
Abstract
Gamma-decanolactone (GD) has been shown to reduce epileptic behavior in different models, inflammatory decreasing, oxidative stress, and genotoxic parameters. This study assessed the GD effect on the pentylenetetrazole (PTZ) model after acute and subchronic treatment. We evaluated the expression of the inflammatory marker cyclooxygenase-2 (COX-2), GluN2B, a subunit of the NMDA glutamate receptor, adenosine A1 receptor, and GD genotoxicity and mutagenicity. Male and female mice were treated with GD (300 mg/kg) for 12 days. On the tenth day, they were tested in the Hot Plate test. On the thirteenth day, all animals received PTZ (90 mg/kg), and epileptic behavior PTZ-induced was observed for 30 min. Pregabalin (PGB) (30 mg/kg) was used as a positive control. Samples of the hippocampus and blood were collected for Western Blotting analyses and Comet Assay and bone marrow to the Micronucleus test. Only the acute treatment of GD reduced the seizure occurrence and increased the latency to the first stage 3 seizures. Males treated with GD for 12 days demonstrated a significant increase in the expression of the GluN2B receptor and a decrease in the COX-2 expression. Acute and subchronic treatment with GD and PGB reduced the DNA damage produced by PTZ in males and females. There is no increase in the micronucleus frequency in bone marrow after subchronic treatment. This study suggests that GD, after 12 days, could not reduce PTZ-induced seizures, but it has been shown to protect against DNA damage, reduce COX-2 and increase GluN2B expression.
Similar content being viewed by others
Avoid common mistakes on your manuscript.
Introduction
Epilepsy is the most common neurological disease globally, and around 50 million people are affected by this [1]. One of the essential characteristics of this pathology is the occurrence of seizures, a behavioral manifestation generated by abnormal neuronal synchronization and activity [2], in addition to cognitive changes, behavioral and psychological impairments [3]. Currently, several cellular processes are associated with epilepsy and have helped to elucidate their pathophysiology.
Neuroinflammation has an essential role in epilepsy pathogenesis. The increase in neuronal activity during seizures could increase chemical mediator release, leading to intense excitability of affected tissue. This intense excitability is responsible for extending and intensify the seizure activity [4,5,6].
The cyclooxygenase-2 (COX-2) is an inducible enzyme involved in the neuroinflammatory process by converting arachidonic acid into prostaglandins. Its expression contributes not only to repair but also to injury and chronicity [7, 8]. This inducible enzyme is expressed in several pathological processes, such as inflammation and pain [9]. Studies that evaluated neuroinflammation in epilepsy models demonstrated that seizure induction up-regulates the expression of COX-2. On the other hand, neuroprotection and increased latency for the seizures can be observed when COX-2 is inhibited [10,11,12].
Anti-inflammatory drugs have a neuroprotective effect on PTZ- induced seizures, and the effects appear to be related to the inhibition of cytokines and the COX-2. After the Kindling PTZ model, a decrease in Interleukin 1β (IL-1β) and Tumor Necrosis Factor α (TNF-α) was demonstrated, as well as less seizure severity in rats treated with dexamethasone [13]. It was also observed that sodium diclofenac (a COX-2 inhibitor) was able to decrease TNF-α and Interleukin 6 (IL-6) in the hippocampus of rats [14].
Several antiseizure drugs are available for therapeutic use. Among the most recent antiseizure drugs, pregabalin (PGB) is a GABA analog approved by the Food and Drug Administration (FDA) in 1999 [15] for refractory epilepsy treatment. It has shown the effects of monotherapy and adjuvant therapy for focal and generalized seizures [16]. PGB is also given to chronic pain, mainly neuropathic pain [17,18,19].
The significant adverse effects of traditional antiseizure drugs, the difficulty of finding treatments that suit all patients, and the cost–benefit of standard treatments, have increased the demand for new drugs. Several natural products can influence GABAergic function. Some compounds have also been shown to reduce the frequency of epileptic seizures in models of epilepsy, also having neuroprotective effects [20]. Lactones are among the organic compounds that have bioactive properties that act on the central nervous system. Such substances have been recognized for some properties such as anesthetic, anti-inflammatory, and hypnotic activity [21,22,23,24].
GD, a synthetic monoterpene lactone, has been evaluated for its neuroprotective ability in different animal models [25,26,27,28]. Studies carried out by our group investigated the effect of GD in a pentylenetetrazole (PTZ) model, both acute and chronic, and seizure-induced by Pilocarpine, 4-aminopyridine, and isoniazid [25, 27, 28]. GD has been shown to reduce seizures and increase the latency for the first seizure (stage 3) in these models, demonstrating a significant neuroprotective effect. Also, an in vitro study conducted by our group demonstrated that GD inhibited TNF-α and the intracellular formation of reactive oxygen species (ROS), decreasing genotoxicity and apoptosis in microglial cells [26].
Although several behavioral studies of the GD effect have been carried out, its ability to modulate neurotransmission systems is still scarce. Pereira et al. 1997 [29] demonstrated GD's inhibitory effect on glutamate binding in cortex samples.
N-methyl-d-aspartate receptors (NMDARs) are ionotropic glutamate receptors (iGluRs) that play essential roles in physiology and pathophysiology of mammalian CNS [30]. Studies have indicated that NMDARs play different roles under different conditions, for instance, under normal activation and overactivation. Under normal activation, activated GluN2B is mainly located in the synapses. In contrast, under overactivation, extrasynaptic GluN2B will be additionally activated [31,32,33,34]. The NMDARs subunits, GluN2A, and GluN2B are the main in the adult forebrain. GluN2B has a bidirectional activity of the signaling of ERK, CREB, BDNF, and PI3K. Moreover, GluN2B may play a more active role in changing the NMDARs effect from normal activation to overactivation [35].
Adenosine is a crucial neuromodulator known for its homeostatic and endogenous anticonvulsant effects on neural network activities. It is deficiency could conduce to excitotoxicity in patients with epilepsy [36,37,38]. The known adenosine anticonvulsant effect is attributed to the adenosine A1 receptor, which is distributed throughout the brain, and in the hippocampus, there is a high density [38,39,40].
Thus, studies aiming to investigate NMDA and Adenosine's role in the GD mechanism of action are necessary. Besides, behavioral studies carried out to date have only evaluated GD's acute effect, and it is also essential to demonstrate its effects after repeated administrations. Therefore, this study aimed to evaluate a possible GD neuroprotective effect after subchronic treatment using the PTZ-induced seizure and compare it to the acute treatment. Also, inflammatory marker expression COX-2, mutagenicity, genotoxicity, and a possible GluN2B and adenosine A1 modulation were investigated.
Material and Methods
Animals
CF-1 mice were obtained from the Reproduction and Research Center (CREAL) at the Federal University of Rio Grande do Sul (UFRGS). To conduct this study, we needed 87 male and 87 female CF-1 mice. Five of each sex were driven to pilot test. All of them were 45 days old. 4–5 animals were housed per box (33 × 17 × 40 cm), with food and water ad libitum, under a 12-h light/dark cycle (lights at 7:00 am) and a constant temperature 23 ± 2 °C at CREAL, UFRGS. The Ethics Committee on Animal Use (CEUA-UFRGS) approved the project, under number 36326/2019. The entire experiment was conducted following the National Council for Animal Experimentation Control (CONCEA), prioritizing animal comfort and respect.
Drugs and Pharmacological Procedures
GD and PTZ were purchased from Sigma-Aldrich (St. Louis, USA). GD was solubilized in tween 80 (5%) and PTZ in saline solution (0.9%). According to our previous studies, a dose of 300 mg/kg of GD was used [24, 41]. A pilot test and previous studies were considered to define the dose of 90 mg/kg PTZ, used as a convulsant agent [24]. PGB (Prefiss®; Aché Laboratórios Farmacêuticos S.A), has been used to treat epilepsy and chronic pain, and in this study, it was used for comparison purposes in subchronic treatment. It was solubilized in tween 80 (5%) and administered at a dose of 30 mg/kg [16]. Tween 80 (5%) and saline (0.9%) were administered as controls. All drugs were administered intraperitoneally (i.p), except PTZ, which was given subcutaneously (s.c) at a volume of 10 ml/kg body weight.
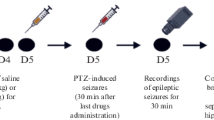
The animals were randomly divided into seven groups: 12 animals for Saline; Sal12; Tween; PGB; GD 12 and GD 1 and 10 animals for Sal 1 group. The experimental design of this study is shown in Fig. 1. The experiment was conducted for 13 days. Pharmacological administrations were always performed in the morning, between 7 am and 11 am. The registration of the animals' weight was every 3 days.

The scheme of experimental design. The mice were daily treated with SAL, Tween, PGB, or GD. On the tenth day, we performed the Hot Plate test. On the thirteenth day, we performed acute treatments with SAL or GD. Immediately after observing epileptic behavior, the animals were killed, and the hippocampus, blood, and marrow samples were collected. i.p (intraperitoneal injections), s.c (subcutaneous injections)
PTZ-Induced Seizure Model and Samples Collection
In the acute trial, Sal or GD was administered 30 min before PTZ injection. For the subchronic evaluation, after 12 days of treatment, the animals were submitted to the PTZ-induced seizure model of the thirteenth day, 1 day after the last drug administration. In the acute and subchronic tests, the animals were observed for 30 min by a blinded researcher who recorded the behavior according to an adapted Racine scale [42]. The adapted scale consists of five stages: 0—no epileptic behavior; 1—Jerkings, playing piano; 2—clonic forelimb seizures lasting less than 3 s; 3—clonic forelimb seizures lasting more than 3 s; 4—generalized seizures with tonic extension episodes and full status epilepticus; 5—death. The latency for the first clonic forelimb seizure (stage 3) and the percentage of occurrence of these seizures were recorded. We used stage 3 seizure for comparison purposes since it is evident and easy to observe in mice. At the end of the treatments, the animals were killed by decapitation, and the hippocampus samples were collected immediately, using surgical instruments on a frozen platform. The hippocampus samples were identified in individual tubes and kept on -80ºC until the tests described in Fig. 1 [43].
Hot Plate Test
The Hot Plaque Test was used to assess central analgesia in treating animals [44], considering that some antiseizure drugs are administered to treat chronic pain [45,46,47]. The test protocol was applied on the tenth day of the experiment, after 30, 60, 90, and 120 min of pharmacological administration. 24 h before the test, the animals were placed under the off plate for habituation [48, 49]. The test was performed with a 55 ± 0.5 °C plate, and the researcher was blinded. The recorded behaviors were: changing feet (tapping), jumping, and standing or licking one of the paws [50]. According to previous studies, the nociceptive limit was 20 s per animal to avoid tissue damage [49, 51, 52].
Western Blotting Assay
Frozen hippocampus samples were homogenized in lysis buffer (150 mM NaCl, 20 mM Tris, 5 mM EDTA, 10% glycerol, 10% NP 40, pH 7.4) centrifuged at 7000 g for 15 min at 4 °C. The supernatant was collected, and the protein concentration was determined by the Bradford method [43] using BSA (1 mg/ml) as standard. A sample of 80 μg of total proteins was separated by electrophoresis on 12% sodium dodecyl sulfate–polyacrylamide gel (SDS-PAGE). After electrophoresis, the proteins were transferred to a nitrocellulose membrane. Membranes were blocked with a 1% BSA for 1 h at room temperature and then were incubated with the primary rabbit polyclonal antibodies overnight at 4 °C. For these experiments, we used ant-GluN2B (190 kDa), anti-adenosine A1 (37 kDa), anti-COX-2 (69 kDa), and anti-βactin (45 kDa), the concentration of antibodies was: 1:1000. After incubation, the membranes were washed three times in TTBS (Tris Buffer Salina with 0,05% Tween 20) and then incubated with peroxidase-bound anti-rabbit secondary antibody (1:5.000) at room temperature for 2 h. The membranes were washed with TBS three times for 10 min. Protein bands were then visualized using an Enhanced Chemiluminescent Reagent (ECL) in the iBright™ Imaging System, then quantified by ImageJ 1.52a software.
Alkaline Comet Assay
The Alkaline Comet assay was carried out to evaluate genotoxic effects following the recommendation described by Tice et al. [53], with minor modification [54]. The samples of peripheral blood and hippocampus were collected immediately after epileptic behavior observation. Blood samples (50 lL) were transferred to heparin tubes (25.000 UI, 10 lL), and the hippocampus samples were transferred to PBS buffer. Cell suspension (10 lL) was embedded in 90 lL low melting point agarose 0.75% (GibcoBRL) and spread onto agarose-precoated microscope slides. After solidification, slides were placed in lysis buffer (2.5 M NaCl, 100 Mm EDTA, and 10 mM Tris, freshly added 1% Triton X-100 (Sigma) and 10% DMSO, pH 10) for 48 h at 4 °C. The slides were incubated in a freshly prepared alkaline buffer (300 mM NaOH and 1 mM EDTA, pH > 13) for 20 min at 4 °C in an electrophoresis cube. An electric current of 300 mA at 25 V (0.90 V/cm) was applied for 15 min to induce DNA electrophoresis. The slides were then neutralized (0.4 M Tris, pH 7.5), stained with silver, and analyzed under a microscope. Images of 100 randomly selected cells (50 cells from each slide) were analyzed from each duplicated sample. Cells were visually scored according to tail size into five classes, ranging from undamaged (0) to maximum damaged (4), resulting in a single DNA-damage score for each sample. Therefore, the damage index (DI) ranged from 0 (undamaged, 100 cells × 0) to 400 (with maximum damage, 100 × 4). Damage frequency (DF) was calculated based on the number of cells with tail versus those with no tail [55].
Micronucleus (MN) Test in Bone Marrow
The MN test was performed according to the US Environmental Protection Agency Gene-Tox Program guidelines [56]. Bone marrow from both femurs was suspended in fetal calf serum, and smears were prepared on clean glass slides. Slides were air-dried, fixed in methanol, stained in 10% Giemsa and coded for blind analysis. The polychromatic erythrocyte/normochromatic erythrocyte (PCE/NCE) ratio was determined in 1000 cells to avoid false-negative results and obtain a value of bone marrow toxicity. The incidence of MN was observed in 2000 PCE for each animal using bright-field optical microscopy at ×1000 magnification.
Statistical Analyses
RM two-way ANOVA followed by Tukey's test was applied to analyze mice's weight. The Generalize Estimate Equation (GEE) was applied to analyze the Hot Plate data. The behavior data and Western Blotting results were analyzed by one-way ANOVA followed by Tukey's test. Alkaline Comet Assay and Micronucleus test were analyzed using the one-way ANOVA followed by Dunnett's test. Two-way ANOVA followed by Tukey's was used to compare sexes, 2 × 2 Fisher's Exact Test was used to compare the occurrence of seizures in percentage (%). GraphPad Prism 7.0 software was used for all variance analyses, and IBM SPSS Statistics 20 Software was used for GEE analysis.
Results
GD Induces Weight Change
In this study, the animal's body weight was recorded throughout the treatment. GD decreased their body weight significantly when compared to the other groups at the third and fourth weights measurements in males [Fig. 2A, F(3,44) = 4.01, P = 0.0132 RM two-way ANOVA] and only on the fourth weights measurements in females [Fig. 2B, F(3,44) = 1.488, P = 0.2309 RM two-way ANOVA].

Weight measurement of males (A) and females (B) during the 12 days of treatment. Weights measurements 1, 2, 3, 4 were performed on experimental days 3, 6, 9, 12, respectively. A F(3, 44) = 4.01, P = 0.0132. B F(3,44) = 1.488, P = 0.2309. RM two-Way ANOVA, n = 12. Results of Tukey’s test is represented as *P < 0.05, **P < 0.01, ***P < 0.001. Values represent means ± SEM
GD Did Not Affect Nociception on the Hot Plate Test
The Hot Plate Test was used to evaluate the antinociceptive activity of GD. This test assesses when animals remain on a metal plate heated until they react to the thermal stimulus. The GEE presented no significant interaction between treatment and time measurements (X2 = 14.963, P = 0.092), as shown in Fig. 3A and B. However, there was an increase in the latency to nociceptive behaviors in females (X2 = 7.774, P = 0.005 data not shown), independent of group and time (X2 = 16.269, P = 0.179 data not shown).

The latency of male (A) and female (B) to Hot Plate response. The evaluations were performed on the tenth day of the treatment, at 30, 60, 90, and 120 min after administering the drugs. No significant interaction between treatment and time measurements was observed (X2 = 14.963, P = 0.092), as shown in A and B. Generalized Estimating Equation, values represent means ± SEM, n = 12
Effect of GD Subchronic and Acute Treatments in Latency to the First Stage 3 Seizure and Their Occurrence
Both male and female mice that received GD, showed no significant difference in the latency to stage 3 seizure [Fig. 4A, males: F(3,41) = 0.09356, P = 0.4322, n = 9–12; females: F(3,44) = 1.205, P = 0.3189, n = 12, one-way ANOVA] there was no sex difference as well [F(1,85) = 3.628, P = 0.0602 two-way ANOVA]. There was no difference in the seizure occurrence between groups compared by 2 × 2 Fisher's Exact Test (Fig. 4B, P > 0.05 for all comparison, n = 9–12), animals that died after the occurrence of stage 3 seizures were excluded from the analysis. Unlike subchronic treatment, a single administration of GD increased the latency for stage 3 seizures in males and in females [Fig. 5A, males: F(3,37) = 9.628, P = < 0.0001, n = 9–12; females: F(3,42) = 8.193, P = 0.0002, n = 10–12], one-way ANOVA, without difference between sex F(1,79) = 0.1398, P = 0.7095 two-way ANOVA. Acute treatment with GD showed a significant decrease in the occurrence of seizures compared to controls in males (Fig. 5B, P = 0.031, 2 × 2 Fisher Exact Test) and females (Fig. 5B, P = 0.005, Fisher Exact Test). However, it was not different to subchronic GD treatment (Fig. 5B, males: P = 0.089 and females: P = 0.096, 2 × 2 Fisher Exact Test). There was no difference between sex (Fig. 5B, P = 1.000). Animals that died after the occurrence of stage 3 seizures were excluded from the analysis. Stage 5 was used to compare the mortality between the treatments. The results showed that GD in repeat doses increased the mortality significantly when compared with PGB in males (Fig. 6A, P = 0.0020, 2 × 2 Fisher's Exact Test) and when compared with all control groups with females (SAL12, P = 0.001, Tween, P = 0.0138, PGB, P = 0.0138, 2 × 2 Fisher's Exact Test). However, the acute dose of GD reduced significantly the animal's mortality compared to control (Fig. 6B, males: P = 0.015, females: P = 0.0001, 2 × 2 Fisher's Exact Test) and also when compared to GD given for 12 days (P < 0.0001 for both sexes, 2 × 2 Fisher's Exact Test). There was no difference between sex in any mortality analyses (P > 0.05, 2 × 2 Fisher's Exact Test).

Stage 3 seizures evaluation in the PTZ-induced seizure model. A Latency to the first stage 3 seizures between GD and controls in subchronic treatment. A F(3,41) = 0.09356, P = 0.4322 n = 9–12 males, F(3,44) = 1.205, P = 0.3189 n = 12 females, one-way ANOVA. There was no difference between sex [F(1,85) = 3.628, P = 0.0602, two-way ANOVA]. Values represent mean ± SEM. B No difference in the seizure occurrence was observed in any pair comparison (P > 0.05, n = 9–12, 2 × 2 contingency table by Fisher's Exact Test). Values represent a percentage

Stage 3 seizures evaluation in the PTZ-induced seizure model. A Latency to the first seizure in acute treatment with GD. B Latency to stage 3 seizures after GD acute treatment compared with control and subchronic GD treatment. [F(3,37) = 9.628, P = < 0.0001, n = 9–12, males, F(3,42) = 8.193, P = 0.0002, n = 10–12, females, on-way ANOVA], comparison between sex [F(1,79) = 0,01,398, P = 0,7095 two-way ANOVA]. Results of Tukey’s test is represented as *P < 0.05, **P < 0.01, ***P < 0.001. Values represent means ± SEM. (B) Occurrence of stage 3 seizures after GD acute treatment compared with control group (P = 0.031, males, P = 0.005, females,), comparison between acute e subchronic GD treatment (P = 0.089, males, P = 0.096, females) as well between sex (P = < 0.05). 2 × 2, contingency table by Fisher’s Exact Test. Values represent percentages

Mortality (stage 5) evaluation in the PTZ-induced seizure model. A GD in repeat dose compared with controls in males (only different of PGB, P = 0.0020) and females (P < 0.05). B The acute dose of GD was compared with control (P = 0.015, males, P = 0.0001, females) and GD in repeat doses (P = 0,0001 for both sexes). Comparison between sex (all mortality analysis, P > 0.05). 2 × 2, contingency table by Fisher's Exact Test. *P < 0.05, **P < 0.01, ***P < 0.001. Values represent percentages
GD Treatments Affect the Expression of the GluN2B Receptor, Adenosine A1 Receptor, and COX-2
Males treated with GD during 12 days, demonstrated a significant increase in the expression of the GluN2B-containing glutamate NMDA receptor compared to saline and Sal12 [Fig. 7B, F(6,22) = 4.725, P = 0.0031, n = 3–6, one-way ANOVA]. There was no difference in females between groups [Fig. 7B, F (6,18) = 0.9703, P = 0.4727, n = 3–5 one-way ANOVA], neither between sex [Fig. 7B, F(1,41) = 1.654, P = 0.2056, n = 3–6, two-way ANOVA].

Effect of the GD acute (Sal1 and GD1) and subchronic (Sal12, Tween, PGB and GD12) treatment on the expression of GluN2B, in male and female mice, after PTZ-induced seizure. The group saline (control) did not receive PTZ administration. A Bands representative of an experiment on males. B Comparative GluN2B expression in males [F(6,22) = 4.725, P = 0.0031, n = 3–6, one-way ANOVA] and females [F (6,18) = 0.9703, P = 0.4727, n = 3–5, one-way ANOVA]. Comparison between sex [F(1,41) = 1.654, P = 0.2056, n = 3–6, two-way ANOVA]. Results of Tukey’s test is represented as *P < 0.05, **P < 0.01, ***P < 0.001. Values represent means ± SEM
When COX-2 expression was evaluated (Fig. 8), the results showed that the group of males treated with GD for 12 days reduced the expression of this protein. This effect was not repeated in the group of females [Fig. 8B, males: F(6,23) = 5.143, P = 0.0018, females: F(6,29) = 0.6614, P = 0.6811, n = 3–6, one-way ANOVA], without a sex difference [Fig. 8B, F(1,52) = 1.253, P = 0.2681, two-way ANOVA].

Effect of the GD acute (Sal1 and GD1) and subchronic (Sal12, Tween, PGB and GD12) treatment on the expression of COX2, in male and female mice, after PTZ-induced seizure. The group called saline (control) did not receive PTZ administration. A Bands representative of an experiment on females. B Comparative COX-2 expression in males [F(6,23) = 5.143, P = 0.0018, n = 3–6, one-way ANOVA] and in females [F(6,29) = 0.6614, P = 0.6811, n = 3–6, one-way ANOVA]. Comparison between sex ( F(1,52) = 1.253, P = 0.2681, two-way ANOVA). Results of Tukey’s test is represented as *P < 0.05, **P < 0.01, ***P < 0.001. Values represent means ± SEM
In addition, adenosine A1 expression was evaluated. In males, no difference was observed [Fig. 9B, F(6,18) = 1.352, P = 0.2860, n = 3–5, one-way ANOVA]. In females, the groups treated with GD12 and PGB showed an increase in the expression of adenosine A1 compared to the GD1 group [Fig. 9B, F(6,19) = 3.324, P = 0.0207, n = 3–5, one-way ANOVA], no difference was observed between sex [Fig. 9B, F(1,37) = 0.1214, P = 0.7295].

Effect of the GD acute (Sal1 and GD1) and subchronic (Sal12, Tween, PGB and GD12) treatment on the expression of adenosine A1, in male and female mice, after PTZ-induced seizure. The group called saline (control) did not receive PTZ administration. A Bands representative of an experiment on males. B Comparative adenosine A1 expression in males [F(6,18) = 1.352, P = 0.2860, n = 3–5, one-way ANOVA], and in females [F(6,19) = 3.324, P = 0.0207, n = 3,5, one-way ANOVA]. Comparison between sex [F(1,37) = 0.1214, P = 0.7295]. Results of Tukey’s test is represented as *P < 0.05, **P < 0.01, ***P < 0.001. Values represent means ± SEM
DNA Damage PTZ-Induced is Mitigated by GD Treatment
The Comet assay was used to assess GD's genotoxic effect and compare the effects between males and females (Figs. 10A, B and 11A, B). As observed in previous studies, PTZ induced DNA damage observed by the increase in the damage index (DI) and damage frequency (DF) (SAL 1 and Sal12) when compared to the saline group, both in males and females.

Comet assay in A peripheral blood and B hippocampus of male mice of the different groups. Damage index was range from 0 (completely undamaged, 100 cells × 0) to 400 (with maximum damage 100 × 4). Damage frequency was calculated based on the number of cells with tail versus those with no tail. *P < 0.05; **P < 0.01; ***P < 0.001 in comparison to Saline group; aP < 0.05; aaP < 0.01; aaaP < 0.001 in comparison to Sal 1 group; bP < 0.05; bbP < 0.01; bbbP < 0.001 in comparison to Sal 12 group. N = 4–6. DI damage index; DF damage frequency

Comet assay in A peripheral blood and B hippocampus of female mice of the different groups. Damage index was range from 0 (completely undamaged, 100 cells × 0) to 400 (with maximum damage 100 × 4). Damage frequency was calculated based on the number of cells with tail versus those with no tail. * P < 0.05; **P < 0.01; ***P < 0.001 in comparison to Saline group; aP < 0.05; aaP < 0.01; aaaP < 0.001 in comparison to Sal 1 group; bP < 0.05; bbP < 0.01; bbbP < 0.001 in comparison to Sal 12 group. N = 4–6. DI damage index; DF damage frequency
Also, GD reduced DNA damage in acute and subchronic treatments. GD decreased both DI and DF in peripheral blood and hippocampus, except DF in the GD12 group that did not decrease significantly in the hippocampus in both sexes. PGB reduced DI and DF in male and female mice in both analyzed tissues in subchronic treatment.
The Treatments Did Not Increase the Micronucleus Frequency
The micronucleus test showed that the treatments did not increase the micronucleus frequency in polychromatic erythrocytes (MNPCE) from bone marrow in female and male mice (Fig. 12). The polychromatic erythrocytes/normochromatic erythrocytes (PCE/NCE) ratio was similar in all groups (data not shown).

The evaluation of the different treatments' mutagenic activity using the micronucleus test in the bone marrow. No significant difference among the groups was found using one-way ANOVA and Tukey's test. N = 5–6. aMNPCE: micronucleus in polychromatic erythrocytes
Discussion
This study aimed to evaluate the effect of repeated GD administration on the seizure model induced by PTZ in male and female mice. The animals received GD 300 mg/kg for 12 consecutive days and had their behavior evaluated after acute administration of PTZ. Throughout the GD treatment, the animals' weights were recorded every 3 days (four measures). The results showed that males had a significant weight reduction concerning the control group at the third measurement. (Fig. 2), while in females, a significant reduction was observed only in the fourth measurement. GD 300 mg, when given acutely, did not show a toxic effect [41]. However, perhaps its repeated administration may interfere with some metabolic pathway or determine more pronounced toxic effects, which should be investigated in the future.
The Hot Plate Test is used to assess the potential antinociceptive effect of compounds by applying a thermal stimulus [50]. The test allows evaluating the supraspinal pain responses since the choice of which paw lift to avoid thermal discomfort requires the nervous system's superior structures [57]. Here, GD did not show a difference in the response compared to the control group, both in males and females neither at different times. Despite this, there was a difference between sexes that was independent of time and treatment group. Stress-induced analgesia in rats was more pronounced in females than in males. Besides that, factors such as body weight as hormonal changes, skin sensitivity, and habituation to the plate were not evaluated in this study. Perhaps they could explain GD's absence in this model [58,59,60].
PGB administered over 10 days showed no difference from the control group in the hot plate latency. A different result was demonstrated by Hamada et al. [61], which observed the antinociceptive effect of acute PGB administration (40 mg/kg) in the same model. £uszczki [62] observed a dose-dependent effect of PGB, in which the dose of 175.26 mg/kg was able to increase the antinociceptive effect by 50%. Thus, the antinociceptive action of PGB seems to be associated with higher doses than tested in this study, explaining the lack in this study.
A previous study showed that GD's acute administration at a dose of 300 mg/kg was able to block seizures induced by a single dose of PTZ [24]. Also, in the PTZ-kindling model, GD is commonly used for drug evaluation, demonstrating a neuroprotective effect [25]. In more recent work, GD reduced acute seizures isoniazid and 4-aminopyridine-induced, and it was able to protect against neuronal damage induced by pilocarpine [21, 22]. Finally, a study of action mechanism suggested that GD's anticonvulsant effect may be related to the possible modulation of the adenosine A1 receptor [43].
On the other hand, in this study, the GD 300 mg/kg repeated administration had not been shown to protect against acute PTZ-induced seizures. There was no significant difference to the control in the latency for the first stage 3 seizures. However, there was an increase in stage 3 seizures occurrence in the GD12 group, which could help understand the possible toxic effect at this dose and time of treatment, as observed in body weight. Perhaps the lack of protection observed in this study is related to the GD action in different molecular targets, which this compound's acute administrations would not modulate. Little is known about the mechanism of GD action. The neurochemical evaluation, carried out employing a binding assay, showed that GD could dose-dependently inhibit the l-[3H]-glutamate specific union, and this action could be involved in the anticonvulsant activity of this compound [29]. Here, GD was tested only at a dose of 300 mg/kg after 12 consecutive days, which could, through repeated administrations, modulate targets other than glutamate receptors.
Our study shows that GD increased the expression of the GluN2B receptors in the male hippocampus compared to the SAL 12 group, while it did not protect against PTZ-induced seizure. This result is a curious finding because Pereira et al. [29], using a binding technique, demonstrated that GD could antagonize glutamate binding in rat cortex receptors. The GluN2B ability to play different brain roles under overactivation and the activation of different signaling pathways could explain the results observed here since, in this study, GD was administered repeatedly for 12 days. For instance, GluN2B bidirectionally regulates BDNF expression, which depends on the activation degree of NMDARs. In a previous study with cortical neurons in vitro, ifenprodil, a selective antagonist of the GluN2B subunit, prevented the BDNF levels from increasing behind moderate activation of GluN2B, induced by a low dose of the NMDA. In contrast, under overactivation conditions, ifenprodil enhanced BDNF expression after high-dose NMDA exposure [34]. In this sense, likely GD administered acutely or repeatedly could stimulate the expression of glutamate receptors differently, including the GluN2B receptor, and thus determine different behavioral effects.
Previous studies have shown that treatment with lactonic compounds can modulate the expression of GluN2B receptors. The increase in the GluN2B receptor expression and phosphorylation was observed in mice hippocampus after chronic treatment with simvastatin, an important lactone used to reduce hepatic cholesterol biosynthesis [63]. Those authors found that increased expression of GluN2B has resulted in the farnesyl-pyrophosphate (FPP) decrease, a mevalonate pathway intermediate. This study has also shown an increase of histone H3K9 and H3K27 acetylation of GluN2B promoter, resulting from this statin treatment. Other studies have indicated epigenetic GluN2B modulation through acetylation and deacetylation of histones. Histone acetylation alters the compact chromatin structure and makes it accessible for DNA regulation of proteins [64]. Fujita et al. [64] observed that vorinostat, an HDAC (a class of histones deacetylase) inhibitor increased the GluN2B expression in rat hippocampus using histone acetylation. Furthermore, Nghia et al. [65] studied chronic treatment with antidepressant imipramine and observed an increase in histones H3K9 and H3K27 in GluN2B promoter implicated by a decrease in HDAC activity. We did not measure histone acetylation in this study. However, the mentioned studies reinforce the importance of rated epigenetic modifications in GluN2B expression, and it is being considered in the subsequent studies.
Some authors have already observed that gonadal hormones, such as 17β-estradiol, play an essential role in the synaptic activity mediated by NMDARs and the receptor itself is express in the hippocampus. These influences may be directly mediated by ERs (estradiol receptors) or indirectly through the modulation of neurotrophic factors such as BDNF [66,67,68]. In the following studies of our group, it will be essential to assess whether this receptor's expression pattern may be under the influence of sex hormones; this can be verified through the dosage of these hormones in females.
Here, we observed a significant decrease in the COX-2 expression in males, but not in females, in groups treated with GD for 12 days. Although our treatment did not change the animal behavior (latency and seizure occurrence), a decrease in COX-2 observed in animals treated with GD may indicate a possible anti-inflammatory effect. This observation was already suggested by Pflüger et al. [26] when they identified that GD inhibited TNF- α (a pro-inflammatory cytokine) in N9 microglial culture. As already mentioned, we did not evaluate the levels of female gonadal hormones. It is known that their role goes beyond reproduction, and we suspect that these may be associated with the expression of COX-2 as well since studies have shown that estrogen can inhibit the production of pro-inflammatory cytokines that cause a direct and indirect impact on the COX-2- induction pathway [69, 70]. This issue is complex and exciting and deserves to be assessed using a specific methodology.
In animals treated 12 days with GD, the adenosine A1 receptor expression was increased compared with animals treated 1 day with GD, in females but not in males. This result corroborates another study carried out by our research group; no changes in the expression of the adenosine A1 receptor were detected in the hippocampus of mice treated with acute GD in the western blotting test [43].
A rapid increase in the adenosine A1 receptor density is seen in animals' hippocampus and cortex after the acute-seizures induction [71,72,73]. This increase might be a receptor adenosine A1 compensatory antiepileptic mechanism, and the tissue attempts to recover and avoid hyperactivity-induced damage [73]. Although it does not be significant compared with the control group, we observed that GD for 12 days seems to increase these receptors' density, especially in females. However, it did not increase latency for the first stage 3 seizures. An interesting observation about the lack of effect on epileptic behavior is the GluN2B receptor increase in male mice treated 12 days with GD. It seems that, although GD may be acting as an adenosine A1 receptor modulator, the GluN2B properties in mediating excitatory currents can be minimizing the chances of observing the effects mediated by adenosine A1 in the epileptic behavior of animals. On male mice, the PGB group shows an adenosine A1 increase. At the moment, this is new information in the literature and could be a property-related mechanism of action.
The antiseizure drugs (AEDs) treatment can have dramatic side effects, even in utero. There is evidence of malformation as neural tube defects, congenital heart disease, and orofacial clefts on children exposed to AEDs by mother treatment during pregnancy, which causes impairments on lifelong [74]. These adverse effects reinforce the need to evaluate the genotoxicity and mutagenicity of candidate compounds to treat epilepsy. Moreover, mutagenic evaluation using a micronucleus test is required to assess the safety of compounds with pharmacological potential and approve new drugs [75]. In the present study, PTZ alone or in combination with GD did not increase the micronucleus frequency, indicating these drugs could not induce chromosomal mutations in the tested conditions. It suggests that the substances do not induce clastogenesis or aneugenesis in bone marrow cells, detectable in the micronucleus test [76,77,78]. Besides, GD decreased DNA damage caused by PTZ in the hippocampus, both in subchronic and acute treatment, which our group had already observed in previous studies with assessment of DNA damage in the total brain of male mice [25], cortex [27], and N9 murine microglial cell line [26]. Here, the evaluation was carried out in both sexes, which is new data regarding the possible protection against DNA damage produced by GD in a seizure model. The results obtained are specific since we evaluated the hippocampus, the most critical target brain structure of PTZ [79]. GD1 decreased DI and DF parameters to detect DNA damage, while GD12 decreased significantly only DI in the hippocampus in male and female mice. Since the acute but not subchronic treatment GD decreased the number of seizures and increased the latency for seizure, this anticonvulsant effect likely contributed to the higher DNA protection in GD1. Indeed, an anti-inflammatory activity in subchronic treatment appears to be more relevant to explain the decrease in DNA damage than Sal 12.
Conclusion
This study showed that GD under subchronic treatment did not protect animals from seizures induced by PTZ. In contrast, this compound confirmed its neuroprotective effect after acute treatment. This activity may be related to the increased expression of GluN2B-containing glutamate NMDA receptors, especially in males. On the other hand, as already demonstrated in previous studies, GD seems to have an anti-inflammatory effect, decreasing COX-2 expression, in addition to protecting against DNA damage generated by PTZ and increasing the adenosine A1 receptor expression in females. For the first time, GD was tested on female mice, showing a different response pattern than males regarding the expression of adenosine A1 receptor, GluN2B receptors, and COX-2, reinforcing the importance of neuropharmacological studies in both sexes. Further studies on the GD neuroprotective profile are needed to complement the data obtained in this investigation.
Data Availability
The datasets generated during or analyzed during the current study are available from the corresponding author on reasonable request.
References
World Health Organization (2019) Epilepsy. https://www.who.int/health-topics/epilepsy. Accessed 30 July 2020
Devinsky O, Vezzani A, O’Brien TJ et al (2018) Epilepsy. Nat Rev Dis Prim. https://doi.org/10.1038/nrdp.2018.24
Fisher RS, Acevedo C, Arzimanoglou A et al (2014) ILAE official report: a practical clinical definition of epilepsy. Epilepsia 55:475–482. https://doi.org/10.1111/epi.12550
Shimada T, Takemiya T, Sugiura H, Yamagata K (2014) Role of inflammatory mediators in the pathogenesis of epilepsy. Mediat Inflamm. https://doi.org/10.1155/2014/901902
Xanthos DN, Sandkühler J (2014) Neurogenic neuroinflammation: inflammatory CNS reactions in response to neuronal activity. Nat Rev Neurosci 15:43–53. https://doi.org/10.1038/nrn3617
Vezzani A, Balosso S, Ravizza T (2019) Neuroinflammatory pathways as treatment targets and biomarkers in epilepsy. Nat Rev Neurol 15:459–472. https://doi.org/10.1038/s41582-019-0217-x
Temp FR, Marafiga JR, Milanesi LH et al (2017) Cyclooxygenase-2 inhibitors differentially attenuate pentylenetetrazol-induced seizures and increase of pro- and anti-inflammatory cytokine levels in the cerebral cortex and hippocampus of mice. Eur J Pharmacol 810:15–25. https://doi.org/10.1016/j.ejphar.2017.05.013
Rawat C, Kukal S, Dahiya UR, Kukreti R (2019) Cyclooxygenase-2 (COX-2) inhibitors: future therapeutic strategies for epilepsy management. J Neuroinflammation 16:197. https://doi.org/10.1186/s12974-019-1592-3
Dhir A (2019) An update of cyclooxygenase (COX)-inhibitors in epilepsy disorders. Expert Opin Investig Drugs 28:191–205. https://doi.org/10.1080/13543784.2019.1557147
Akula KK, Dhir A, Kulkarni SK (2008) Rofecoxib, a selective cyclooxygenase-2 (COX-2) inhibitor increases pentylenetetrazol seizure threshold in mice: Possible involvement of adenosinergic mechanism. Epilepsy Res. https://doi.org/10.1016/j.eplepsyres.2007.10.008
Dhir A, Naidu PS, Kulkarni SK (2007) Neuroprotective effect of nimesulide, a preferential COX-2 inhibitor, against pentylenetetrazol (PTZ)-induced chemical kindling and associated biochemical parameters in mice. Seizure. https://doi.org/10.1016/j.seizure.2007.05.016
Katyal J, Kumar H, Gupta YK (2015) Anticonvulsant activity of the cyclooxygenase-2 (COX-2) inhibitor etoricoxib in pentylenetetrazole-kindled rats is associated with memory impairment. Epilepsy Behav. https://doi.org/10.1016/j.yebeh.2014.12.032
Guzzo FE, Rodrigues MK et al (2018) Effect of dexamethasone on seizures and inflammatory profile induced by kindling seizure model. J Neuroimmunol 325:92–98. https://doi.org/10.1016/j.jneuroim.2018.10.005
Vieira V, Glassmann D, Marafon P et al (2016) Effect of diclofenac sodium on seizures and inflammatory profile induced by kindling seizure model. Epilepsy Res 127:107–113. https://doi.org/10.1016/j.eplepsyres.2016.08.020
Blommel ML, Blommel AL (2007) Pregabalin: an antiepileptic agent useful for neuropathic pain. Am J Heal Pharm 64:1475–1482. https://doi.org/10.2146/ajhp060371
Nader MA, Ateyya H, El-shafey M, El-sherbeeny NA (2018) Neurochemistry international sitagliptin enhances the neuroprotective effect of pregabalin against pentylenetetrazole-induced acute epileptogenesis in mice : implication of oxidative, inflammatory, apoptotic and autophagy pathways. Neurochem Int 115:11–23. https://doi.org/10.1016/j.neuint.2017.10.006
Kruszewski SP, Shane JA (2007) Pregabalin in central neuropathic pain associated with spinal cord injury: a placebo-controlled trial. Neurology 68:2158–2160. https://doi.org/10.1212/01.wnl.0000269479.84817.0a
Ra M, Straube S, Pj W et al (2010) Pregabalin for acute and chronic pain in adults (review). Cochrane Libr. https://doi.org/10.1002/14651858.CD007076.pub2
Wiffen PJ, McQuay HJ, Edwards J, Moore RA (2005) Gabapentin for acute and chronic pain (review). Cochrane Database Syst Rev. https://doi.org/10.1002/14651858.CD005452
Woodbury A, Yu SP, Wei L, García P (2013) Neuro-modulating effects of honokiol: a review. Front Neurol. https://doi.org/10.3389/fneur.2013.00130
Ferrendelli JA, Holland KD, McKeon AC, Covey DF (1989) Comparison of the anticonvulsant activities of ethosuximide, valproate, and a new anticonvulsant, thiobutyrolactone. Epilepsia 30:617–622. https://doi.org/10.1111/j.1528-1157.1989.tb05482.x
Holland KD, McKeon AC, Canney DJ et al (1992) Relative anticonvulsant effects of GABAmimetic and GABA modulatory agents. Epilepsia 33:981–986. https://doi.org/10.1111/j.1528-1157.1992.tb01747.x
Duke JA (1992) Handbook of biologically active phytochemicals and their activities. CRC Press, Boca Raton, pp 3–183
Viana CCS, de Oliveira PA, da Brum LF et al (2007) Gamma-decanolactone effect on behavioral and genotoxic parameters. Life Sci 80:1014–1019. https://doi.org/10.1016/j.lfs.2006.11.042
De Oliveira PA, Lino FL, Cappelari SE et al (2008) Effects of gamma-decanolactone on seizures induced by PTZ-kindling in mice. Exp Brain Res 187:161–166. https://doi.org/10.1007/s00221-008-1295-y
Pflüger P, Viau CMA, Coelho VR et al (2016) Gamma-decanolactone inhibits iNOS and TNF-alpha production by lipopolysaccharide-activated microglia in N9 cells. Eur J Pharmacol 780:38–45. https://doi.org/10.1016/j.ejphar.2016.03.029
Pflüger P, Regner GG, Coelho VR et al (2018) Gamma-decanolactone improves biochemical parameters associated with pilocarpine-induced seizures in male mice. Curr Mol Pharmacol 11:162–169. https://doi.org/10.2174/1874467210666171002114954
Pflüger P, Coelho VR, Regner GG et al (2018) Neuropharmacological profile of gamma-decanolactone on hemically-induced seizure in mice. Cent Nerv Syst Agents Med Chem 18:222–227. https://doi.org/10.2174/1871524918666180611103802
Pereira P, Elisabetsky E, Souza DO (1997) Effect of γ-decanolactone on glutamate binding in the rat cerebral cortex. Neurochem Res 22:1507–1510. https://doi.org/10.1023/A:1021962714034
Wyllie DJA, Livesey MR, Hardingham GE (2013) Influence of GluN2 subunit identity on NMDA receptor function. Neuropharmacology 74:4–17. https://doi.org/10.1016/j.neuropharm.2013.01.016
Kim MJ, Dunah AW, Wang YT, Sheng M (2005) Differential roles of NR2A- and NR2B-containing NMDA receptors in ras-ERK signaling and AMPA receptor trafficking. Neuron 46:745–760. https://doi.org/10.1016/j.neuron.2005.04.031
Lai TW, Zhang S, Wang YT (2014) Excitotoxicity and stroke: identifying novel targets for neuroprotection. Prog Neurobiol 115:157–188. https://doi.org/10.1016/j.pneurobio.2013.11.006
Waxman EA, Lynch DR (2005) N-methyl-D-aspartate receptor subtype mediated bidirectional control of p38 mitogen-activated protein kinase. J Biol Chem 280:29322–29333. https://doi.org/10.1074/jbc.M502080200
Zhou X, Ding Q, Chen Z et al (2013) Involvement of the GluN2A and GluN2B subunits in synaptic and extrasynaptic N -methyl- D -aspartate receptor function and neuronal excitotoxicity. J Biol Chem 288:24151–24159. https://doi.org/10.1074/jbc.M113.482000
Sun Y, Xu Y, Cheng X et al (2018) The differences between GluN2A and GluN2B signaling in the brain. J Neurosci Res 96:1430–1443. https://doi.org/10.1002/jnr.24251
Aronica E, Sandau US, Iyer A, Boison D (2013) Glial adenosine kinase—a neuropathological marker of the epileptic brain. Neurochem Int 63:688–695. https://doi.org/10.1016/j.neuint.2013.01.028
Boison D (2013) Role of adenosine in status epilepticus: a potential new target? Epilepsia 54:20–22. https://doi.org/10.1111/epi.12268
Ghislandi AB, Garcez ML, Zambon GM et al (2020) Adenosine and NMDA receptors modulate neuroprotection-induced NMDA preconditioning in mice. J Mol Neurosci 2:590–599
Vianna EP, Ferreira AT, Jos M (2005) Modulation of seizures and synaptic plasticity by adenosinergic receptors in an experimental model of temporal lobe epilepsy induced by pilocarpine in rats. Epilepsia 46:166–173. https://doi.org/10.1111/j.1528-1167.2005.01027.x
Zhou Q, Zhu S, Guo Y et al (2018) Adenosine A1 receptors play an important protective role against cognitive impairment and long-term potentiation inhibition in a pentylenetetrazol mouse model of epilepsy. Mol Neurobiol. https://doi.org/10.1007/s12035-017-0571-x
Coelho De Souza GP, Elisabetsky E, Nunes DS et al (1997) Anticonvulsant properties of γ-decanolactone in mice. J Ethnopharmacol 58:175–181. https://doi.org/10.1016/S0378-8741(97)00102-5
Da Silva LF, Pereira P, Elisabetsky E (1998) A neuropharmacological analysis of PTZ-induced kindling in mice. Gen Pharmacol. https://doi.org/10.1016/S0306-3623(97)00423-0
Pflüger P, Gregory G, Luft J et al (2020) Gamma-decanolactone attenuates acute and chronic seizures in mice: a possible role of adenosine A 1 receptors. Behav Pharmacol. https://doi.org/10.1097/FBP.0000000000000554
Pérez-Saad H, Buznego MT (2008) Behavioral and antiepileptic effects of acute administration of the extract of the plant Cestrum nocturnum Lin (the lady of the night). Epilepsy Behav. https://doi.org/10.1016/j.yebeh.2007.12.012
Gilron I, Baron R, Jensen T (2015) Neuropathic pain: principles of diagnosis and treatment. Mayo Clin Proc 90:532–545. https://doi.org/10.1016/j.mayocp.2015.01.018
Enke O, New HA, New CH et al (2018) Anticonvulsants in the treatment of low back pain and lumbar radicular pain: a systematic review and meta-analysis. CMAJ 190:E786–E793. https://doi.org/10.1503/cmaj.171333
St. John Smith E (2018) Advances in understanding nociception and neuropathic pain. J Neurol 265:231–238. https://doi.org/10.1007/s00415-017-8641-6
Silva JC, Raquel S, De LG et al (2013) Modelos experimentais para avaliação da atividade antinociceptiva de produtos naturais : uma revisão. Rev Bras Farm 94:18–23
Scarabelot VL, Medeiros LF, de Oliveira C et al (2016) Melatonin alters the mechanical and thermal hyperalgesia induced by orofacial pain model in rats. Inflammation 39:1649–1659. https://doi.org/10.1007/s10753-016-0399-y
Bannon AW, Malmberg AB (2007) Models of nociception: hot-plate, tail-flick, and formalin tests in rodents. Curr Protoc Neurosci Chapter 8:1–16. https://doi.org/10.1002/0471142301.ns0809s41
Leite JPV (2009) Fitoerapia: bases científicas e tecnológicas. Atheneu, São Paulo
Scarabelot VL, de Oliveira C, Medeiros LF et al (2019) Transcranial direct-current stimulation reduces nociceptive behavior in an orofacial pain model. J Oral Rehabil 46:40–50. https://doi.org/10.1111/joor.12726
Tice RR, Agurell E, Anderson D et al (2000) Single cell gel/comet assay: guidelines for in vitro and in vivo genetic toxicology testing. Environ Mol Mutagen 35:206–221. https://doi.org/10.1002/(SICI)1098-2280(2000)35:3%3c206::AID-EM8%3e3.0.CO;2-J
Antunes FTT, Angelo SG, Dallegrave E et al (2020) Recombinant peptide derived from the venom the Phoneutria nigriventer spider relieves nociception by nerve deafferentation. Neuropeptides. https://doi.org/10.1016/j.npep.2019.101980
Lazzarotto L, Pflüger P, Regner GG et al (2020) Lacosamide improves biochemical, genotoxic, and mitochondrial parameters after PTZ-kindling model in mice. Fundam Clin Pharmacol. https://doi.org/10.1111/fcp.12598
Mavournin KH, Blakey DH, Cimino MC et al (2008) The in vivo micronucleus assay in mammalian bone marrow and peripheral blood. A report of the U.S. Environmental Protection Agency Gene-Tox Program. Mutat Res Rev Genet Toxicol 239:29–80
Gregory NS, Harris AL, Robinson CR et al (2013) An overview of animal models of pain: disease models and outcome measures. J Pain 14:1255–1269. https://doi.org/10.1016/j.jpain.2013.06.008
Gunn A, Bobeck EN, Weber C, Morgan MM (2011) The influence of non-nociceptive factors on hot-plate latency in rats. J Pain. https://doi.org/10.1016/j.jpain.2010.06.011
Vendruscolo LF, Pamplona A, Takahashi RN (2004) Strain and sex differences in the expression of nociceptive behavior and stress-induced analgesia in rats. Brain Res 1030:277–283. https://doi.org/10.1016/j.brainres.2004.10.016
Kavaliers M, Douglas DC (1991) Sex differences in opioid and non-opioid mediated predator-induced analgesia in mice. Brain Res 568:173–177
Hamada NM, Ashour RH, Shalaby AA, El-Beltagi HM (2018) Calcitonin potentiates the anticonvulsant and antinociceptive effects of valproic acid and pregabalin in pentylenetetrazole-kindled mice. Eur J Pharmacol 818:351–355. https://doi.org/10.1016/j.ejphar.2017.11.003
Łuszczki JJ (2010) Dose-response relationship analysis of pregabalin doses and their antinociceptive effects in hot-plate test in mice. Pharmacol Rep 62:942–948. https://doi.org/10.1016/S1734-1140(10)70355-8
Chen T, Zhang B, Li G et al (2016) Simvastatin enhances NMDA receptor GluN2B expression and phosphorylation of GluN2B and GluN2A through increased histone acetylation and Src signaling in hippocampal CA1 neurons. Neuropharmacology 107:411–421. https://doi.org/10.1016/j.neuropharm.2016.03.028
Fujita Y, Morinobu S, Takei S et al (2012) Vorinostat, a histone deacetylase inhibitor, facilitates fear extinction and enhances expression of the hippocampal NR2B-containing NMDA receptor gene. J Psychiatr Res 46:635–643. https://doi.org/10.1016/j.jpsychires.2012.01.026
Nghia NA, Hirasawa T, Kasai H et al (2015) Long-term imipramine treatment increases N-methyl-d-aspartate receptor activity and expression via epigenetic mechanisms. Eur J Pharmacol 752:69–77. https://doi.org/10.1016/j.ejphar.2015.02.010
Morissette M, Le Saux M, D’Astous M et al (2008) Contribution of estrogen receptors alpha and beta to the effects of estradiol in the brain. J Steroid Biochem Mol Biol 108:327–338. https://doi.org/10.1016/j.jsbmb.2007.09.011
McCarthny CR, Du X, Wu YC, Hill RA (2018) Investigating the interactive effects of sex steroid hormones and brain-derived neurotrophic factor during adolescence on hippocampal NMDA receptor expression. Int J Endocrinol. https://doi.org/10.1155/2018/7231915
Adkins JM, Lynch JF, Hagerdorn P et al (2019) Anterior cingulate cortex and dorsal hippocampal glutamate receptors mediate generalized fear in female rats. Psychoneuroendocrinology 107:109–118. https://doi.org/10.1016/j.psyneuen.2019.05.009
Ospina JA, Brevig HN, Krause DN et al (2020) Estrogen suppresses IL-1 1beta -mediated induction of COX-2 pathway in rat cerebral blood vessels. Am J Physiol Heart Circ Physiol 4625:2010–2019
Stacey W, Bhave S, Uht RM (2016) Mechanisms by which 17 β -estradiol ( E2) suppress neuronal cox-2 gene expression. PLoS ONE. https://doi.org/10.1371/journal.pone.0161430
Vanore G, Giraldez L, Arnaiz GRDL, Girardi E (2001) Seizure activity produces differential changes in adenosine A 1 receptors within rat hippocampus. Neurochem Res 26:225–230
Boison D (2005) Adenosine and epilepsy: from therapeutic rationale to new therapeutic strategies. Neuroscientist 11:25–36. https://doi.org/10.1177/1073858404269112
Davoudi M, Shojaei A, Reza M et al (2013) Comparison between standard protocol and a novel window protocol for induction of pentylenetetrazol kindled seizures in the rat. Epilepsy Res 106:54–63. https://doi.org/10.1016/j.eplepsyres.2013.03.016
Kardoost M, Hajizadeh-Saffar E, Ghorbanian MT et al (2019) Genotoxicity assessment of antiepileptic drugs (AEDs) in human embryonic stem cells. Epilepsy Res. https://doi.org/10.1016/j.eplepsyres.2019.106232
Coelho VR, Prado LS, Rossato R et al (2018) A 28-day sub-acute genotoxic and behavioural assessment of garcinielliptone FC. Basic Clin Pharmacol Toxicol 123:207–212. https://doi.org/10.1111/bcpt.13010
Hayashi M (2016) The micronucleus test-most widely used in vivo genotoxicity test. Genes Environ 38:4–9. https://doi.org/10.1186/s41021-016-0044-x
Knasmüller S, Fenech M (2019) The micronucleus assay in toxicology. Royal Society of Chemistry, Cambridge
Sommer S, Buraczewska I, Kruszewski M (2020) Micronucleus assay: the state of art, and future directions. Int J Mol Sci 21:7–9. https://doi.org/10.3390/ijms21041534
Samokhina E, Samokhin A (2018) Neuropathological profile of the pentylenetetrazol (PTZ) kindling model. Int J Neurosci 128:1086–1096. https://doi.org/10.1080/00207454.2018.1481064
Funding
This research received financial support from Conselho Nacional de Desenvolvimento Científico e Tecnológico (CNPq, 307515/2019-2), and Federal University of Rio Grande do Sul (UFRGS, 36326).
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
There is no conflict of interest.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
About this article

Cite this article
dos Santos, F.M., Pflüger, P.F., Lazzarotto, L. et al. Gamma-Decanolactone Alters the Expression of GluN2B, A1 Receptors, and COX-2 and Reduces DNA Damage in the PTZ-Induced Seizure Model After Subchronic Treatment in Mice. Neurochem Res 46, 2066–2078 (2021). https://doi.org/10.1007/s11064-021-03345-7
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s11064-021-03345-7