
Abstract
Epigallocatechin gallate (EGCG) is one of polyphenol that is abundant in green tea. It has anti-oxidative activity and exerts neuroprotective effects in ischemic brain damage. Ischemic conditions induce oxidative stress and result in cell death. Thioredoxin is a small redox protein that plays an important role in the regulation of oxidation and reduction. This study was designed to investigate the regulation of thioredoxin by EGCG in ischemic brain damage. Middle cerebral artery occlusion (MCAO) was performed to induce focal cerebral ischemia in male Sprague–Dawley rats. The EGCG (50 mg/kg) or was administered before MCAO surgical operation. Neurological behavior test, reactive oxygen species (ROS), and lipid peroxidation (LPO) measurement were performed 24 h after MCAO. The cerebral cortex was isolated for further experiments. EGCG alleviated MCAO-induced neurological deficits and increases in ROS and LPO levels. EGCG also ameliorated the decrease in thioredoxin expression by MCAO. This finding was confirmed using various techniques such as Western blot analysis, reverse transcription PCR, and immunofluorescence staining. Results of immunoprecipitation showed that MCAO decreases the interaction between apoptosis signal-regulating kinase 1 (ASK1) and thioredoxin, while EGCG treatment attenuates this decrease. EGCG also attenuated decrease of cell viability and thioredoxin expression in glutamate-exposed neuron in a dose-dependent manner. It alleviated the increase of caspase-3 by glutamate exposure. However, this effect of EGCG on caspase-3 change was weakened in thioredoxin siRNA-transfected neurons. These findings suggest that EGCG exerts a neuroprotective effect by regulating thioredoxin expression and modulating ASK1 and thioredoxin binding in ischemic brain damage.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Epigallocatechin gallate (EGCG) is a polyphenolic component that is abundant in green tea, as well as in vegetables and fruits [1, 2]. EGCG has beneficial properties against harmful conditions including oxidative stress, metabolic syndrome, and cancer [3,4,5]. EGCG penetrates the blood brain barrier and easily distributes in brain tissue when administered in various routes such as oral or intraperitoneal injection [6,7,8]. Therefore, it is considered a useful neuroprotective agent. EGCG also exerts neuroprotective effects on various neurological diseases such as Alzheimer's disease, Parkinson's disease, traumatic brain injury, and stroke [9,10,11,12].
Stroke is one of the most serious neurological diseases and is a major cause of human death [13, 14]. In particular, ischemic stroke has various causes such as glutamate toxicity, excessive calcium accumulation, and oxidative stress and can result in permanent neurological disorders [15]. Oxidative stress is a major harmful factor in various diseases and disorders and is a cause of many pathological changes [16]. As a self-protective action against oxidative stress, there are various molecular biological mechanisms in living organisms [17]. Thioredoxin is a small 12 kDa protein that is widely distributed in most cells and is involved in antioxidant defense signaling pathways [18, 19]. It has two redox-active cysteine residues that reduce the oxidized protein by catalyzing the cysteine-thiol disulfide exchange [20]. Thioredoxin acts as an antioxidant by removing reactive oxygen species (ROS). It is associated with growth factors, inflammatory reactions, DNA synthesis, and protein folding [21,22,23,24]. In neurons, thioredoxin regulates signal transduction and neuronal growth [25]. In addition, overexpression of thioredoxin was reported to inhibit inflammation or apoptosis through antioxidative effects against neuronal damage caused by ischemic stroke [26, 27]. Apoptosis signaling kinase 1 (ASK1) is an essential protein in apoptosis signaling pathways and is activated in response to cytotoxic stresses such as inflammation and ROS [28, 29]. It is involved in the JNK and p38 MAP kinase cascades and induces various stress responses like apoptosis [28]. Thioredoxin binds to ASK1 and promotes ASK1 ubiquitination, thereby inhibiting apoptosis signaling pathways [30]. We previously demonstrated the decrease of thioredoxin expression and the reduction of interaction between thioredoxin and ASK1 in ischemic damage [31, 32]. EGCG shows a protective effect against oxidative damage caused by hydrogen peroxide and mediates antioxidant defense through modulation of thioredoxin and thioredoxin reductase [33]. We hypothesized that EGCG exerts neuroprotective effects through regulation of thioredoxin and modulation of interaction between thioredoxin and ASK1 in the ischemic cerebral cortex. Thus, we investigated whether EGCG modulates thioredoxin expression and regulates thioredoxin and ASK1 binding in cerebral ischemia and glutamate-induced neuronal damage.
Materials and Methods
Experimental Animal Preparation and Drug Treatment
Male Sprague–Dawley rats (n = 89, 210 ± 10 g) were obtained from Samtako Co. (Animal breeding center, Osan, Korea). Rats were housed in controlled light condition (12/12 h light and dark) and constant temperature (22 ± 1 °C), and freely provided with feed and water. All the experimental procedures were conducted in accordance with the guideline for Institutional Animal Care and Use Committee of Gyeongsang National University (Approval number: GNU-190218-R0008). Animals were randomly divided into four groups: phosphate buffered saline (PBS)-treated with sham, EGCG-treated with sham, PBS-treated with middle cerebral artery occlusion (MCAO), and EGCG-treated with MCAO animals. For accurate assessment, this study excluded dead animals, subarachnoid hemorrhagic animals, and animals that showed no signs of ischemic damage after MCAO surgery. Nine animals were excluded from this study according to the assessment. The excluded animals appeared only MCAO-operated animals, they were 7 animals in PBS-treated group and 2 animals in EGCG-treated group. In this study, 80 animals were used and 24 rats were assigned to each group. EGCG (Sigma-Aldrich, St. Louis, MO, USA) was dissolved in PBS and injected into the abdominal cavity just before MCAO surgery according to a previously described method [34]. EGCG was single administrated at 50 mg/kg. PBS animals were injected with the same volume of PBS only.
Middle Cerebral Artery Occlusion
Rats were injected with Zoletil (50 mg/kg, Virbac, Carros, France) to induce anesthesia and were subjected to MCAO as a previously reported method [35]. Animals were laid in supine position and a midline neck skin incision was made to expose common carotid artery. The right common carotid artery (CCA), external carotid artery (ECA), and internal carotid artery (ICA) were separated from the surrounding tissues and nerves. The CCA was temporarily blocked with a microvascular clip to block blood flow, and then the distal part of the ECA was ligated and cut. A 4-0 monofilament nylon suture with blunt end by heating was inserted from the stump of ECA to ICA and carefully advanced to origin of middle cerebral artery. The length of inserted nylon suture was approximately 22–24 mm and middle cerebral artery was completely blocked. The inserted nylon suture was ligated with ECA and the incised skin was sutured. Sham-operation was performed with the same surgical method except for the insertion of nylon suture. Rats were placed on a heating pad until recovery from anesthesia and returned to their cage. Food and water were freely provided. Rats were subjected to neurological deficit scoring test, corner test, and adhesive-remove test 24 h after MCAO.
Neurological Deficit Scoring Test
Neurological deficits were scored by a previously described method [36]. Score was determined according to the following 5-point scale: no neurological deficit (0), incomplete extension of the contralateral forelimb (1), involuntary contralateral side circling (2), involuntary longitudinal rotation, falling down, and hypersensitive reflex (3), no movement and fainting (4).
Corner Test
Corner test was performed as a previously reported manual [37]. Animals were removed from the cage and placed between two identical boards (30 × 20 × 1 cm3) with a small opening and a 30° corner. They were facing a corner and started moving into a small open corner. When the rats reached to the corner, both vibrissae were stimulated by two boards and turned back from the corner. Only the complete turn was recorded as a response and the test was conducted ten times per trial. Incomplete turning or stepping backward was excluded from the record. Rats were trained every day for 7 days before MCAO surgery. Animals that rotated same ratio in the left and right directions were used for the experiment. Sham-operated animals had no preference in the direction of turning. Animals with right MCAO damage preferred to circle to the right, which is the ipsilateral side of the MCAO damaged brain. The resulting data is displayed as the number of right turns in ten trials.
Adhesive Removal Somatosensory Test
The adhesive removal test was performed according to a previously described method [38]. Animals were transferred to a test cage and habituated for 60 s, and temporarily taken out from the cage to attach on adhesive tape. Adhesive circular tapes (12 mm in diameter) were firmly attached on both hairless part of the forelimb and animals were placed in a test cage. Rats removed the tapes in response to tactile stimuli. The time for removing adhesive tapes was measured and the time was recorded up to 180 s. Rats were trained for 3 days before MCAO surgery to limit the individual variations and rats that removed adhesive tapes within 15 s were used for the experiment.
Reactive Oxygen Species (ROS) Assay
ROS assays were performed to measure intracellular ROS levels in cerebral cortex using a reactive oxygen species assay kit according to manual of manufacturer (Elabscience Biotechnology Inc. Houston, TX, USA). Cerebral cortex was isolated, cut into small pieces, and reacted with the provided enzyme solution at 37 °C for 30 min to obtain a single cell sample. Single cells were washed with PBS and incubated with 5 mM 2′,7′-dichlorodihydrofluorescein diacetate (DCFH-DA) at 37 °C for 1 h. DCFH-DA and cells mixtures were converted to 2′,7′-dichlorofluorescein (DCF) by reacting with the intracellular ROS that expressed fluorescence. These cells were centrifuged at 1000 g for 10 min and washed with PBS for 5 min. Samples were centrifuged again and cell pellet was collected. Fluorescence expression was measured with a Multiskan EX spectrophotometer (Thermo Fisher Scientific, Waltham, MA, USA) at 500 nm excitation wavelength and at 525 nm emission wavelength. Result data were represented as relative DCF pmol/mg of tissue.
Lipid Peroxidation (LPO) Assay
LPO assay was performed by the measurement of malondialdehydride (MDA), a marker of membrane polyunsaturated fatty acid peroxidation [39]. It was performed by lipid peroxidation colorimetric assay kit (BioVision Inc., Milpitas, CA, USA) according to manufacturer’s instruction. Cerebral cortex was homogenized with MDA lysis buffer containing butylated hydroxytoluene. Homogenates were centrifuged at 13,000 g for 10 min and supernatants were collected. Samples were treated with thiobarbituric acid, incubated at 95 °C for 1 h, and cooled at 4 °C for 10 min. Absorbance at 532 wavelength was measured from samples by spectrophotometer. Result data were expressed as MDA nmol/mg of protein.
Fluoro-Jade B (FJB) Staining
FJB staining was carried out to detect the degeneration of neuronal cells in cerebral cortex. Brain tissues were fixed in 4% paraformaldehyde for 24 h and fixed tissues were washed with tap water for overnight. They were dehydrated by gradient ethyl alcohol (70% to 100%) and cleaned by xylene. Brain tissues were embedded in paraffin block and sliced into 4 μm-thickness by rotary microtome (Leica, Wetzlar, Germany). Tissue sections were dried in slide warmer (Thermo Fisher Scientific) and deparaffinized with xylene, rehydrated with gradient ethyl alcohol (100% to 70%) and washed in distilled water. Sections were immersed with 0.06% potassium permanganate solution for 10 min to quench the fluorescence background. They were stained with 0.0001% FJB in 0.1% acetic acid solution for 30 min, washed with distilled water, and completely dried. Nuclei were counterstained with 4′,6-diamidine-2-phenylindole (DAPI, Sigma-Aldrich) and slides were mounted with a non-fluorescence dibutylphthalate polystyrene xylene mounting media (Sigma-Aldrich). Tissue slides were observed with a confocal laser scanning microscope (FV 1000, Olympus) and micrographs were taken. Data were expressed as the ratio of FJB positive cells to DAPI positive cells.
Two-Dimensional Gel Electrophoresis
The cerebral cortex tissues were isolated from the brain tissue and stored at − 70 °C for further experiments. Tissue samples were homogenized with lysis buffer (8 M urea, 4% CHAPS, ampholyte and 40 mM Tris–HCl) and gently vortexed for overnight at 4 °C. Homogenates were centrifuged at 20,000 g for 20 min at 4 °C and the supernatant was mixed with 10% trichloroacetic acid for 30 min. Mixtures were centrifuged at 20,000 g for 20 min at 4 °C and precipitated protein pellet was collected. The protein pellets were washed with 90% acetone and dissolved in sample buffer [8 M urea, 4% CHAPS, 0.2% amphoteric solution, 40 mM Tris–HCl, 2 μg/ml dithiothreitol (DTT)]. Protein concentration was determined by Bradford assay kit (Bio-Rad, Hercules, CA, USA) following the provided manufacturer’s instruction. Total proteins (50 µg) were added to rehydration buffer [8 M urea, 2% CHAPS, 20 mM DTT, 0.5% immobilized pH gradient (IPG) buffer, bromophenol blue] and applied to IPG gel strips (pH range 4–7, 17 cm, Bio-Rad) for 13 h at room temperature. Rehydrated IPG gel strips containing protein samples were loaded on an Ettan IPGphor 3 System (GE Healthcare, Little Chalfont, Buckinghamshire, UK) and electrophoresed according to the following protocol: 250 V for 15 min, 10,000 V for 3 h, and then 10,000 V to 50,000 V. First dimensional electrophoresed IPG gel strips were immersed in a equilibration buffer (6 M urea, 30% glycerol, 2% sodium dodecyl sulfate, 50 mM Tris–HCl, bromophenol blue) containing 1% DTT for 10 min and immersed with an identical equilibration buffer containing 2.5% iodoacetamide for 10 min. IPG gel strips were placed on a 12% sodium dodecyl sulphate-polyacrylamide gel electrophoresis (SDS-PAGE) gel and electrophoresed at 10 mA using Protein-II XI electrophoresis equipment (Bio-Rad) at 15 °C until the bromophenol blue dye reached the bottom end of the gel. Electrophoresed gels were immersed in fixation solution (12% acetic acid, 50% methanol) for 2 h and washed twice for 20 min with 50% ethyl alcohol. They were sensitized by immersing in 0.02% sodium thiosulfate for 1 min, washed with distilled water, stained with a silver nitrate solution (0.2% silver nitrate, 0.0003% formaldehyde) for 20 min, and washed with distilled water. The development reaction was completed by immersion in 0.2% sodium carbonate containing 0.0002% formaldehyde and stopped with 1% acetic acid solution. Gel images were taken with an Agfar ARCUS 1200™ scanner (Agfar-Gevaert, Mortsel, Belgium) and analyzed with PDQuest 2D analysis software (Bio-Rad) to quantify the protein spot intensity of the gel. Matrix-assisted laser desorption ionization time (MALDI-TOF) was performed to identify the proteins. Each protein spots were incised from the gel, desalted with 30 mM potassium hexacyanoferate containing 100 mM sodium thiosulfate, and washed with 10% acetic acid in 50% methanol. They were treated with 50 mM ammonium bicarbonate and acetonitrile, dried in a vacuum centrifuge (Biotron, Seoul, Korea) for 20 min, and dissolved in reduction solution (10 mM DTT in 0.1 M ammonium bicarbonate) for 45 min at 56 °C. They were treated with 0.1 M ammonium bicarbonate and acetonitrile, dried in vacuum centrifuge for 20 min, and treated with digestion solution (12.5 ng/ml trypsin, 0.1% octyl beta-d glycopyranside in 50 mM ammonium bicarbonate) for 12 h at 37 °C. Proteins were extracted by 1% trifluoroacetic acid in 66% acetonitrile and dried in a vacuum centrifuge. They were dissolved with 1% trifluoroacetic acid in 66% acetonitrile and matrix solution (16 mg/ml alpha-cyano-4-hydroxinic acid, 4 mg/ml nitrocellulose in acetone). Each protein was placed on a MALDI-TOF metal plate and MALDI-TOF was performed by a Voyager-DE STR (Applied Biosystem, Foster City, CA, USA). These results were investigated with NCBI and MS-FIT protein sequence databases.
Western Blot Analysis
Cerebral cortices were homogenized with lysis buffer [1% Triton X-100, 1 mM EDTA in PBS (pH 7.4)] containing 200 µM phenylmethylsulfonyl fluoride. Homogenates were reacted for 1 h at 4 °C and sonicated with on ice using an ultrasonic probe. Samples were centrifuged at 15,000 g for 1 h at 4 °C and supernatant was collected. Concentrations of protein samples were determined by bicinchoninic acid protein assay kit (Pierce, Rockford, IL, USA) according to the protocol provided by the manufacturer. Protein samples (30 μg) were heated for 3 min at 100 °C for protein denaturation and cooled on ice. They were loaded on 10% SDS-PAGE gel, electrophoresed at 10 mA for 30 min, and continuously performed at 20 mA for 90 min using a Mini-PROTEAN 3 Cell (Bio-Rad). They were transferred to a polyvinylidenedifluoride membrane (Millipore, Billerica, MA, USA) at 120 V for 2 h. Membrane was incubated with 5% skim milk in Tris-buffered saline containing 0.1% Tween-20 (TBST) for 1 h to block non-specific binding and washed 3 times for 10 min with TBST. They were incubated with the following primary antibodies: anti-thioredoxin, anti-caspase-3, cleaved caspase-3 (1:1000 in TBS, Cell Signaling Technology, Beverly, MA, USA), and anti-β-actin (1:1000 in TBS, Santa Cruz Biotechnology, Santa Cruz, CA, USA) for overnight at 4 °C, and washed with TBST. They were subsequently incubated with horseradish peroxidase-conjugated anti-rabbit IgG or anti-mouse IgG (1:5000 in TBST, Cell Signaling Technology) for 2 h, washed with TBST, and reacted with enhanced chemiluminescence detection reagents (GE Healthcare) to detect protein expression. They were exposed on Fuji medical X-ray film (Fuji Film, Tokyo, Japan) to visualize protein bands. Films were developed with developer, washed with distilled water, fixed with fixation solution, washed with distilled water, and dried. Relative protein band intensities were normalized by β-actin band signal intensity. Protein levels were expressed as a ratio of specific protein band intensity to β-actin band intensity. The band intensity was analyzed by SigmaGel 1.0 (Jandel Scientific, San Rafael, CA, USA) and SigmaPlot 4.0 (SPSS Inc., Point Richmond, CA, USA).
Reverse-Transcript Polymer Chain Reaction (RT-PCR)
Cerebral cortices were isolated from brain tissues, immediately frozen, and keep at − 70 °C before total RNA extraction. Total RNA was extracted using Trizol Reagent (Life Technologies, Rockville, MD, USA) and 1 mg of RNA was used for reverse-transcription. Complementary DNA (cDNA) was prepared with Superscript III first-strand system (Invitrogen, Carlsbad, CA, USA) according to the manufacturer’s instruction. Specific gene codes were detected by the following primers: thioredoxin product (188 bp), forward primer: 5′-TTCTTTCATTCCCTCTGTG-3′, reverse primer: 5′-TCCGTAATAGTGGCTTCG-3′; β-actin (238 bp), forward primer: 5′-GGGTCAGAAGGACTCCTACG-3′, reverse primer: 5′-GGTCTCAAACATGATCTGGG-3′. Genes were amplified by PCR with following protocols: initial denaturation for 5 min at 94 °C; template denaturation for 30 s at 94 °C, primer annealing for 30 s at 54 °C, extension 1 min at 72 °C for 30 cycles; and a final extension for 10 min at 72 °C. PCR products were mixed with Loading STAR dye (Dyne Bio, Seongnam, Korea) and loaded on a 1% agarose gel. They were electrophoresed by Mupid-2 plus (Takara bio, Shiga, Japan) and visualized under ultraviolet light exposure.
Immunofluorescence Staining
Paraffin tissues were deparaffinized and hydrated with xylene and gradient ethyl alcohol series, dipped in with water, and rinsed with PBS. Tissues were incubated with 1% normal goat serum in PBS for 1 h at room temperature to block non-specific reaction. They were incubated with anti-thioredoxin antibody (diluted 1:100 in 1% normal goat serum; Cell Signaling Technology) overnight at 4 °C and washed with PBS. Tissues were incubated with anti-rabbit fluorescein isothiocyanate (FITC)-conjugated secondary antibody (1:100 in PBS; Santa Cruz Biotechnology) in the dark chamber for 1 h at room temperature and washed with PBS. They were counterstained with 4,6-diamidino-2-phenylindole (DAPI, Santa Cruz Biotechnology) to observe nuclei, mounted with UltraCruz mounting medium (Santa Cruz Biotechnology), and cover slipped. Immunofluorescence reactions of FITC and DAPI were observed and microscopic images were taken with a confocal microscope (FV-1000, Olympus, Tokyo, Japan). Five regions of the cerebral cortex were randomly selected and the number of FITC-positive cells or DAPI-positive cells was counted using Image-Pro Plus software (Media Cybernetics, Silver Spring, MD, USA). These results were represented as the ratio of thioredoxin-positive cells and DAPI-positive cells.
Immunoprecipitation Assay
Extracted proteins (200 μg) were cleaned with Protein A/G agarose beads (Santa Cruz Biotechnology) to prevent non-specific binding proteins in agarose beads. They were reacted with anti-thioredoxin antibody (Santa Cruz Biotechnology) for overnight at 4 °C with gentle shaking and mixed with Protein A/G agarose beads for 2 h at 4 °C. These complexes were rinsed with radioimmunoprecipitation assay buffer (Sigma-Aldrich), denatured with sample buffer for 3 min at 4 °C, and cooled on ice. They were electrophoresed on a 10% SDS-PAGE gel and transferred to PVDF membrane. Membranes were washed with TBST, incubated with anti-ASK1 antibody (1:1000 diluted with TBST), and continuously processed with the same protocol as described in the Western blot analysis.
Hippocampal Cell Line Culture
Immortalized mouse hippocampal neuron cell line (HT22, passages 7–12) was cultured in Dulbecco's modified Eagle's medium (DMEM, Gibco BRL, Gaithersburg, MD, USA) without l-glutamate. DMEM was added with 10% fetal bovine serum (FBS), penicillin (100 units/ml), and streptomycin (100 mg/ml) as a growth supplement and antibiotics. Cells were seeded at a density of 5 × 103 per well in 96 well culture plate (Thermo Fisher Scientific) for MTT assay and 2 × 106 in 100 mm diameter culture dish (Nalge Nunc International, Rochester, NY, USA) for further studies. Cells were cultured in a controlled environment with a 5% CO2 humidified atmosphere and a constant temperature (37 °C). They were cultured until the confluence rate reached 70%. Glutamate (5 mM) was added to DMEM to induce excitatory toxicity [40] and EGCG (10, 20, 40 μM) was treated 1 h before glutamate treatment. After 24 h of glutamate treatment, cells were scraped and stored at − 70 °C before further experiment.
Cell Viability Assay
A 3-(4,5-Dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT) assay was carried out to investigate cell viability. Cells were seeded in 24-well plates (5 × 104 cells/300 μl) and grown for 24 h. Glutamate and EGCG were treated in plates as previously mentioned. Cells were incubated with 0.1% MTT solution in PBS for 4 h at 37 °C. MTT solution was discarded and cells were cleaned with PBS. MTT solution was discarded and the cells were rinsed with PBS. Metabolized MTT formazan was formed in viable cells and dissolved in 300 μl of DMSO. Sample absorption rate was measured at 570 nm wavelength using spectrophotometer, and cell viability was expressed as percentage (%) of absorption rate. The cell viability of the PBS group was evaluated as 100%.
siRNA Transfection
siRNA transfection process was carried out to knockdown thioredoxin expression in cultured cells. Cells were incubated with Neurobasal A media 24 h before siRNA transfection. Neurobasal A media with Lipofectamine 2000TM (Thermo Fisher Scientific) was incubated for 5 min, mixed with thioredoxin siRNA, and incubated for 20 min. The prepared siRNA mixture was treated in a cell medium at a final concentration of 30 nM and transfected for 12 h. After siRNA transfection, the cell culture media was changed and treated with 5 mM of glutamate and 10, 20, 40 μM of EGCG. Cells were scraped and preserved at − 70 °C until further experiment conducted.
Statistical Analysis
All experimental data are expressed as the mean ± standard deviation (S.D.).Normality of data distribution and equal variance of data were confirmed by Shapiro–Wilk test and Lev ene’s test, respectively. One-way ANOVA or two-way ANOVA followed by post-hoc Scheffe’s test was applied to data which satisfying the normal distribution and equal variance. Two-way ANOVA followed by Dunnett’s multiple comparison test was applied to data which showing the effect of EGCG in dose-dependent manner. Mann–Whitney U test was applied to data which is not satisfying the normal distribution and equal variance. p < 0.05 was considered to be statistically significant.
Results
Neuroprotective and Antioxidative Effects of EGCG in MCAO Animal
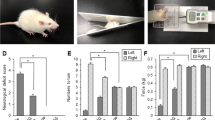
Neurological deficit scores were evaluated for neurobehavioral deficits in MCAO animals. Sham-operated animals had normal posture without neurological defects. However, serious behavioral defects such as falling, hypersensitivity reflection, and fainting were observed in PBS-treated animals with MCAO. However, mild defects such as involuntary rotation were observed in EGCG-treated animals. EGCG treatment alleviated these defects caused by MCAO. Neurological deficit scores were divided from 0 to 4 point, they were 3.63 ± 0.49 and 2.17 ± 0.70 in PBS- and EGCG-treated animals with MCAO animals, respectively (Fig. 1a). Corner tests showed the degree of biased behavior due to loss of response to stimuli. The rate of rotation was similar in the two directions in sham-operated animals. However, right-biased rotation was observed in PBS-treated animals with MCAO. This biased behavior was significantly reduced by EGCG treatment. The number of right turns was 9.20 ± 1.04 and 7.10 ± 0.58 in PBS- and EGCG-treated animals with MCAO, respectively (Fig. 1b). The significance of these results is less than 0.01. The adhesion-removal test showed the response time for tactile stimulus of both forepaws. Sham animals removed both forepaws very quickly. However, adhesion removal time was delayed in MCAO animals. The removal times in MCAO animals were 163.6 ± 15.5 and 83.6 ± 16.8 s in PBS- and EGCG-treated groups, respectively (Fig. 1c). These results are significant, and the significance is less than 0.01. It was significantly shorter in EGCG-treated animals than in PBS-treated animals. The DCF and MDA assays were performed to measure oxidative stress, showing increased DCF and MDA levels in MCAO-operated animals compared to sham-operated animals. This increase was alleviated in EGCG-treated animals. DCF levels were 4.82 ± 1.36 and 2.46 ± 1.08 pmol/mg protein in PBS- and EGCG-treated animals, respectively (Fig. 1d). MDA levels were 2.75 ± 0.61 in PBS and 1.50 ± 0.68 pmol/mg protein in EGCG-treated animals (Fig. 1e). The significance of these results is less than 0.01. We observed FJB-positive cells to detect degenerated neurons in the cerebral cortex. These positive cells were observed in ischemic regions in MCAO animals. EGCG treatment significantly reduced the number of FJB-positive cells compare to PBS-treated animals (Fig. 2a). No positive cells were detected in sham-operated animals. The ratio of FJB positive cells to DAPI-positive cells were 0.54 ± 0.18 and 0.34 ± 0.09 in PBS- and EGCG-treated animals with MCAO, respectively (Fig. 2b).

Neurological deficit score (a), corner test (b), adhesive-removal test (c), reactive oxygen species (ROS) (d), and lipid peroxidation (e) analyses in phosphate buffered saline (PBS) + middle cerebral artery occlusion (MCAO), epigallocatechin gallate (EGCG) + MCAO, PBS + sham, and EGCG + sham animals. EGCG improved neurological deficit and biased right turning behavior in ischemic brain damage (a–c). Data (n = 24) of a, b and c are graphed as median value (bold black line), upper and lower quartiles (box boundaries), highest and lowest values (whiskers), and black dot (outlier). Mann–Whitney test. EGCG alleviated the increases in DCF and MDA in ischemic-damaged cerebral cortex (d, e). Data (n = 4) of d and e are shown as the mean ± S.D. #p < 0.001, two-way ANOVA followed by Scheffe’s test

Representative images of Fluoro-Jade B (FJB, green) and DAPI (nuclei marker, blue) staining in the cerebral cortex of phosphate buffered saline (PBS) + middle cerebral artery occlusion (MCAO), epigallocatechin gallate (EGCG) + MCAO, PBS + sham, and EGCG + sham animals. FJB-positive cells were increased in the cerebral cortex with ischemic damage. EGCG alleviated these increases (a, b). Arrows indicate FJB-positive cells. Scale bar: 100 μm. Data (n = 4) are shown as the mean ± S.D. *p < 0.001, two-way ANOVA followed by Scheffe’s test
Regulation of Thioredoxin Expression by EGCG in MCAO Animals
We observed the change of thioredoxin expression between PBS- and EGCG-treated animal with MCAO (Fig. 3a). The matched peptide mass with extracted protein sample was 8/87 and the sequence coverage was 42%. Thioredoxin expression decreased in MCAO-operated animals, while EGCG treatment alleviated this decrease. Thioredoxin levels were 0.21 ± 0.07 and 0.58 ± 0.11 in PBS- and EGCG-treated animals with MCAO, respectively (Fig. 3d). Thioredoxin expression did not change in sham-operated animals regardless of EGCG treatment. Western blot and reverse transcription-PCR analyses confirmed the decrease of thioredoxin expression in MCAO animals (Fig. 3b, c). The level of thioredoxin proteins was 0.55 ± 0.13 in PBS- and 0.79 ± 0.13 in EGCG-treated animals with MCAO (Fig. 3e). Thioredoxin mRNA level was 0.60 ± 0.11 in PBS-treated and 1.21 ± 0.21 in EGCG-treated aniamls with MCAO, respectively (Fig. 3f). Immunofluorescence staining showed a significant reduction in the number of thioredoxin-positive cells in PBS-treated animals with MCAO. However, this decrease was alleviated by EGCG (Fig. 4a). We evaluated thioredoxin level as the ratio of thioredoxin-positive cells to DAPI-positive cells. The thioredoxin levels were 0.22 ± 0.02 in PBS- and 0.44 ± 0.08 in EGCG-treated animals with MCAO (Fig. 4b).

Image of thioredoxin (TRX) protein spots using proteomics (a, d), Western blot (b, e), and reverse transcription-PCR analyses (c, f) in phosphate buffered saline (PBS) + middle cerebral artery occlusion (MCAO), epigallocatechin gallate (EGCG) + MCAO, PBS + sham, and EGCG + sham animals. Squares of A photos indicate TRX protein spot. TRX protein level is expressed as a ratio of spot intensity in each group relative to that in the PBS + sham group (d). TRX protein and mRNA levels are expressed as the intensity of TRX to that of β-actin (e, f). Data (n = 4) are shown as the mean ± S.D. *p < 0.01 and **p < 0.001, two-way ANOVA followed by Scheffe’s test

Images of immunofluorescence staining with thioredoxin (TRX, green) and DAPI (nuclei marker, blue) in the cerebral cortex of phosphate buffered saline (PBS) + middle cerebral artery occlusion (MCAO), epigallocatechin gallate (EGCG) + MCAO, PBS + sham, and EGCG + sham animals (a). Quantified number of TRX-positive cells in the cerebral cortex region (b). Arrows indicate TRX-positive cells. Data (n = 4) are shown as the mean ± S.D.*p < 0.001, two-way ANOVA followed by Scheffe’s test. Scale bars = 100 µm
Modulation of Thioredoxin and ASK1 Interaction by EGCG in MCAO Animals
The results of immunoprecipitation showed the changes of thioredoxin and ASK1 interaction in MCAO animals with EGCG treatment. These protein interaction levels decreased in MCAO animals compared to sham animals, and these reductions were alleviated by EGCG treatment. The level of interaction was similar in sham-operated animals regardless of PBS or EGCG treatment (Fig. 5a). The interaction levels in MCAO animals were 0.35 ± 0.10 and 0.51 ± 0.08 in PBS- and EGCG-treated groups, respectively (Fig. 5b).

Immunoprecipitation result of thioredoxin (TRX) and apoptosis signal-regulating kinase 1 (ASK1) interaction in the cerebral cortex of phosphate buffered saline (PBS) + middle cerebral artery occlusion (MCAO), epigallocatechin gallate (EGCG) + MCAO, PBS + sham, and EGCG + sham animals (a). Densitometric results are represented as intensity ratio of identified protein to IgG intensity (b). Data (n = 4) are represented as the mean ± S.D. *p < 0.01, two-way ANOVA followed by Scheffe’s test
Regulation of Thioredoxin Expression by EGCG in Glutamate-Exposed HT22 Cells
Glutamate toxicity decreases the cell viability and induces serious cell death in HT22 cells. EGCG treatment alleviated these damages in a dose-dependent manner (Fig. 6a). The cell viability was evaluated as 100% in the PBS group and was 34.7 ± 4.5% in the glutamate-only-treated group. The survival rates of combined treatment with EGCG 10, 20, and 40 μM were 43.3 ± 3.3, 64.6 ± 4.1, and 76.9 ± 5.5%, respectively (Fig. 6b). These results are significant, and the significance is less than 0.01. Glutamate exposure decreased the expression of thioredoxin, while EGCG treatment attenuated this reduction in a dose-dependent manner (Fig. 6c). The thioredoxin protein level in the glutamate-only-treated group was 0.06 ± 0.02. They were 0.18 ± 0.07, 0.22 ± 0.07, and 0.46 ± 0.10 in the combined group with EGCG 10, 20, and 40 μM, respectively (Fig. 6d). We used a thioredoxin siRNA transfection technique to reveal the anti-apoptotic effect of EGCG through regulation of thioredoxin expression.

Cell viability (a, b) and Western blot analysis (c, d) of thioredoxin (TRX) in HT22 cells. Epigallocatechin gallate (EGCG, 10, 20, 40 µM) was applied for 1 h and glutamate (Glu, 5 mM) was subsequently treated. Cell images were obtained by phase-contrast microscopy to confirm the morphological changes (a). MTT assay was performed to access cell viability (b). Densitometric results are expressed as the intensity of thioredoxin to that of β-actin (d). Data (n = 5) are shown as the mean ± S.D. *p < 0.01 and **p < 0.001, two-way ANOVA followed by Dunnett’s multiple comparison. Scale bar = 100 µm
Modulation of Caspase-3 Expression by EGCG in Thioredoxin siRNA-Transfected HT22 Cells
Figure 7 showed the thioredoxin, caspase-3, and cleaved caspase-3 expressions in thioredoxin siRNA-transfected cells. The expression of thioredoxin was down-regulated in the glutamate-only-treated group, and EGCG co-treatment alleviated this decrease (Fig. 7a). The thioredoxin levels in non-transfected cells were 0.08 ± 0.03 and 0.17 ± 0.02 in glutamate-only- and EGCG co-treated group, respectively (Fig. 7b). In addition, the expressions of caspase-3 and cleaved caspase-3 in glutamate-treated groups increased compared to the PBS-treated group. Caspase-3 and cleaved caspase-3 expressions increased in siRNA transfection groups compared to non-transfection groups. EGCG treatment alleviated the increase in the glutamate-only-treated group regardless of siRNA transfection (Fig. 7c). The caspase-3 levels in non-transfected cells were 0.46 ± 0.09 and 0.24 ± 0.06 in the glutamate-only and EGCG co-treated group, respectively. In transfected cells, they were 0.96 ± 0.14 and 0.81 ± 0.13 in the glutamate-only and EGCG co-treated group (Fig. 7d). The cleaved caspase-3 levels in non-transfected cells were 1.01 ± 0.12 and 0.63 ± 0.08 in the glutamate-only and EGCG co-treated group. In siRNA transfected cells, they were 1.57 ± 0.20 and 1.12 ± 0.08 in the glutamate-only and EGCG co-treated group, respectively (Fig. 7d).

Western blot analysis of thioredoxin (TRX, a, b), caspase-3, and cleaved caspase-3 (c, d) in siRNA transfected cells. Epigallocatechin gallate (EGCG, 10, 20, 40 µM) was applied for 1 h and followed with glutamate (Glu, 5 mM). Densitometric results are expressed as the ratio of TRX, caspase-3, cleaved caspase-3 intensity to that of β-actin. Data (n = 5) are shown as the mean ± S.E.M. *p < 0.05, **p < 0.001 in un-transfected group, #p < 0.05, ##p < 0.001 in transfected group, two-way ANOVA followed by Scheffe’s test
Discussion
EGCG is a powerful antioxidant that acts as a neuroprotective agent for neurodegenerative diseases, including Alzheimer’s disease, Parkinson’s disease, and stroke [9, 10, 41]. EGCG has a protective effect against oxidative stress-induced neuronal damage through ROS elimination characteristics [42, 43]. We previously reported that EGCG improves neurological deficits and reduces infarct lesion in ischemic brain damage [10]. The present study additionally demonstrated that EGCG regulates the expression of thioredoxin and modulates the binding of thioredoxin and ASK 1. EGCG also exhibits the property of scavenging ROS and LPO. The thioredoxin system regulates reduction and oxidation homeostasis in mammalian cells and consists of thioredoxin, thioredoxin reductase, and nicotinamide adenine dinucleotide phosphate [44]. Thioredoxin acts as a strong antioxidant and exerts stress response properties [45, 46]. Thioredoxin protects various organs such as the heart, kidney, and brain against ischemic damage [26, 47, 48]. Oxidative stress is known as a major pathological cause of ischemia. The inhibition of thioredoxin expression by siRNA transfection increases oxidative stress, activates ASK1 cell death signaling pathway, and exacerbates ischemic brain damage [49, 50]. We previously showed decrease in thioredoxin expression in MCAO-induced damage [31]. We confirmed the decrease of thioredoxin expression in an ischemic-injured cerebral cortex model. Moreover, we identified decrease in thioredoxin by glutamate exposure in cerebral cortex of neonatal rats [51]. Overexpression or administration of recombinant thioredoxin showed a significant reduction in oxidative stress and a decrease in apoptosis in neurons [27, 52]. These results demonstrate that thioredoxin plays an important role in neuroprotection through maintenance of the redox cycle in ischemic brain injury.
EGCG restores neurons of the cerebral cortex damaged by MCAO [10]. The present study additionally showed that EGCG alleviates the increase in ROS and LPO levels and the decrease in thioredoxin expression caused by MCAO. Previous studies demonstrated that EGCG attenuates ROS and LPO levels by modulating enzymic antioxidants including superoxide dismutase, glutathione, and glutathione peroxidase [53]. EGCG also prevents oxidative stress by regulating thioredoxin system in liver [33]. Based on these results, we investigated whether EGCG affects the interaction of thioredoxin and ASK1 in apoptotic signaling pathways. ASK1 acts as a major regulator of oxidative stress-related apoptosis through activation of JNK and p38-MAPK pathways [54]. The inactive ASK1 or the ASK1 knockout condition has resistivity against the apoptosis induced by oxidative stress [55]. Thioredoxin acts as a negative regulator of ASK1 activation by binding to the N-terminal site of ASK1 protein [54]. However, apoptotic stimuli such as tumor necrosis factor and ROS dissociate thioredoxin from ASK1 to activate the apoptosis signaling pathway [30]. We have previously reported that ischemic brain damage decreases thioredoxin expression and reduces the binding between thioredoxin and ASK1 [32]. Maintaining the interaction between thioredoxin and ASK1 is an important mechanism to prevent apoptosis caused by oxidative stress. Our results showed that MCAO damage reduces thioredoxin and ASK1 binding. EGCG treatment relieves dissociation of thioredoxin from ASK1 due to MCAO damage. These findings demonstrate that regulation of thioredoxin and ASK1 binding by EGCG contributes to neuroprotective effects of EGCG against ischemic damage.
Thioredoxin is widely distributed in various tissues, especially in red blood cells [56]. We found small particles of thioredoxin-positive signals in all experimental animal groups, which were considered as erythrocytes in blood. We also counted thioredoxin and DAPI-double positive signals in the cerebral cortex. Interestingly, we found that the positive signal size of thioredoxin differs in animal group. We have previously reported histopathological changes in the MCAO damaged cerebral cortex [10]. Ischemic damage induces pyramidal cell distortion with contracted nucleus, while EGCG noticeably attenuates these histopathological changes [10]. The result of immunofluorescent staining showd a decrease in the number of thioredoxin-positive cells due to MCAO damage, and EGCG alleviated this decrease. The size of thioredoxin positive signal decreased in ischemic damaged cells, while EGCG attenuated this decrease. The size change of the positive signal can be explained with the shape change due to the MCAO damage. These changes are mitigated by EGCG treatment. EGCG also exerts neuroprotective effects on glutamate-treated neurons. The high extracellular concentration of glutamate causes accumulation of ROS and lipid peroxidation through interaction with glutamate/cystine antiporter systems [57]. Glutamate toxicity reduces cell viability, and EGCG administration improves this reduction in a dose-dependent manner. Moreover, EGCG alleviates the decrease in thioredoxin expression caused by glutamate toxicity, and the effect of EGCG was dose dependent. This study showed that thioredoxin is involved in the neuroprotective effects of EGCG using an siRNA transfection technique. EGCG exerts anti-apoptotic effects through regulation of thioredoxin expression. EGCG alleviates the increase in caspase-3 expression caused by glutamate toxicity, but thioredoxin siRNA transfection ameliorates this impact of EGCG treatment. The cleavage of caspase-3 is an important event in apoptosis, and is accepted as a biomarker of apoptosis in cerebral ischemia [58]. In this study, glutamate treatment increases cleaved caspase-3 expression and EGCG alleviates the increase of cleaved caspase-3 by glutamate toxicity. However, the transfection of thioredoxin siRNA decreases the alleviative effect of EGCG. These results demonstrated that thioredoxin acts as an important protein for the neuroprotective effect of EGCG. EGCG attenuates apoptosis through the regulation of thioredoxin expression. Thus, we can suggest that the anti-apoptotic effect of EGCG in glutamate-exposed neuron is mediated by the regulation of thioredoxin expression. Our results demonstrated that EGCG has neuroprotective and anti-apoptotic effects against ischemic damage, and thioredoxin is involved in these actions of EGCG. However, despite the inhibition of thioredoxin, the protective effect of EGCG against apoptosis was not reduced completely. EGCG exerts a neuroprotective effect through the regulation of various factors as well as thioredoxin. Although more researches are required to identify other factors, this study can demonstrate the protective mechanism of EGCG by regulation of thioredoxin expression in neuronal cell damage caused by ischemia. In conclusion, our findings suggest that EGCG has a neuroprotective effect on ischemic brain damage and glutamate toxicity by modulating thioredoxin expression and regulating the interaction between thioredoxin and ASK1.
Data Availability
Data will be made available on reasonable request.
References
Arts IC, van de Putte B, Hollman PC (2000) Catechin contents of foods commonly consumed in The Netherlands. 1. Fruits, vegetables, staple foods, and processed foods. J Agric Food Chem 48:1746–1751
Bettuzzi S, Brausi M, Rizzi F, Castagnetti G, Peracchia G, Corti A (2006) Chemoprevention of human prostate cancer by oral administration of green tea catechins in volunteers with high-grade prostate intraepithelial neoplasia: a preliminary report from a one-year proof-of-principle study. Cancer Res 66:1234–1240
Liao B, Ying H, Yu C, Fan Z, Zhang W, Shi J, Ying H, Ravichandran N et al (2016) (−)-Epigallocatechin gallate (EGCG)-nanoethosomes as a transdermal delivery system for docetaxel to treat implanted human melanoma cell tumors in mice. Int J Pharm 512:22–31
Suzuki T, Pervin M, Goto S, Isemura M, Nakamura Y (2016) Beneficial effects of tea and the green tea catechin epigallocatechin-3-gallate on obesity. Molecules 21:1305
Saeki K, Hayakawa S, Nakano S, Ito S, Oishi Y, Suzuki Y, Isemura M (2018) In vitro and in silico studies of the molecular interactions of epigallocatechin-3-O-gallate (EGCG) with proteins that explain the health benefits of green tea. Molecules 23:1295
Suganuma M, Okabe S, Oniyama M, Tada Y, Ito H, Fujiki H (1998) Wide distribution of [3H](−)-epigallocatechin gallate, a cancer preventive tea polyphenol, in mouse tissue. Carcinogenesis 19:1771–1776
Pervin M, Unno K, Nakagawa A, Takahashi Y, Iguchi K, Yamamoto H, Hoshino M, Hara A et al (2017) Blood brain barrier permeability of (−)-epigallocatechin gallate, its proliferation-enhancing activity of human neuroblastoma SH-SY5Y cells, and its preventive effect on age-related cognitive dysfunction in mice. Biochem Biophys Rep 9:180–186
Wei BB, Liu MY, Zhong X, Yao WF, Wei MJ (2019) Increased BBB permeability contributes to EGCG-caused cognitive function improvement in natural aging rats: pharmacokinetic and distribution analyses. Acta Pharmacol Sin 40:1490–1500
Xu Y, Zhang Y, Quan Z, Wong W, Guo J, Zhang R, Yang Q, Dai R, McGeer PL, Qing H (2016) Epigallocatechin gallate (EGCG) inhibits alpha-synuclein aggregation: a potential agent for Parkinson’s disease. Neurochem Res 41:2788–2796
Park DJ, Kang JB, Koh PO (2020) Epigallocatechin gallate alleviates neuronal cell damage against focal cerebral ischemia in rats. J Vet Med Sci 82:639–645
Wu Y, Cui J (2020) (−)-Epigallocatechin-3-gallate provides neuroprotection via AMPK activation against traumatic brain injury in a mouse model. Naunyn Schmiedebergs Arch Pharmacol 393:2209–2220
Zhang S, Zhu Q, Chen JY, Ouyang D, Lu JH (2020) The pharmacological activity of epigallocatechin-3-gallate (EGCG) on Alzheimer’s disease animal model: a systematic review. Phytomedicine 79:153316
Kissela BM, Khoury JC, Alwell K, Moomaw CJ, Woo D, Adeoye O, Flaherty ML, Khatri P et al (2012) Age at stroke: temporal trends in stroke incidence in a large, biracial population. Neurology 79:1781–1787
Tibæk M, Dehlendorff C, Jørgensen HS, Forchhammer HB, Johnsen SP, Kammersgaard LP (2016) Increasing incidence of hospitalization for stroke and transient ischemic attack in young adults: a registry-based study. J Am Heart Assoc 5:e003158
Khoshnam SE, Winlow W, Farzaneh M, Farbood Y, Moghaddam HF (2017) Pathogenic mechanisms following ischemic stroke. Neurol Sci 38:1167–1186
Liguori I, Russo G, Curcio F, Bulli G, Aran L, Della-Morte D, Gargiulo G, Testa G et al (2018) Oxidative stress, aging, and diseases. Clin Interv Aging 13:757–772
Pamplona R, Costantini D (2011) Molecular and structural antioxidant defenses against oxidative stress in animals. Am J Physiol Regul Integr Comp Physiol 301:R843-863
Rozell B, Hansson HA, Luthman M, Holmgren A (1985) Immunohistochemical localization of thioredoxin and thioredoxin reductase in adult rats. Eur J Cell Biol 38:79–86
Lu J, Holmgren A (2014) The thioredoxin antioxidant system. Free Radic Biol Med 66:75–87
Nakamura H, Nakamura K, Yodoi J (1997) Redox regulation of cellular activation. Annu Rev Immunol 15:351–369
Gasdaska JR, Berggren M, Powis G (1995) Cell growth stimulation by the redox protein thioredoxin occurs by a novel helper mechanism. Cell Growth Differ 6:1643–1650
Wiesel P, Foster LC, Pellacani A, Layne MD, Hsieh CM, Huggins GS, Strauss P, Yet SF et al (2000) Thioredoxin facilitates the induction of heme oxygenase-1 in response to inflammatory mediators. J Biol Chem 275:24840–24846
Berndt C, Lillig CH, Holmgren A (2008) Thioredoxins and glutaredoxins as facilitators of protein folding. Biochim Biophys Acta 1783:641–650
Muri J, Heer S, Matsushita M, Pohlmeier L, Tortola L, Fuhrer T, Conrad M, Zamboni N et al (2018) The thioredoxin-1 system is essential for fueling DNA synthesis during T-cell metabolic reprogramming and proliferation. Nat Commun 9:1851
Bai J, Nakamura H, Kwon YW, Hattori I, Yamaguchi Y, Kim YC, Kondo N, Oka S et al (2003) Critical roles of thioredoxin in nerve growth factor-mediated signal transduction and neurite outgrowth in PC12 cells. J Neurosci 23:503–509
Hattori I, Takagi Y, Nakamura H, Nozaki K, Bai J, Kondo N, Sugino T, Nishimura M et al (2004) Intravenous administration of thioredoxin decreases brain damage following transient focal cerebral ischemia in mice. Antioxid Redox Signal 6:81–87
Zhou F, Gomi M, Fujimoto M, Hayase M, Marumo T, Masutani H, Yodoi J, Hashimoto N et al (2009) Attenuation of neuronal degeneration in thioredoxin-1 overexpressing mice after mild focal ischemia. Brain Res 1272:62–70
Sekine Y, Takeda K, Ichijo H (2006) The ASK1-MAP kinase signaling in ER stress and neurodegenerative diseases. Curr Mol Med 6:87–97
Iriyama T, Takeda K, Nakamura H, Morimoto Y, Kuroiwa T, Mizukami J, Umeda T, Noguchi T et al (2009) ASK1 and ASK2 differentially regulate the counteracting roles of apoptosis and inflammation in tumorigenesis. EMBO J 28:843–853
Liu Y, Min W (2002) Thioredoxin promotes ASK1 ubiquitination and degradation to inhibit ASK1-mediated apoptosis in a redox activity-independent manner. Circ Res 90:1259–1266
Koh PO (2010) Proteomic analysis of focal cerebral ischemic injury in male rats. J Vet Med Sci 72:181–185
Sung JH, Gim SA, Koh PO (2014) Ferulic acid attenuates the cerebral ischemic injury-induced decrease in peroxiredoxin-2 and thioredoxin expression. Neurosci Lett 566:88–92
Dong R, Wang D, Wang X, Zhang K, Chen P, Yang CS, Zhang J (2016) Epigallocatechin-3-gallate enhances key enzymatic activities of hepatic thioredoxin and glutathione systems in selenium-optimal mice but activates hepatic Nrf2 responses in selenium-deficient mice. Redox Biol 10:221–232
Choi YB, Kim YI, Lee KS, Kim BS, Kim DJ (2004) Protective effect of epigallocatechin gallate on brain damage after transient middle cerebral artery occlusion in rats. Brain Res 1019(1–2):47–54
Longa EZ, Weinstein PR, Carlson S, Cummins R (1989) Reversible middle cerebral artery occlusion without craniectomy in rats. Stroke 20:84–91
Jin Z, Liang J, Wang J, Kolattukudy PE (2015) MCP-induced protein 1 mediates the minocycline-induced neuroprotection against cerebral ischemia/reperfusion injury in vitro and in vivo. J Neuroinflam 12:39
Zhang L, Schallert T, Zhang ZG, Jiang Q, Arniego P, Li Q, Lu M, Chopp M (2002) A test for detecting long-term sensorimotor dysfunction in the mouse after focal cerebral ischemia. J Neurosci Methods 117:207–214
Schallert T, Upchurch M, Wilcox RE, Vaughn DM (1983) Posture-independent sensorimotor analysis of inter-hemispheric receptor asymmetries in neostriatum. Pharmacol Biochem Behav 18:753–759
Esterbauer H, Schaur RJ, Zollner H (1991) Chemistry and biochemistry of 4-hydroxynonenal, malonaldehyde and related aldehydes. Free Radic Biol Med 11:81–128
Kim H, Choi J, Ryu J, Park SG, Cho S, Park BC, Lee DH (2009) Activation of autophagy during glutamate-induced HT22 cell death. Biochem Biophys Res Commun 388:339–344
Walker JM, Klakotskaia D, Ajit D, Weisman GA, Wood WG, Sun GY, Serfozo P, Simonyi A et al (2015) Beneficial effects of dietary EGCG and voluntary exercise on behavior in an Alzheimer’s disease mouse model. J Alzheimers Dis 44:561–572
Jung JY, Han CR, Jeong YJ, Kim HJ, Lim HS, Lee KH, Park HO, Oh WM et al (2007) Epigallocatechin gallate inhibits nitric oxide-induced apoptosis in rat PC12 cells. Neurosci Lett 411:222–227
Meng L, Liu J, Wang C, Ouyang Z, Kuang J, Pang Q, Fan R (2021) Sex-specific oxidative damage effects induced by BPA and its analogs on primary hippocampal neurons attenuated by EGCG. Chemosphere 264:128450
Maulik N, Das DK (2008) Emerging potential of thioredoxin and thioredoxin interacting proteins in various disease conditions. Biochim Biophys Acta 1780:1368–1382
Makino Y, Okamoto K, Yoshikawa N, Aoshima M, Hirota K, Yodoi J, Umesono K, Makino I et al (1996) Thioredoxin: a redox-regulating cellular cofactor for glucocorticoid hormone action Cross talk between endocrine control of stress response and cellular antioxidant defense system. J Clin Invest 98:2469–2477
Das KC, Das CK (2000) Thioredoxin, a singlet oxygen quencher and hydroxyl radical scavenger: redox independent functions. Biochem Biophys Res Commun 277:443–447
Kasuno K, Nakamura H, Ono T, Muso E, Yodoi J (2003) Protective roles of thioredoxin, a redox-regulating protein, in renal ischemia/reperfusion injury. Kidney Int 64:1273–1282
Turoczi T, Chang VW, Engelman RM, Maulik N, Ho YS, Das DK (2003) Thioredoxin redox signaling in the ischemic heart: an insight with transgenic mice overexpressing Trx1. J Mol Cell Cardiol 35:695–704
Li L, Zhu K, Liu Y, Wu X, Wu J, Zhao Y, Zhao J (2015) Targeting thioredoxin-1 with siRNA exacerbates oxidative stress injury after cerebral ischemia/reperfusion in rats. Neuroscience 284:815–823
Wu X, Li L, Zhang L, Wu J, Zhou Y, Zhou Y, Zhao Y, Zhao J (2015) Inhibition of thioredoxin-1 with siRNA exacerbates apoptosis by activating the ASK1-JNK/p38 pathway in brain of a stroke model rats. Brain Res 1599:20–31
Kang JB, Park DJ, Koh PO (2019) Identification of proteins differentially expressed by glutamate treatment in cerebral cortex of neonatal rats. Lab Anim Res 26(35):24
Ma YH, Su N, Chao XD, Zhang YQ, Zhang L, Han F, Luo P, Fei Z et al (2012) Thioredoxin-1 attenuates post-ischemic neuronal apoptosis via reducing oxidative/nitrative stress. Neurochem Int 60:475–483
Srividhya R, Jyothilakshmi V, Arulmathi K, Senthilkumaran V, Kalaiselvi P (2008) Attenuation of senescence-induced oxidative exacerbations in aged rat brain by (−)-epigallocatechin-3-gallate. Int J Dev Neurosci 26:217–223
Soga M, Matsuzawa A, Ichijo H (2012) Oxidative stress-induced diseases via the ASK1 signaling pathway. Int J Cell Biol 2012:439587
Tobiume K, Matsuzawa A, Takahashi T, Nishitoh H, Morita K, Takeda K, Minowa O, Miyazono K et al (2001) ASK1 is required for sustained activations of JNK/p38 MAP kinases and apoptosis. EMBO Rep 2:222–228
Gromer S, Urig S, Becker K (2004) The thioredoxin system-from science to clinic. Med Res Rev 24:40–89
Albrecht P, Lewerenz J, Dittmer S, Noack R, Maher P, Methner A (2010) Mechanisms of oxidative glutamate toxicity: the glutamate/cystine antiporter system xc- as a neuroprotective drug target. CNS Neurol Disord Drug Targets 9:373–382
Namura S, Zhu J, Fink K, Endres M, Srinivasan A, Tomaselli KJ, Yuan J, Moskowitz MA (1998) Activation and cleavage of caspase-3 in apoptosis induced by experimental cerebral ischemia. J Neurosci 18:3659–3668
Funding
This research was supported by the National Research Foundation of Korea (NRF) grant funded by the Korea government [MEST][NRF-2018R1D1A1B07044074].
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
The authors declare no conflicts of interest to disclose.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
About this article

Cite this article
Park, DJ., Kang, JB., Shah, MA. et al. Epigallocatechin Gallate Alleviates Down-Regulation of Thioredoxin in Ischemic Brain Damage and Glutamate-Exposed Neuron. Neurochem Res 46, 3035–3049 (2021). https://doi.org/10.1007/s11064-021-03403-0
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s11064-021-03403-0