan M (2018) Role of autophagy in sevoflurane-induced neurotoxicity in neonatal rat hippocampal cells. Brain Res Bull 140:291–298
Tang XL, Wang X, Fang G, Zhao YL, Yan J, Zhou Z, Sun R, Luo AL, Li SY (2021) Resveratrol ameliorates sevoflurane-induced cognitive impairment by activating the SIRT1/NF-κB pathway in neonatal mice. J Nutr Biochem 90:108579
Yue T, Shanbin G, Ling M, Yuan W, Ying X, Ping Z (2015) Sevoflurane aggregates cognitive dysfunction and hippocampal oxidative stress induced by β-amyloid in rats. Life Sci 143:194–201
Zheng Y, Shi G, Cai J, Yang J, Zhang Y, Gong Y, Liu Q, Yu D, Zhang Z (2020) Di-(2-ethyl hexyl) phthalate induces necroptosis in chicken cardiomyocytes by triggering calcium overload. J Hazard Mater 387:1696
Tang X, Zhang X, Li S, Chi X, Luo A, Zhao Y (2020) NR2B receptor- and calpain-mediated KCC2 cleavage resulted in cognitive deficiency exposure to isoflurane. Neurotoxicology 76:75–83
Lin H, Kang Z, Li S, Zeng J, Zhao J (2021) Sarm1 is essential for anesthesia-induced neuroinflammation and cognitive impairment in aged mice. Cell Mol Neurobiol. Online ahead of print
Yagami T, Yamamoto Y, Koma H (2019) Pathophysiological roles of intracellular proteases in neuronal development and neurological diseases. Mol Neurobiol 56:3090–3112
Kim H, Zamel R, Bai XH, Lu C, Keshavjee S, Keshavjee S, Liu M (2018) Ischemia-reperfusion induces death receptor-independent necroptosis via calpain-STAT3 activation in a lung transplant setting. Am J Physiol Lung Cell Mol Physiol 315:L595-l608
Wang WY, Xie L, Zou XS, Li N, Yang YG, Wu ZJ, Tian XY, Zhao GY, Chen MH (2021) Inhibition of extracellular signal-regulated kinase/calpain-2 pathway reduces neuroinflammation and necroptosis after cerebral ischemia-reperfusion injury in a rat model of cardiac arrest. Int Immunopharmacol 93:7377
Cai Z, Jitkaew S, Zhao J, Chiang HC, Choksi S, Liu J, Ward Y, Wu LG, Liu ZG (2014) Plasma membrane translocation of trimerized MLKL protein is required for TNF-induced necroptosis. Nat Cell Biol 16:55–65
Yang SH, Lee DK, Shin J, Lee S, Baek S, Kim J, Jung H, Hah JM, Kim Y (2017) Nec-1 alleviates cognitive impairment with reduction of Aβ and tau abnormalities in APP/PS1 mice. EMBO Mol Med 9:61–77
Yang R, Hu K, Chen J, Zhu S, Li L, Lu H, Li P, Dong R (2017) Necrostatin-1 protects hippocampal neurons against ischemia/reperfusion injury via the RIP3/DAXX signaling pathway in rats. Neurosci Lett 651:207–215
You Z, Savitz SI, Yang J, Degterev A, Yuan J, Cuny GD, Moskowitz MA, Whalen MJ (2008) Necrostatin-1 reduces histopathology and improves functional outcome after controlled cortical impact in mice. J Cereb Blood Flow Metab 28:1564–1573
Li J, McQuade T, Siemer AB, Napetschnig J, Moriwaki K, Hsiao YS, Damko E, Moquin D, Walz T, McDermott A, Chan FK, Wu H (2012) The RIP1/RIP3 necrosome forms a functional amyloid signaling complex required for programmed necrosis. Cell 150:339–350
Lule S, Wu L, Sarro-Schwartz A, Edmiston WJ 3rd, Izzy S, Songtachalert T, Ahn SH, Fernandes ND, Jin G, Chung JY, Balachandran S, Lo EH, Kaplan D, Degterev A, Whalen MJ (2021) Cell-specific activation of RIPK1 and MLKL after intracerebral hemorrhage in mice. J Cereb Blood Flow Metab 41:1623–1633
Brandeis R, Brandys Y, Yehuda S (1989) The use of the Morris Water Maze in the study of memory and learning. Int J Neurosci 48:29–69
Ma H, Yao L, Pang L, Li X, Yao Q (2016) Tetrandrine ameliorates sevoflurane-induced cognitive impairment via the suppression of inflammation and apoptosis in aged rats. Mol Med Rep 13:4814–4820
Peng S, Li P, Liu P, Yan H, Wang J, Lu W, Liu C, Zhou Y (2020) Cistanches alleviates sevoflurane-induced cognitive dysfunction by regulating PPAR-γ-dependent antioxidant and anti-inflammatory in rats. J Cell Mol Med 24:1345–1359
Notaras M, van den Buuse M (2019) Brain-derived neurotrophic factor (BDNF): novel insights into regulation and genetic variation. Neuroscientist 25:434–454
Colucci-D’Amato L, Speranza L, Volpicelli F (2020) Neurotrophic factor BDNF, physiological functions and therapeutic potential in depression, neurodegeneration and brain cancer. Int J Mol Sci 21:7777
Fan D, Li J, Zheng B, Hua L, Zuo Z (2016) Enriched environment attenuates surgery-induced impairment of learning, memory, and neurogenesis possibly by preserving BDNF expression. Mol Neurobiol 53:344–354
Yoo JM, Lee BD, Sok DE, Ma JY, Kim MR (2017) Neuroprotective action of N-acetyl serotonin in oxidative stress-induced apoptosis through the activation of both TrkB/CREB/BDNF pathway and Akt/Nrf2/Antioxidant enzyme in neuronal cells. Redox Biol 11:592–599
Dong Y, Hong W, Tang Z, Gao Y, Wu X, Liu H (2020) Sevoflurane leads to learning and memory dysfunction via breaking the balance of tPA/PAI-1. Neurochem Int 139:4789
Baek SY, Li FY, Kim DH, Kim SJ, Kim MR (2020) Enteromorpha prolifera extract improves memory in scopolamine-treated mice via downregulating amyloid-β expression and upregulating BDNF/TrkB pathway. Antioxidants (Basel) 9:620
Funding
This work was supported by the National Natural Science Foundation of China (No. 81771134), the Natural Science Foundation of Hebei Province (No. H2018206305), The Hebei Provincial government funded the specialty capacity building and specialty leader training program.
Author information
Authors and Affiliations
Contributions
CY designed the work and wrote the paper, QZ and JZ performed the research, JY and WL analyzed and interpreted the research data, YL and QW revised the paper.
Corresponding author
Ethics declarations
Conflict of interest
The authors declare that they have no conflict of interest.
Ethical Approval
The experimental design and scheme have been approved by the Ethics Committee of the Third Hospital of Hebei Medical University [G A2017-026-1].
Consent for Publication
Agree.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
About this article

Cite this article
Yin, C., Zhang, Q., Zhao, J. et al. Necrostatin-1 Against Sevoflurane-Induced Cognitive Dysfunction Involves Activation of BDNF/TrkB Pathway and Inhibition of Necroptosis in Aged Rats. Neurochem Res 47, 1060–1072 (2022). https://doi.org/10.1007/s11064-021-03505-9
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s11064-021-03505-9