
Abstract
Methylmercury (MeHg) exposure and its harmful effects on the developing brain continue to be a global environmental health concern. Decline in mitochondrial function is central to the toxic effects of MeHg and pathogenesis of mitochondria-related diseases including Parkinson's disease (PD). LRRK2 (Leucine-rich repeat kinase 2) mutation is one of the most common genetic risk factors for PD. In this study, we utilize an acute toxicity model of MeHg exposure in the model organism Caenorhabditis elegans (C. elegans) to compare lifespan, developmental progression, mitochondrial membrane potential and reactive oxygen species (ROS) between the wild-type N2 strain, wild-type LRRK2 transgenic strain (WLZ1), and mutant LRRK2(G2019S) transgenic strain (WLZ3). Additionally, the expression levels of skn-1 and gst-4 were investigated. Our results show that acute MeHg exposure (5 and 10 µM) caused a significant developmental delay in the N2 and WLZ3 worms. Notably, the worms expressing wild-type LRRK2 were resistant to 5 µM MeHg- induced developmental retardation. ROS levels in response to MeHg exposure were increased in the N2 worms, but not in the WLZ1 or WLZ3 worms. The mitochondrial membrane potential was decreased in the N2 worms but increased in the WLZ1 and WLZ3 worms following MeHg exposure. Furthermore, MeHg exposure increased the expression of skn-1 in N2, but not in WLZ1 worms. Although skn-1 expression was increased in the WLZ3 worms following MeHg exposure, gst-4 expression was not induced. Both skn-1 and gst-4 had higher basal expression levels in LRRK2s transgenic than wild-type N2 worms. Knocking down of skn-1 with feeding RNAi had a significant developmental effect in WLZ1 worms; however, the effect was not found in WLZ3 worms. These results suggest that mitochondrial dysfunction and a defect in the SKN-1 signaling in the LRRK2 G2019S worms contribute to the severe developmental delay, establishing a modulatory role of LRRK2 mutation in MeHg-induced acute toxicity.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Methylmercury (MeHg) exposure through consumption of fish continues to be an environmental health issue, given the ubiquitous presence of the chemical in the environment [1,2,3]. Environmental mercury derived from the earth’s crust undergoes global recycling. Human activity has a major impact on global mercury distribution [4]. The increased use of fossil fuels culminates in the release of large amounts of mercury into the atmosphere, which can be converted into more toxic forms in aquatic environment, namely MeHg. Furthermore, global warming and overfishing has led to a shift in the dietary structure of marine predators, contributing to high levels of mercury in marine animals at the top of the food chain, thus increasing the risk for human exposure [5]. The major toxic target of MeHg is the brain, especially in developing children [6]. Developmental MeHg exposure may cause long-lasting adverse health effects [7, 8].
Impairment in mitochondria function is involved in MeHg-induced neurotoxicity [9]. Mitochondrial membrane potential is maintained by a proton gradient produced by proton pumps in the respiratory complexes, in which some sulfhydryl groups are potentially modified by mercury [10]. Acute MeHg exposure concentration-dependently decreased mitochondrial membrane potential in astrocytes [11], which is in line with in vivo studies showing that MeHg exposure (5 µM in culture medium) reduced the intra-mitochondria fluorescence level of the mitochondrial dye, MitoTracker Red [12], the uptake of which into the organelle is driven by the mitochondrial membrane potential. In addition, the mitochondria are an important source for generation of reactive oxygen species (ROS) during mitochondrial respiration. Mitochondrially generated ROS are involved in cellular signaling at the physiological range. However, inadequately high amounts of ROS are a potential source for oxidative damage to biomolecules, contributing to the pathogenesis of many age-related degenerative diseases. MeHg exposure increased production of ROS in in vitro mitochondrial preparations [13]. The increase in mitochondrial ROS could result from the disruption of the electron transport chain [14]. In addition, the toxicity of MeHg is related to its ability to inhibit enzymes important for the maintenance of cellular redox balance, many of which are indispensable for mitochondrial function, such as glutathione peroxidases (GPx) and thioredoxin reductases (TrxRs) [15], establishing a critical role of mitochondria dysfunction and oxidative stress in MeHg toxicity [16].
The model organism Caenorhabditis elegans (C. elegans) has been shown to be an alternative genetic model for accessing mechanisms of MeHg toxicity [12, 17]. C. elegans has a cluster of genes highly homologous to mammalian counterparts that are involved in transportation and detoxification of MeHg [18, 19]. For example, the transcriptional factor for induction of phase II detoxification enzymes, nuclear factor erythroid 2-related factor 2 (Nrf-2), plays a crucial role in mitigation of MeHg toxicity [20, 21]. Under basal conditions, Nrf-2 is regulated by its relatively quick turnover rate through ubiquitination and proteasomal degradation. Exposure to xenobiotics increases Nrf-2 levels and its activity to mobilize transcription of genes with important functions in detoxification and antioxidant defense [22]. Recent advances suggest that Nrf-2 is not only a central regulator for detoxification of xenobiotics, but is also involved in mitochondrial function as well as the development of neurodegenerative diseases [23]. Deficiency in skn-1, the C. elegans homolog of nrf-2, increased dopaminergic neurodegeneration in the animals exposed to MeHg [19]. Glutathione S-transferase (GST) activity is inversely correlated with in vivo mercury body burden. One of skn-1 targets is GST-4, which catalyzes MeHg-glutathione conjugation, facilitating MeHg excretion [24].
Our previous study showed that the neurotoxic effects of MeHg on dopaminergic (DAergic) neurons in C. elegans were modified by the human gene LRRK2 (Leucine-rich repeat kinase 2) [25]. LRRK2 and its various mutants are associated with increased risk of Parkinson's disease (PD) [26]. The LRRK2 G2019S mutation is one of the most common pathogenic factors for the development of PD [27]. Oxidative stress and impairment in mitochondrial function are the pathogenic mechanisms associated with the LRRK2 G2019S mutation [28, 29], both of which are believed to be the fundamental processes which are involved in neurodegenerative disorders [30].
As the mitochondria function is both a toxic target of MeHg and a pathogenic endpoint of the LRRK2 mutations [9, 27], we hypothesized here that the toxic effect of MeHg on mitochondrial function might be modulated by LRRK2. In this study, we took advantage of human LRRK2 transgenic worms to investigate several aspects of MeHg toxicity, including mitochondrial membrane potential, ROS and the expression of skn-1 and gst-4 in a C. elegans model.
Materials and Methods
Caenorhabditis elegans Strains
The C. elegans strains including the wild-type N2 strain, wild-type LRRK2 transgenic worms (WLZ1) and mutant LRRK2(G2019S) transgenic worms (WLZ3) were obtained from the Caenorhabditis Genetic Center (University of Minnesota). Worms were cultured at 20 °C on the nematode growth medium (NGM) plates seeded with OP50 food.
MeHg Exposure
Caenorhabditis elegans has four larvae stages before developing into reproductive adult stage: larvae stage 1 to larvae stage 4 (L1 to L4). Worms hatched from eggs are at L1 stage. The L1 stage worms were harvested from eggs by bleaching worms at gravid stage. The eggs were incubated at 20 °C to get symphonized L1 stage worms. 1,000 L1 stage worms were exposed to MeHg (0, 5, 10 µM) for 1 h in NGM medium (3 g NaCl, 2.5 g peptone, 975 ml H2O, 1 ml cholesterol (5 mg/ml in ethanol), 1 ml nystatin, 1 ml 1 M CaCl2, 1 ml 1 M MgSO4, 25 ml 1 M pH6 KPO4), as reported in our previous study [25]. The doses used in the current study were based on the data on acute toxicity of MeHg reported in our previous study [17].
Lifespan and Development Assessments
Worms were washed with M9 buffer three times following MeHg exposure. They were transferred each day to 30 mm OP50-seeded NGM plates for evaluation of the live status during a period of 24 days. Worms that died of failing to lay eggs or on the border of the plates were not included in the lifespan analyses. For developmental stage assessment, following MeHg exposure, worms were allowed to be fed on 60 mm plates for 24 h. The criteria for evaluation of worm developmental stages were based on the morphology of vulval development, as described in our previous study [31].
Measurement of Mitochondrial Membrane Potential and ROS
For measurement of mitochondrial membrane potential, 5,000 L1 stage worms per group were incubated with 100 nM TMRE (Tetramethylrhodamine ethyl ester) (Cayman Chemical) in 100 µl NGM medium, and then treated with MeHg for 1 h. The fluorescence level was measured at pre- and post- 1 h exposure with the microplate reader (SpectraMax® M3, Molecular Devices) at excitation/emission: 530/580 nm. For ROS, 5,000 L1 stage worms were incubated with 10 µM 2ʹ,7ʹ-Dichlorofluorescin Diacetate (DCF-DA) (Sigma) for 0.5 h in 100 µl NGM medium, followed by 1 h MeHg exposure. The fluorescence level was measured at excitation/emission: 485/535 nm. To accurately measure ROS and mitochondrial membrane potential in live worms is a challenge as the fluorescent probes may not be absorbed well into the body of worms. To overcome this, we used NGM buffer to increase the absorption of probes into the worms. The fluorescent probes for mitochondrial membrane potential and ROS were majorly located in the intestine. We incubated worms in NGM buffer rather than M9 buffer in order for that there is an adequate amount of probes was absorbed in the intestine, which was shown in one of our previous studies [32]. We performed a series of experiments to optimize the conditions, such as washing the probes after the incubation; however, in these cases the fluorescence levels in the experimental groups with worms trended toward that of the blank control without worm present. Therefore, we used a large number of worms for each group.
Gene Expression Assay
The total mRNA was extracted from 1000 worms per group following MeHg exposure. Worms were washed with M9 for three times, and treated with 500 µl TRIzol (ambion, Life Technologies, Grand Island, NY USA). The samples were centrifuged at 16,000 g at 4 °C for 10 min, followed by addition of 100 µl Chloroform (Sigma). The mixture was briefly vortexed three times and centrifuged at 16,000 g at 4 °C for 10 min. The top 50 µl layer of colorless liquid was carefully transferred to a RNAase free 1.5 ml tube. This was followed by the protocol of the RNA cleaning kit (NEB, Monarch RNA Cleanup Kit, Ipswich, MA, USA). All mRNA samples were freshly reverse-transcribed into cDNA with High Capacity cDNA Reverse Transcription kit (Applied Biosystems, Foster City, CA, USA). The housekeeping gene ama-1 was used as loading control [32]. Probes for qPCR were obtained from TaqMan Gene Expression assays (ThermoFisher): skn-1 (Ce02407445_g1), gst-4 (Ce02458730_g1), and ama-1 (Ce02462735_g1).
Feeding RNAi
Synchronized N2, WLZ1 and WLZ3 worms were fed for 60 h with RNAi bacteria (L4440 for control, T19E7.2a for skn-1 RNAi) that were freshly cultured on 60 mm NGM plates with isopropylthio-β-galactoside (IPTG, 1 mM) and carbenicillin (100 µg/ml), followed by harvest of eggs laid by adult worms in 1 h. The L1 stage worms hatched from the eggs in 16 h, and then were treated with MeHg. After 24-h on RNAi bacteria food, the development stage of worms was assessed. In the newly hatched L1 stage worms, the RNAi efficacy for N2, WLZ1 and WLZ3 strains were 86% (± 19%), 77% (± 11%) and 76% (± 14%), respectively (AVOVA test, p = 0.685).
Statistical Analyses
For lifespan data, comparison of median survivals was carried out with Log-rank (Mantel-Cox) test. The Chi-square test was used to analyze the percentage of development stages. The fluorescence data of mitochondrial membrane potential and ROS levels were standardized to the percentage of mean values of the control. Comparison of fluorescence data (Ftreatment/Fcontrol) and gene expression data were analyzed with one-way ANOVA and post hoc Tukey’s multiple comparison analyses. The 2−△△CT method was used to calculate the relative mRNA levels [33]. Two-way ANOVA analysis of Ft/F0 data was used to compare the interactive effect of strain and MeHg. All experiments were repeated for three times. All analyses were performed with Graphpad 8 (La Jolla, CA, USA). Numeric data was expressed as mean ± SD. P < 0.05 was considered as the significant level.
Results
To assess the toxicity of acute MeHg exposure, wild-type N2 worms and human LRRK2 transgenic worms WLZ1 (wild-type LRRK2), and WLZ3 (G2019S mutant LRRK2) were exposed to MeHg (0, 5, 10 µM) for 1 h. Lifespan analysis showed that acute MeHg exposure did not change the lifespan of the N2, WLZ1, or WLZ3 worms (Fig. 1). However, the developmental speed of these worms was significantly affected during the following 24 h period post MeHg exposure (Fig. 2). In N2 worms, both 5 µM and 10 µM MeHg exposure significantly increased the percentage of younger L1 and L2 stage worms (more than 20%, Fig. 2a) 24 h post exposure, while the percentage of younger L2 stage worms in the control was less than 10%. Furthermore, the percentage of younger worms (L1 and L2 stages) in the 10 µM MeHg group was significantly higher than in the 5 µM MeHg group, establishing a dose-dependent effect.

Lifespan of worms is not decreased following acute MeHg exposure. L1 stage N2 (a), WLZ1 (b) and WLZ3 (c) worms were exposed to MeHg (0, 5, 10 µM) for 1 h in NGM medium. Following exposure, 30 ~ 50 worms of each group were picked each day into new 35 mm NGM plates for evaluation of live status (n = 3). Lifespan analysis was made with Log-rank (Mantel-Cox) test (d)

Growth speed of worms is decreased following acute MeHg exposure. L1 stage N2 (a), WLZ1 (b) and WLZ3 (c) worms were exposed to MeHg (0, 5, 10 µM) for 1 h in NGM medium. The developmental stages of worms 24 h post exposure were evaluated in a population of 80–120 worms per group (n = 3). Comparisons between each group were made with Chi-square test. *P < .05, **P < .01, ***P < .001
Both WLZ1 and WLZ3 worms developed slowly than N2 worms after vehicle treatment (Chi-square test, p < 0.001). In the WLZ1 worms, MeHg exposure significantly impeded the developmental progression (Fig. 2b). The percentage of older L4 stage worms in the 10 µM MeHg group was significantly lower than in control worms. Although the percentage of L4 stage worms in the 5 µM MeHg group was lower than in the control worms, it failed to attain statistically significant difference (p = 0.075, Chi-square test). In WLZ3 worms, both 5 µM and 10 µM MeHg significantly increased the percentage of L1 stage worms, which were altogether absent from the control group (Fig. 2c). Notably, all worms of the WLZ3 strain failed to reach the older L4 stage 24 h post MeHg exposure (Fig. 2c), attesting to a developmental delay.
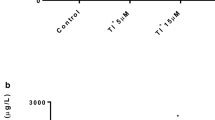
The mitochondria are a crucial target of MeHg-induced toxicity [9, 16]. In N2 worms, 5 µM MeHg exposure significantly decreased mitochondrial membrane potential (Fig. 3a). However, the mitochondrial membrane potential in worms following 10 µM MeHg exposure was statistically indistinguishable from the controls. In WLZ1 worms, the effect of 5 µM MeHg exposure was comparable to that in N2 worms, showing a significant decline in mitochondrial membrane potential. Intriguingly, 10 µM MeHg exposure significantly increased mitochondrial membrane potential, although the size of effect was small (103.3% of the control, Fig. 3b). In the WLZ3 worms, 5 µM MeHg exposure increased mitochondrial membrane potential, but the effect was statistically indistinguishable from the control (p = 0.065, Tukey’s test). However, 10 µM MeHg significantly increased mitochondrial membrane potential in WLZ3 worms (Fig. 3c). Two-way ANOVA analyses showed that there was a significant interactive effect between strain and MeHg on mitochondrial membrane potential (F = 175.200, p < 0.001).

MeHg exposure increases mitochondrial membrane potential in the WLZ1 and WLZ3 worms. L1 stage N2 (a), WLZ1 (b) and WLZ3 (c) worms were incubated with 100 nM TMRE and exposed to MeHg (0, 5, 10 µM) for 1 h. The florescence levels of worms were measured following MeHg exposure. Analysis was made with one-way ANOVA and Tukey’s multiple comparisons test (n = 3). *P < .05, **P < .01, ***P < .001. d, two-way ANOVA analyses of fluorescence data (Ft/F0)
Mitochondrial dysfunction may cause imbalance of ROS, leading to oxidative damage to the animal following MeHg exposure. Additionally, an increase in ROS levels may result from elevated efficiency in mitochondrial respiration that stimulated by a drop in mitochondria membrane potential and energy production [34]. In N2 worms, both 5 µM and 10 µM MeHg significantly increased ROS levels (Fig. 4a). However, ROS levels in the WLZ1 worms were significantly decreased following 10 µM MeHg exposure, compared to both control and 5 µM MeHg-exposed worms (Fig. 4b). In WLZ3 worms, 5 µM MeHg failed to increase ROS levels at the end of 1 h exposure, but 10 µM MeHg significantly decreased ROS levels (Fig. 4c). Two-way ANOVA analyses showed that there was a significant interactive effect between strain and MeHg on the ROS level (F = 31.220, p < 0.001).

MeHg exposure decreases ROS in the WLZ1 and WLZ3 worms. L1 stage N2 (a), WLZ1 (b) and WLZ3 (c) worms were incubated with 10 µM DCF-DA for 0.5 h, followed by exposure to MeHg (0, 5, 10 µM) for 1 h. The florescence levels of worms were measured following MeHg exposure. Analysis was made with one-way ANOVA and Tukey’s multiple comparisons test (n = 3). *P < .05, **P < .01, ***P < .001. d, two-way ANOVA analyses of fluorescence data (Ft/F0)
To investigate the response of detoxifying pathways upon MeHg exposure in N2, WLZ1 and WLZ3 worms, gene expression levels of skn-1 and gst-4 were compared in worms following acute MeHg exposure. Statistical analyses showed that 5 µM MeHg significantly increased the expression of skn-1 (Fig. 5a), but 10 µM MeHg failed to do so in N2 worms. In the WLZ1 worms, neither 5 µM nor 10 µM MeHg had a significant effect on the expression of skn-1 (Fig. 5b). However, in the WLZ3 worms, both 5 µM and 10 µM MeHg significantly increased the expression of skn-1 (Fig. 5c). There was a significant effect between MeHg and strain on the expression of skn-1 (F = 8.225, p = 0.0006).

skn-1 mRNA levels are increased by MeHg exposure in the N2 and WLZ3 worms. The mRNA levels of skn-1 in N2 (a), WLZ1 (b) and WLZ3 (c) were compared following acute MeHg exposure for 1 h. The relative mRNA levels to the housekeeping gene ama-1 was calculated with 2 delta Ct method. Analysis of relative mRNA expression levels were made with one-way ANOVA and Tukey’s multiple comparisons test (n = 3). *P < .05. d Two-way ANOVA analyses of gene expression data
The expression of gst-4 was significantly increased by both 5 µM and 10 µM MeHg in the N2 worms (Fig. 6a). There was no statistically significant difference between 5 µM and 10 µM MeHg. In the WLZ1 worms, 10 µM MeHg significantly increased the expression of gst-4, which was higher than both the control and 5 µM MeHg-exposed worms (Fig. 6b). In the WLZ3 worms, neither 5 µM MeHg nor 10 µM altered the expression of gst-4 (Fig. 6c). There was a significant effect between MeHg and strain on the expression of gst-4 (F = 9.874, p = 0.0002).

gst-4 mRNA levels are increased by MeHg exposure in the N2 and WLZ1 worms. The mRNA levels of gst-4 in N2 (a), WLZ1 (b) and WLZ3 (c) were compared following acute MeHg exposure. The relative mRNA levels to the housekeeping gene ama-1 was calculated with 2 delta Ct method. Analysis of relative mRNA expression levels were made with one-way ANOVA and Tukey’s multiple comparisons test (n = 3). *P < .05, **P < .01. d Two-way ANOVA analyses of gene expression data
To compare the basal expression levels of skn-1 and gst-4 in the N2, WLZ1 and WLZ3 worms, mRNA samples of synchronized L1 stage worms without MeHg exposure were collected for gene expression assays. Statistical analyses showed that the basal skn-1 level was significantly higher in the WLZ1 worms than in the N2 and WLZ3 worms (Fig. 7a). Furthermore, the basal expression level of gst-4 was significantly higher in both the WLZ1 and WLZ3 worms than in the N2 worms (Fig. 7b).

The basal expression levels of skn-1 and gst-4 mRNA are higher in the WLZ1 worms than the N2 worms. The mRNA levels of skn-1 (a) and gst-4 (b) were compared in N2, WLZ1 and WLZ3 worms at L1 stage without MeHg treatment. Analysis of relative mRNA expression levels were made with one-way ANOVA and Tukey’s multiple comparisons test (n = 3). *P < .05, **P < .01
To further investigate the role of skn-1 in MeHg-induced development retardation, worms were fed with RNAi HT115 strain for 60 h. The next generation of synchronized worms was exposed to MeHg. In N2 (Fig. 8a), WLZ1 (Fig. 8b) and WLZ3 worms (Fig. 8c), MeHg caused a significant developmental delay both in worms fed with L4440 or skn-1 RNAi bacteria (P < 0.05, Chi-square test). WLZ1 worms fed with skn-1 RNAi bacteria showed a significant effect of RNAi both in the control and treatment groups (Fig. 8b); however, in N2 worms, skn-1 RNAi caused a significant effect only in the control group (Fig. 8a). In WLZ3 worms, although MeHg exposure caused a significant effect (p < 0.05, Chi-square test), knocking down skn-1 had no significant effect on worms’ development (Fig. 8c).

Knocking down skn-1 alters C. elegans sensitivity to MeHg-induced developmental retardation. N2 (a), WLZ1 (b) and WLZ3 (c) worms fed with RNAi bacteria were assessed for developmental stages 24 h following MeHg exposure. The L4440 RNAi HT115 strain was used as control. The developmental stages of worms were evaluated in a sample of 100–150 worms per group (n = 3).**P < 0.01, ***P < 0.001
Discussion
We showed that worms’ developmental stage was differentially affected by acute MeHg exposure (Fig. 2). Notably, WLZ1 worms with wild-type LRRK2 expression appeared resistant to MeHg (5 µM)-induced developmental delay (Fig. 2). MeHg exposure decreased mitochondrial membrane potential in N2 worms, yet it increased in LRRK2 transgenic worms (Fig. 3). The strain-specific responses of ROS to MeHg suggest mitochondrial dysfunction in the worms expressing the human LRRK2 genes (Fig. 4). In addition, skn-1 expression was noted in N2 and WLZ3 worms exposed to MeHg, but not in WLZ1 worms expressing the wild-type LRRK2, the latter showing a significantly higher level of skn-1 expression at basal conditions. In contrast to the N2 and WLZ1 worms, the expression of gst-4 in the WLZ3 worms was not induced in response to MeHg exposure. gst-4 levels had higher basal expression levels in the LRRK2 transgenic vs. the wild-type N2 worms (Fig. 7). Knocking down skn-1 in WLZ1 worms decreased development speed (Fig. 8). These results revealed that mitochondrial dysfunction in LRRK2 (G2019S) worms might contribute to the severe developmental delay, establishing a modulatory role of the LRRK2 mutation in MeHg-induced developmental delay. Furthermore, the relative higher expression level of skn-1 and gst-4 in wild-type LRRK2 transgenic worms might potentiate the detoxication process following acute MeHg exposure. The failed response of gst-4 to MeHg exposure in the LRRK2 (G2019S) worms suggests that the mutant LRRK2 may cause a defect in SKN-1 signaling.
The toxicity of MeHg in children is characterized by delayed achievement of developmental milestones following MeHg exposure [6]. The manifestation of developmental MeHg toxicity is modified by genetic factors that are involved in cellular detoxification processes [24]. Herein, we showed that acute MeHg exposure in the early developmental stages of C. elegans significantly decreased the developmental progression, and human wild-type LRRK2 transgenic worms were somewhat resistant to the effect of a moderate dose of MeHg (5 µM) on development progression (Fig. 2). Further, it has been shown that wild-type LRRK2 transgenic worms have slightly yet significantly longer lifespan than wild-type N2 and LRRK2(G2019S) transgenic worms following chronic treatment with rotenone (25 µM), an inhibitor of complex I of the mitochondrial respiratory chain [28]. In addition, in the G2019S mutant of LRRK2 dopaminergic neurotoxicity is exacerbated in response to mitochondria dysfunction by rotenone exposure [28]. In the acute MeHg toxicity model reported herein, we further corroborated that wild-type LRRK2 afforded a protective effect against MeHg-induced toxicity, possibly by preservation of mitochondrial function and antioxidant capacity, while the G2019S mutant LRRK2 exhibited impaired mitochondrial function and antioxidant signaling.
We further showed that the mitochondrial membrane potential was deferentially regulated in N2 and WLZ1 vs. WLZ3 worms (Fig. 3). Our results are in agreement with previous studies on the propensity of MeHg to compromise mitochondrial membrane polarity by disrupting the proton pump activity of the respiratory complexes and the integrity of the mitochondrial membrane [14, 35]. The failed response of mitochondrial membrane potential after 1 h exposure to 10 µM MeHg could result from a stronger inhibitory effect of MeHg on ATP production which, in turn, would trigger respiratory control in the mitochondria to keep the membrane polarity for restoration of ATP supply. Although a dynamic change in mitochondria membrane potential by MeHg could provide an unequivocal clue for the dose-dependent responsiveness to this organometal, the similar pattern between N2 and WLZ1 worms suggests that there could be a potentiated feedback-control mechanism to counter the depolarization effect of MeHg in the mitochondria. It is also noteworthy that the effect was stronger in WLZ1 worms. Our results are different from previous studies showing that higher MeHg doses had a stronger effect on depolarization of mitochondrial membrane potential [11, 35], likely because our results were based on in vivo study in live animals and a short exposure time of 1 h. Nonetheless, the current study is consistent with a previous report showing that 5 µM MeHg decreased the fluorescence level of mitochondrial dye by decreasing mitochondrial membrane potential [12].
The relatively unchanged mitochondrial membrane potential in the WLZ3 worms might be related to the fact that the mutant LRRK2(G2019S) might have had a depolarization effect, as revealed in previous studies [36, 37]. Accordingly, the effect of MeHg exposure at 5 µM might not be able to further reduce the mitochondrial membrane potential, as this could trigger the opening of the mitochondrial permeability transition pore and ensuing cell death [38]. Ca2+ entry into the mitochondrial matrix driven by mitochondrial membrane potential is tightly coupled to many processes in the organelle: regulation of metabolic enzymes, ROS production, and calcium signaling [39]. It is well documented that MeHg exposure impairs mitochondrial buffering capacity for Ca2+ by inducing Ca2+ efflux from the mitochondria [40]. The collapse of mitochondrial membrane potential is believed to be the primary cause of MeHg-induced calcium efflux [40]. It is possible that during the early phases of MeHg exposure, a mild depolarization triggered calcium efflux and restoration of mitochondrial membrane potential. Therefore, the increase in mitochondria membrane potential should be a temporarily adaptive change, and a longer exposure period to MeHg, for example, more than 1 h exposure to 10 µM, would eventually cause the collapse of mitochondrial membrane potential. Additionally, the increase in the mitochondrial membrane potential after 1 h exposure to 10 µM MeHg might be caused by a more potent effect of the higher dose on mitochondrial Ca2+ efflux [40], to counterbalance the depolarization effect of MeHg. It has been previously proposed that a mild decrease in mitochondrial membrane potential may be beneficial to the health of the mitochondria [41]. However, a large increase in mitochondrial membrane potential is deemed detrimental [42], contributing to the more severe developmental damage inherent to the LRRK2(G2019S) worms (Fig. 2).
The increase in ROS levels by MeHg might result from a feedback stimulatory effect on the mitochondrial respiratory rate in response to a drop in ATP production, a defect in the electron transport, or depletion of antioxidant defense system in the organelle (or a combination thereof) [9, 13, 16, 42]. Therefore, further investigations into the kinetics of mitochondrial parameters are warranted to better characterize sequential events that contribute to ROS production. We speculated that the increased ROS production could occur in the initial phase of the mitochondrial response to MeHg, as the driving force for mitochondrial respiration, namely, mitochondrial membrane potential, had already been reduced or unchanged following 1 h exposure to the organometal (Fig. 3). The diverse responses of ROS between the N2 and LRRK2 transgenic worms suggests that LRRK2 had a significant effect on mitochondrial function (Fig. 4), which might be caused by altered capacity for Ca2+ buffering in the LRRK2 transgenic worms. Consistent with this observation, in primary mouse cortical neurons, expression of the mutant LRRK2 (G2019S) upregulated the mitochondrial calcium uniporter (MCU) and the mitochondrial calcium uptake 1 protein (MICU1), along with increased mitochondrial Ca2+ uptake [43].
The most severe developmental delay was inherent to LRRK2(G2019S) transgenic worms, as these worms failed to reach the L4 stage, compared to the N2 and wild-type LRRK2 transgenic worms (Fig. 2). In a study comparing the nrf-2 expression in dermal fibroblasts from individuals of the mutant LRRK2(G2019S), it has been shown that the expression of nrf-2 was invoked in the G2019S LRRK2 carriers experiencing PD symptoms compared to non-carriers and G2019S LRRK2 carriers absent PD symptoms [44]. Herein, we showed that the expression of skn-1 was significantly increased in LRRK2(G2019S) transgenic worms (Fig. 5), establishing that the antioxidant defense system was upregulated in response to MeHg toxicity. However, gst-4 was not significantly induced in response to MeHg (Fig. 6), which could lead to a sustained damage that manifested in developmental delay in the LRRK2(G2019S) transgenic worms as compared to wild-type LRRK2 transgenic worms. In the N2 and wild-type LRRK2 transgenic worms, 10 µM MeHg failed to induce expression of skn-1 (Fig. 5). However, gst-4 expression was significantly increased in these worms when exposed to 10 µM MeHg (Fig. 6). SKN-1 levels are negatively regulated by degradation of the protein in basal conditions [20]. It is most likely that SKN-1 is preferentially regulated by its turnover rate rather than transcriptional program. The negative regulation, rather than the energy-consuming transcriptional program, could facilitate a fast response for mobilization of target genes to encounter toxic effects of xenobiotics. In the WLZ3 worms, although skn-1 expression was significantly higher, gst-4 expression was not increased (Fig. 5c and 6c), suggesting the SKN-1 signaling may be disrupted in these animals. Furthermore, the more severe developmental effect of skn-1 knockdown in WLZ1 worms compared with N2 worms suggests that the increased expression of skn-1 in WLZ1 worms may play a significant regulatory role in the worms (Fig. 7 and Fig. 8), which may not be interrupted by MeHg exposure.
Although LRRK2(G2019S) is the most common genetic risk factor for PD, most PD cases are idiopathic, suggesting an environmental origin of pathogenic factors for PD. Exposure to neurotoxic chemicals is believed to be involved in the etiology of idiopathic PD. In the acute MeHg toxicity model reported herein, we showed that the LRRK2(G2019S) transgenic worms had a significant developmental delay, impairment in mitochondrial functions and disrupted antioxidant signaling following MeHg exposure. Our results highlight a mechanistic link between genetic and environmental factors in mitochondrial toxicity and neurodegenerative diseases. Further studies on the mechanisms of altered mitochondrial functions by LRRK2 are warranted in the context of MeHg as well as other environmental scenarios associated with increased risk for PD.
The datasets generated during and/or analyzed during the current study are available from the corresponding author on reasonable request.
References
Sheehan MC, Burke TA, Navas-Acien A, Breysse PN, McGready J, Fox MA (2014) Global methylmercury exposure from seafood consumption and risk of developmental neurotoxicity: a systematic review. Bull World Health Organ 92:254–269f
Aschner M, Clarkson TW (1989) Methyl mercury uptake across bovine brain capillary endothelial cells in vitro: the role of amino acids. Pharmacol Toxicol 64:293–297
Aschner M, Eberle NB, Goderie S, Kimelberg HK (1990) Methylmercury uptake in rat primary astrocyte cultures: the role of the neutral amino acid transport system. Brain Res 521:221–228
Pacyna JM, Sundseth K, Pacyna EG, Jozewicz W, Munthe J, Belhaj M, Aström S (2010) An assessment of costs and benefits associated with mercury emission reductions from major anthropogenic sources. J Air Waste Manag Assoc 60:302–315
Schartup AT, Thackray CP, Qureshi A, Dassuncao C, Gillespie K, Hanke A, Sunderland EM (2019) Climate change and overfishing increase neurotoxicant in marine predators. Nature 572:648–650
Myers GJ, Davidson PW (1998) Prenatal methylmercury exposure and children: neurologic, developmental, and behavioral research. Environ Health Perspect 106(Suppl 3):841–847
Davidson PW, Myers GJ, Weiss B, Shamlaye CF, Cox C (2006) Prenatal methyl mercury exposure from fish consumption and child development: a review of evidence and perspectives from the Seychelles Child Development Study. Neurotoxicology 27:1106–1109
Weiss B, Clarkson TW, Simon W (2002) Silent latency periods in methylmercury poisoning and in neurodegenerative disease. Environ Health Perspect 110(Suppl 5):851–854
Roos D, Seeger R, Puntel R, Vargas Barbosa N (2012) Role of calcium and mitochondria in MeHg-mediated cytotoxicity. J Biomed Biotechnol 2012:248764
Nesci S, Trombetti F, Pirini M, Ventrella V, Pagliarani A (2016) Mercury and protein thiols: stimulation of mitochondrial F(1)F(O)-ATPase and inhibition of respiration. Chem Biol Interact 260:42–49
Yin Z, Milatovic D, Aschner JL, Syversen T, Rocha JB, Souza DO, Sidoryk M, Albrecht J, Aschner M (2007) Methylmercury induces oxidative injury, alterations in permeability and glutamine transport in cultured astrocytes. Brain Res 1131:1–10
Ke T, Aschner MJN (2019) Bacteria affect Caenorhabditis elegans responses to MeHg toxicity. Neurotoxicology 75:129–135
LeBel CP, Ali SF, McKee M, Bondy SC (1990) Organometal-induced increases in oxygen reactive species: the potential of 2’,7’-dichlorofluorescin diacetate as an index of neurotoxic damage. Toxicol Appl Pharmacol 104:17–24
Mori N, Yasutake A, Marumoto M, Hirayama K (2011) Methylmercury inhibits electron transport chain activity and induces cytochrome c release in cerebellum mitochondria. J Toxicol Sci 36:253–259
Barbosa NV, Nogueira CW, Nogara PA, de Bem AF, Aschner M, Rocha JBT (2017) Organoselenium compounds as mimics of selenoproteins and thiol modifier agents. Metallomics 9:1703–1734
Farina M, Aschner M, Rocha JB (2011) Oxidative stress in MeHg-induced neurotoxicity. Toxicol Appl Pharmacol 256:405–417
Ke T, Bornhorst J, Schwerdtle T, Santamaría A, Soare FAA, Rocha JB, Farina M, Bowman AB, Aschner MJNR (2020) Therapeutic Efficacy of the N, N′ Bis-(2-Mercaptoethyl) Isophthalamide Chelator for Methylmercury Intoxication in Caenorhabditis elegans.1–12
Caito SW, Zhang Y, Aschner M (2013) Involvement of AAT transporters in methylmercury toxicity in Caenorhabditis elegans. Biochem Biophys Res Commun 435:546–550
Vanduyn N, Settivari R, Wong G, Nass R (2010) SKN-1/Nrf2 inhibits dopamine neuron degeneration in a Caenorhabditis elegans model of methylmercury toxicity. Toxicol Sci 118:613–624
Unoki T, Akiyama M, Kumagai Y, Gonçalves FM, Farina M, da Rocha JBT, Aschner M (2018) Molecular pathways associated with methylmercury-induced Nrf2 modulation. Front Genet 9:373
Ni M, Li X, Yin Z, Jiang H, Sidoryk-Wegrzynowicz M, Milatovic D, Cai J, Aschner M (2010) Methylmercury induces acute oxidative stress, altering Nrf2 protein level in primary microglial cells. Toxicol Sci 116:590–603
Hayes JD, Dinkova-Kostova AT (2014) The Nrf2 regulatory network provides an interface between redox and intermediary metabolism. Trends Biochem Sci 39:199–218
Bento-Pereira C, Dinkova-Kostova AT (2020) Activation of transcription factor Nrf2 to counteract mitochondrial dysfunction in Parkinson’s disease. Med Res Rev 41(2):785–802
Vorojeikina D, Broberg K, Love TM, Davidson PW, van Wijngaarden E, Rand MD (2017) Editor’s highlight: glutathione S-transferase activity moderates methylmercury toxicity during development in Drosophila. Toxicol Sci 157:211–221
Ke T, Santamaria A, Rocha JBT, Tinkov AA, Lu R, Bowman AB, Aschner M (2020) The role of human LRRK2 in methylmercury-induced inhibition of microvesicle formation of cephalic neurons in Caenorhabditis elegans. Neurotoxicol Res 38:751–764
Tolosa E, Vila M, Klein C, Rascol O (2020) LRRK2 in Parkinson disease: challenges of clinical trials. Nat Rev Neurol 16:97–107
Howlett EH, Jensen N, Belmonte F, Zafar F, Hu X, Kluss J, Schule B, Kaufman BA, Greenamyre JT, Sanders LH (2017) LRRK2 G2019S-induced mitochondrial DNA damage is LRRK2 kinase dependent and inhibition restores mtDNA integrity in Parkinson’s disease. Hum Mol Genet 26:4340–4351
Saha S, Guillily MD, Ferree A, Lanceta J, Chan D, Ghosh J, Hsu CH, Segal L, Raghavan K, Matsumoto K, Hisamoto N, Kuwahara T, Iwatsubo T, Moore L, Goldstein L, Cookson M, Wolozin B (2009) LRRK2 modulates vulnerability to mitochondrial dysfunction in Caenorhabditis elegans. J Neurosci 29:9210–9218
Loeffler DA, Klaver AC, Coffey MP, Aasly JO, LeWitt PA (2017) Increased oxidative stress markers in cerebrospinal fluid from healthy subjects with Parkinson’s disease-associated LRRK2 gene mutations. Front Aging Neurosci 9:89
Lionaki E, Markaki M, Palikaras K, Tavernarakis N (2015) Mitochondria, autophagy and age-associated neurodegenerative diseases: new insights into a complex interplay. Biochim Biophys Acta 1847:1412–1423
Ke T, Antunes Soares FA, Santamaría A, Bowman AB, Skalny AV, Aschner M (2020) N, N’ bis-(2-mercaptoethyl) isophthalamide induces developmental delay in Caenorhabditis elegans by promoting DAF-16 nuclear localization. Toxicol Rep 7:930–937
Ke T, Tsatsakis A, Santamaría A, Soare FAA, Tinkov AA, Docea AO, Skalny A, Bowman AB, Aschner MJN (2020) Chronic exposure to methylmercury induces puncta formation in cephalic dopaminergic neurons in Caenorhabditis elegans. Neurotoxicology 7:105–113
Livak KJ, Schmittgen TD (2001) Analysis of relative gene expression data using real-time quantitative PCR and the 2(T)(-Delta Delta C) method. Methods 25:402–408
Shabalina IG, Nedergaard J (2011) Mitochondrial ('mild’) uncoupling and ROS production: physiologically relevant or not? Biochem Soc Trans 39:1305–1309
Hare MF, Atchison WD (1992) Comparative action of methylmercury and divalent inorganic mercury on nerve terminal and intraterminal mitochondrial membrane potentials. J Pharmacol Exp Ther 261:166–172
Papkovskaia TD, Chau KY, Inesta-Vaquera F, Papkovsky DB, Healy DG, Nishio K, Staddon J, Duchen MR, Hardy J, Schapira AH, Cooper JM (2012) G2019S leucine-rich repeat kinase 2 causes uncoupling protein-mediated mitochondrial depolarization. Hum Mol Genet 21:4201–4213
Mortiboys H, Johansen KK, Aasly JO, Bandmann O (2010) Mitochondrial impairment in patients with Parkinson disease with the G2019S mutation in LRRK2. Neurology 75:2017–2020
Scorrano L, Petronilli V, Bernardi P (1997) On the voltage dependence of the mitochondrial permeability transition pore. A critical appraisal. J Biol Chem 272:12295–12299
Duchen MR (2000) Mitochondria and calcium: from cell signalling to cell death. J Physiol 529(Pt 1):57–68
Levesque PC, Atchison WD (1991) Disruption of brain mitochondrial calcium sequestration by methylmercury. J Pharmacol Exp Ther 256:236–242
Padalko VI (2005) Uncoupler of oxidative phosphorylation prolongs the lifespan of Drosophila. Biochemistry (Mosc) 70:986–989
Marty MS, Atchison WD (1997) Pathways mediating Ca2+ entry in rat cerebellar granule cells following in vitro exposure to methyl mercury. Toxicol Appl Pharmacol 147:319–330
Verma M, Callio J, Otero PA, Sekler I, Wills ZP, Chu CT (2017) Mitochondrial calcium dysregulation contributes to dendrite degeneration mediated by PD/LBD-associated LRRK2 mutants. J Neurosci 37:11151–11165
Delcambre S, Ghelfi J, Ouzren N, Grandmougin L, Delbrouck C, Seibler P, Wasner K, Aasly JO, Klein C, Trinh J, Pereira SL, Grünewald A (2020) Mitochondrial mechanisms of LRRK2 G2019S penetrance. Front Neurol 11:881
Acknowledgements
This work was supported by the National Institutes of Health to MA and ABB (NIEHS R01ES007331). Some strains were provided by the CGC, which is funded by NIH Office of Research Infrastructure Programs (P40 OD010440).
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
The authors declare no conflict of interest.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
About this article

Cite this article
Ke, T., Rocha, J.B.T., Tinkov, A.A. et al. The Role of Human LRRK2 in Acute Methylmercury Toxicity in Caenorhabditis elegans. Neurochem Res 46, 2991–3002 (2021). https://doi.org/10.1007/s11064-021-03394-y
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s11064-021-03394-y