
Abstract
Lack of blood or glucose supply is the most common pathological factor in the brain. To cope with such an energy stress, initiating programmed autophagic processes in neurons is required. However, the mechanisms controlling neuronal autophagy during starvation remain far from clear. Here, we report an essential role of 14-3-3γ in starvation-activated neuronal autophagic influx signaling and elucidate the underlying mechanism. Double-fluorescent immunostaining demonstrates that 14-3-3γ protein elevation is well co-localized with Beclin-1 and LC3 elevation in cortical neurons in ischemic brains. Starvation treatment activates autophagic influx and upregulates Beclin-1 and only the γ isoform of 14-3-3 in N2a cells and cultured cortical neurons. Suppressing overall 14-3-3 function by difopein overexpression or knocking-out the γ isoform of 14-3-3 is sufficient to abolish starvation-induced Beclin-1 induction and LC3 activation while overexpressing 14-3-3γ but no other 14-3-3 isoform significantly upregulate Beclin-1-LC3 signaling. Upon starvation, 14-3-3γ binds more p-β-catenin but less Beclin-1. Finally, overexpressing 14-3-3γ reactivates β-catenin-suppressed Beclin-1-LC3 signaling in neuronal cells. Taken together, our data reveal that starvation-induced 14-3-3γ is required for β-catenin-Beclin-1-LC3-autophagy in starved neurons in vitro and in vivo, which may provide insights in the treatment of neurologic diseases such as stoke.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Human brain accounts 2% of body weight but consumes 20% of ATP. Insufficient energy supply in neurons due to lack of blood or glucose supply commonly occurs in the brain as results of cardio-cerebral vascular, metabolic or neurological diseases. Upon early brain ischemic injury, autophagy is an important cellular mechanism to supply energy via self-digestion in order to avoid cell death [1, 2].
14-3-3 is group of highly conserved proteins most abundant in the brain. 14-3-3 proteins function mainly as scaffold proteins and serve as signaling hubs [3]. In mammalian brains including both neurons and astrocytes, five (β, ε, η, γ, ζ) of the seven 14-3-3 isoforms are highly expressed [4,5,6,7]. Previous studies have shown that different 14-3-3 isoforms may play a specific role in different cellular context or pathological conditions. For examples, the γ isoform of 14-3-3 plays a major role in regulating ischemic signaling and protecting neurons [5] or astrocytes [4] while the η isoform is mainly associated with neuronal development [6]. On the other hand, a distinct isoform of 14-3-3 (e.g., 14-3-3γ) may exert multiple functions (e.g., preventing cell death or scar formation) under similar pathological conditions (e.g., ischemia) via binding different ligands (e.g., Bad, NF-κB, GFAP) and mediating related signaling pathways [4, 7, 8]. Therefore, identifying specific role of 14-3-3 isoforms and related signaling pathways may provide more precise potential therapeutic targets.
Autophagic processes are highly conserved cellular events, however, the regulatory machinery is complicated and remains far from clear. Autophagic influx or signaling is activated and becomes therapeutic targets for various diseases such as stroke [9] and cancers [10]. Beclin-1, a mammalian ortholog of the yeast autophagy-related gene 6 (Atg6), binds to Bcl-2 or PI3K class III. Upon starvation or other stimuli, Beclin-1 is upregulated/activated and then binds to VPS34, leading to the initiation of membrane/organelle isolation and phagophore formation [11]. Further membrane expansion/fusion and autophagosome formation requires microtubule-associated proteins 1A/1B light chain 3B (LC3). Newly synthesized LC3 (pre-LC3) is initially hydrolyzed by Atg4 to LC3-I, which is further activated by Atg3/7 and conjugated to phosphatidylethanolamine (PE-LC3-II). PE-LC3-II in the isolated membrane further recruits various other Atg proteins and promotes the formation of autolysosome [12]. In yeast cells, 14-3-3β prevents cell death induced by various autophagy stimuli including leucine-starvation [13]. In cancer cells, 14-3-3 (mainly ε) binds to Beclin-1 to control the balance between autophage and tumorigenesis [14]. Until now, direct functional study of 14-3-3 proteins in neuronal autophage is scarce.
In the present study, we first demonstrated that 14-3-3 proteins promoted starvation-induced autophagy in neuronal cells via upregulating Beclin-1 but not directly binding to it. Further, the γ but no other 14-3-3 isoform played a major role in regulating neuronal autophagic signaling upon starvation or ischemia in vitro and in vivo.
Materials and Methods
Plasmids and Antibodies
Plasmids pGFP-LC3 and pcDNA-Beclin-1 were provided by Dr. Xingding Zhang (Suzhou Univeresity); pcDNA-14-3-3 isoforms ( γ, τ and ζ) and pYFP-difopein (DFP, dimeric 14-3-3 peptide inhibitor) were provided by Dr. Haian Fu (Emory University). pcDNA-β-catenin was provided by Dr. Yasuyuki Fujita (University College London). pDEST27-GST-14-3-3γ was constructed in our laboratory by PCR cloning the full length of 14-3-3γ cDNA using the pcDNA-14-3-3γ template (Forward primer: 5′-ttttctagatatggtggaccgcgagcaactg-3′; Reverse primer: 5′-tttagatct ctaattgttgccttcgccgccatc-3′). The PCR products were cut with XbaI and BglII and ligated into XbaI/BgaII-linearized pDEST27 vector. pLentiCRISPRv2-sg-14-3-3γ for knocking out mouse 14-3-3γ were constructed as previously described [15, 16]. DNA oligos encoding specific sgRNA targeting to mouse 14-3-3γ genome was selected from Dr. Feng Zhang’s Mouse GeCKO v2 Library (cat#1000000052; targeting sequence, #1, TCGAGATGGTCCGAGCCTAC and #2, CGTTCTTGTAGGCCACCGAC). All plasmids were confirmed by sequencing. Antibodies against to 14-3-3β, ε, η, γ, τ and ζ (Immuno-Biological Laboratories, TakasakiShi, Japan), p-β-catenin Ser33/37/Thr41, LC3B, Beclin-1 and β-actin (Cell Signaling Technology, Boston, MA, USA) and GST or GFP (Santa Cruz Biotechnology, Santa Cruz, CA, USA) were purchased.
Middle Cerebral Artery Occlusion/Reperfusion (MCAo/Re) and Double-Fluorescent Immunostaining
Ischemic reperfusion in mice was conducted as previously described [17]. Animal experiments were approved and performed in accordance with the guidelines of the Animal Care and Use Committees of Tongji Medical College, Huazhong University of Science and Technology. Briefly, adult C57BL/6J mice (25–35 g, Beijing Vital River Laboratory Animal Technology Company, Beijing, China) were anesthetized (7% chloral hydrate, 350 mg/kg) and rectal temperature was maintained at 37 ± 0.5 °C. A silicone-coated monofilament nylon suture (0.22–0.23 mm in diameter) was gently introduced into the internal carotid artery through the external carotid artery stump and advanced to the anterior cerebral artery until a slight resistance was felt. After 1 h of occlusion, the suture was withdrawn to allow reperfusion. Successful ischemic reperfusion in mice was confirmed by measuring modified neurological severity scores and brain infarct volume (2, 3, 5-triphenyl-tetrazolium-chloride staining). Double-fluorescent staining of paraffin-embedded mouse brain tissues was performed as previously described [17]. At least three whole-brain slice across the ischemic infract from each brain (n = 5) were used. After primary and corresponding secondary antibody incubation, brain slices were photographed with the same parameters under a conventional microscope. Fluorescent intensity of 14-3-3 (green) and Beclin-1 or LC3 (red) staining in each neurons was estimated by using the software Image-Pro Plus 6.0 (Media Cybernetics Inc., Rockville, USA) [18]. Pearson’s correlation was used for correlation study between 14-3-3 and Beclin-1 or LC3 in ischemic neurons.
N2a Cell Cultures, Transfection and Starvation
N2a cells were cultured with 1:1 Dulbecco’s modified Eagle’s medium (DMEM) and OPTI-MEM supplemented with 5% fetal bovine serum (FBS) (Gemini, CA, USA) as reported [18]. Transient transfection of N2a cells with Neofect™ DNA transfection reagent (Neofect Biotech Co., Ltd, Beijing, China) was conducted at 24 h after initial seeding according to the manufacturer’s instructions. Fresh Earle’s balanced salt solution (EBSS, GIBCO BRL, USA) media was used to induce N2a cell starvation at 24 h after transient transfection or initial seeding. Cells were washed with EBSS media three times and then incubated with EBSS media for various time points.
Primary Cultures of Mouse Cerebral Cortical Neurons and SNLYSO Staining
Primary cultures of rat cerebral cortical neurons were set as reported previously [5, 6]. Briefly, cerebral cortices isolated from E16 rat embryos (SD mice) were digested with trypsin and dissociated by several passages through a 10-ml pipette. Dispersed cells were filtered with a 200-mesh filer and then centrifuged. Dissociated neurons were resuspended with DMEM-10% FBS and plated onto 35-mm culture dishes or 96-well plates coated with poly-d-lysine. At 4 h of initial seeding, culture media were replaced with neurobasal medium supplemented with 2% B27 and 2 mM l-glutamine (Invitrogen, Grand Island, NY, USA). At 7 days in vitro (7 DIV), cultured neurons were washed with EBSS media three times and starvation was induced by EBSS incubation. To probe autolysosome [19], cultured neurons in 96-well plates were incubated with EBSS media first and then incubated with fluorescent SNLYSO sensor in neurobal media for another 4 h. After washing, fluorescent SNLYSO signal of each well was detected at emission 465 nm/excitation 613 nm with a fluorescent microreader according to the manufacturer’s instructions (Catalog #E0010, Chengdu SinoPharmTech Co., Ltd., Chengdu, China). Relative SNLYSO signal was calculated by an average fluorescent signals from 6 parallel wells/group and normalized to that of EBSS-0 h control.
GST-Pull Down, Co-Immunoprecipitation and Western Blotting Analysis
GST-pull down, co-immunoprecipitation and Western blotting analysis were performed as previously described [18]. Briefly, total proteins were extracted from transfected N2a cells with binding buffer (150 mM NaCl, 0.5% NP-40, 50 mM Tris–Hcl, pH 7.5, 50 mM NaF and protease inhibitors) and 400 µg of soluble proteins were incubated with Glutathione Sepharose beads (GE healthcare, USA) or 14-3-3γ antibody with gentle rotation at 4 °C for overnight. After three times of washing, GST-pull down precipitates were boiled in 2 × SDS–PAGE gel loading buffer and the dissociate proteins in supernatants were collected and subjected for Western blotting analysis. For conventional Western blotting analysis, total soluble proteins were extracted from cultures with radioimmunoprecipitation assay lysis buffer (Applygen Technologies Incorporation, Beijing, China) containing phenylmethanesulfonyl fluoride (Sigma, USA). Equal amount of total proteins was subjected to Western blotting analysis as previously described [20]. After primary antibody incubation, the NC membrane was then incubated with corresponding IRDye 800CW/680CW-conjugated goat anti-rabbit or anti-mouse IgG (LI-COR Biosciences, USA). Labeled bands were visualized and quantified by using the Odyssey Infrared Imaging System (LI-COR Biosciences, USA).
GFP-LC3 Punctate Quantification
N2a cells were transiently transfected with pGFP-LC3 with or without other plasmids for 24 h. Then, the cultures were subjected to EBSS treatment and starved cells were stained with Hoechst 33342 at various time points of starvation. N2a cells expressing GFP-LC3 were randomly photographed under 400×-magnifications with same conditions. Cells with five or more GFP-LC3 vacuole dots (puncta) were considered autophagy-positive. An average percentage of puncta-positive cells from nine fields/culture (total cells > 500) was calculated and used for statistical analysis.
Statistical Analysis
All experiments were repeated independently for at least three times. The values were expressed as mean ± SEM and statistics were performed with a 2-way ANOVA followed by the Student–Newman–Keuls test. P < 0.05 was considered to be significant.
Results
14-3-3γ Elevation Positively Correlates to Becli-1 and LC3 Elevation in Ischemic Neurons In Vivo
Ischemic insult is the most common pathological factor causing brain energy failure. We examined the relationships between 14-3-3 protein levels and autophagy markers (Beclin-1 and LC3) [11, 12] in ischemic brains (Fig. 1). Double-fluorescent immunostaining showed evident 14-3-3γ and Beclin-1 (Fig. 1a, right upper corner showing enlarged cells in the rectangular square) or LC3 (Fig. 1c) signals in cortical neurons (indicated by arrows) in ipsilateral cortex (Ipsi). Further, elevated intensity of 14-3-3γ staining was positively correlated to that of Beclin-1 (r = 0.7837, P < 0.0001, Fig. 1b) and LC3 (r = 0.9259, P < 0.0001, Fig. 1d) in surviving neurons in the Ipsi brain. Thus, ischemia-induced 14-3-3 upregulation was correlated with ischemia-induced autophagy in cortical neurons in the brain.

Elevation of 14-3-3 proteins positively correlates to Beclin-1 and LC3 in cortical neurons after ischemic stroke in mice. a Representative micrographs of double-fluorescent immuostaining of 14-3-3γ and Beclin-1 in mouse cerebral cortex after MCAo 1 h/Re 24 h. Bar 50 µm. Boxes indicate the regions of interest and are enlarged in upper right corner. Arrows indicate neurons. b Correlation between 14-3-3γ intensities and Beclin-1 intensities in individual neurons in the Ipsi of mouse brains after MCAo 1 h/Re 24 h (r = 0.7837, P < 0.0001, N = 23). c Representative micrographs of double-fluorescent immuostaining of 14-3-3γ and LC3 in mouse cerebral cortex after MCAo 1 h/Re 24 h. Bar 50 µm. d Correlation between 14-3-3γ intensities and LC3 intensities in individual neurons in the Ipsi of mouse brains after MCAo 1 h/Re 24 h (r = 0.9259, P < 0.0001, N = 31)
EBSS Starvation-Induced Autophagic Influx Signaling and 14-3-3 Proteins in N2a Cells and Cortical Neurons
To investigate the causative role of 14-3-3 proteins in neuronal autophagy, we established neuronal autophagic modes. Subjected to 2 and 3 h of EBSS incubation (starvation), the number of N2a cells with GFP-LC3-puncta (representing autophagosome, indicated by arrowheads) was largely increased (Fig. 2a, b). Consistently, Western blotting analysis demonstrated that autophagic marker Beclin-1 was significantly upregulated in N2a cells at various time points of starvation (Fig. 2c). Similar to N2a cells, EBSS also activated autophagic influx in cultured cortical neurons. Fluorescent SNLYSO signals (a specific probe for autolysosome) were steadily and significantly increased along 2 to 6 h of EBSS incubation (Fig. 2d), reflecting fluent autophagic influx in EBSS-treated neurons. Consistently, Beclin-1 and cleaved LC3-II were steadily and significantly increased in cultured neurons along with EBSS incubation time (Fig. 2e).

EBSS-starvation upregulates Beclin-1 and activates LC-3-mediated autophagic influx in neuronal cells. a Starvation-induced LC3 fluorescent puncta in autophagosomes in N2a cells. N2a cells were transiently transfected with pGFP-LC3 plasmids for 24 h and then incubated with EBSS media for various time. Representative fluorescent micrographs showed that GFP-LC3 puncta (representing autophagosomes, indicated by arrowheads) was increased in N2a cells upon 2 and 3 h of EBSS incubation. Bar 20 µm. b Statistical analysis demonstrated that the percentage of GFP-LC3 puncta-positive cells (with more than 5 puncta in a cell) were significantly increased at 2 and 3 h of EBSS incubation. **P < 0.01 and ***P < 0.001 vs. 0 h control (n = 3). c Western blotting analysis of Beclin-1 in EBSS-treated N2a cells. Statistical analysis demonstrated that relative level of Beclin-1/β-actin was significantly increased at 2, 3 and 6 h of EBSS. **P < 0.01 and ***P < 0.001 vs. 0 h control (n = 3). d Starvation-induced autolysosome in cultured cortical neurons. Primary cultures of mouse cerebral cortical neurons (7 DIV) were incubated with EBSS media for various time and then stained with specific autolysosome SNLYSO sensor for 4 h. After washing, fluorescent SNLYSO signal in living neurons in each well was measured with a fluorescent microplate reader at emission 465 nm/excitation 613 nm. Relative SNLYSO signal was normalized with media blank. **P < 0.01 and ***P < 0.001 vs. 0 h control (n = 3). e Representative Western blotting results and statistical analysis of Beclin-1 and LC-3-II/I expression expression in cultured neurons upon EBSS treatment. **P < 0.01 and ***P < 0.001 vs. 0 h control (n = 3)
In N2a- and neuron-starvation models, we examined the expression of six 14-3-3 isoforms. Results of Western blotting analysis demonstrated that only the γ isoform of 14-3-3 proteins were evidently upregulated in N2a cells (Fig. 3a, b, Supplemental Figure 1) and primary cultures of cortical neurons (Fig. 3c, d, Supplemental Figure 2) at various time points of EBSS incubation. These data together demonstrated that 14-3-3γ and the canonical autophagic influx (i.e., Beclin-1-LC3-autophagosome-autolysosome) were concurrently upregulated/activated in neurons upon EBSS-induced starvation. In addition, prominent 14-3-3γ elevation occurred after EBSS-1 h (Fig. 3c) while elevation/activation of Beclin-1 or LC3-II occurred after EBSS-2 h (Fig. 2e). The earlier and robust 14-3-3γ induction suggests that 14-3-3 protein is an upstream regulator of canonical autophagic signaling.

EBSS-starvation selectively induces 14-3-3γ proteins in N2a cells and cultured neurons. a Representative Western blotting results showed that 14-3-3 isoforms were differential expressed in EBSS-treated N2a cells. Only the γ isoform of 14-3-3 was elevated evidently in N2a cells upon starvation. b Statistical analysis demonstrated that 14-3-3γ was significantly increased at 1, 2 and 3 h of EBSS incubation compared to 0 h control. **P < 0.01 and ***P < 0.001 vs. 0 h control (n = 3). c Representative Western blotting results showed that only the γ isoform of 14-3-3 was elevated evidently in primary cultures of cerebral cortical neurons upon EBSS treatment. d Statistical analysis demonstrated that 14-3-3γ was significantly increased at 1, 2, 3 and 6 h of EBSS incubation compared to 0 h control. **P < 0.01 and ***P < 0.001 vs. 0 h control (n = 3)
14-3-3γ is the Major Isoform Activating Neuronal Autophagic Signaling upon Starvation
The selective 14-3-3γ induction and its correlation with autophagic markers suggested a major role of 14-3-3γ in regulating neuronal autophagy upon starvation. We then examined the causative role of 14-3-3γ in neuronal autophagy. Representative florescent micrographs and statistical analysis demonstrated that overexpression of the γ but not τ or ζ isoform of 14-3-3 significantly increased GFP-LC3 puncta-positive N2a cells upon starvation (Fig. 4a). Consistently, 14-3-3γ overexpression evidently increased Beclin-1 and cleaved LC3-II levels in N2a cells upon starvation (Fig. 4b, Supplemental Figure 3). On the contrary, 14-3-3γ knockout (KO-1 and KO-2) in N2a cells clearly decreased Beclin-1 and LC3-II levels as compared to its control (WT, 14-3-3γ wild type) (Fig. 4c, Supplemental Figure 4). In addition, 14-3-3γ could rescue DFP-suppressed autophagic signaling. Overexpressing DFP (specific 14-3-3 blocking peptide) evidently reduced Beclin-1 and LC3-II in starved N2a cells (Fig. 4d, Supplemental Figure 5) while co-overexpression of 14-3-3γ effectively reversed DFP-reduced Beclin-1 and LC3-II in starved N2a cells (Fig. 4e, Supplemental Figure 6). These data together demonstrated that 14-3-3γ played an essential role in controlling Beclin-1-LC3-autophage activation in neuronal cells upon starvation.

Effects of 14-3-3γ overexpression and knockout on starvation-induced Beclin-1-LC3-autophagy in neuronal cells. a Effects of 14-3-3 isoforms overexpression on GFP-LC3 puncta formation in EBSS-treated N2a cells. N2a cells were co-transfected with equal amounts of pGFP-LC3 and pcDNA-14-3-3γ or pcDNA-14-3-3τ or pcDNA-14-3-3ζ plasmids for 24 h and then subjected to EBSS incubation. Representative micrographs and statistical analysis demonstrated that only 14-3-3γ overexpression significantly increased autophagosome formation in starved N2a cells. *P < 0.05 and **P < 0.01 (n = 3). Bar 20 µm. b Representative Western blotting results showed that 14-3-3γ overexpression evidently increased GFP-LC3-II and Beclin-1 expression in N2a cells upon EBSS incubation. The experiments were repeated three times. c Representative Western blotting results showed that 14-3-3γ knockout (ΚΟ) prominently reduced GFP-LC3-II and Beclin-1 in N2a cells upon EBSS incubation. The experiments were repeated three times. d Representative Western blotting results showed that overexpressing specific 14-3-3 blocking peptide DFP evidently reduced GFP-LC3-II and Beclin-1 in N2a cells upon EBSS incubation. The experiments were repeated three times. e Representative Western blotting results showed that overexpressing 14-3-3γ reversed DFP-reduced GFP-LC3-II and Beclin-1 in N2a cells upon EBSS incubation. The experiments were repeated three times
14-3-3γ Promotes Beclin-1-LC3 Signaling via Binding More p-β-Catenin but Less Beclin-1 in Neuronal Cells
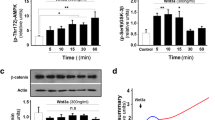
Considering a major role of 14-3-3 proteins in protein–protein interactions, we further examined the interactions between 14-3-3γ and Beclin-1 in starved N2a cells as 14-3-3-Beclin-1 interactions are recently considered a key regulatory mechanism in 14-3-3-mediated autophagy [14]. Unexpectedly, GST-pull down assay showed that 14-3-3γ-bound Beclin-1 was decreased in starved N2a cells (Fig. 5a). Then, we tested whether 14-3-3γ may exert its function via binding to transcriptional factor β-catenin, which is also up-regulated in ischemic neurons [5]. Indeed, co-immunoprecipitation showed that 14-3-3γ bound more endogenous p-β-catenin in starved N2a cells (Fig. 5b).

14-3-3γ binds to more p-β-catenin and antagonizes β-catenin-inhibited Beclin-1-LC3 signaling in starved neuronal cells. a Representative Western blotting results showed the interaction of 14-3-3γ and Beclin-1 in starved N2a cells. N2a cells were co-transfected with pGST-14-3-3γ and pcDNA-Beclin-1 plasmids for 24 h and then subjected to EBSS incubation. Four hundred micrograms of total soluble proteins from each culture were subjected to GST pull-down followed by Western blotting analysis. WCL whole cell lysate. b Representative Western blotting results showed the interaction of 14-3-3γ and endogenous p-β-catenin. c Representative Western blotting results showed that β-catenin overexpression reduced Beclin-1 and GFP-LC3-II in EBSS-treated N2a cells. d, e Representative Western blotting results showed that 14-3-3γ or Beclin-1 overexpression reactivated GFP-LC3 cleavage in the presence of β-catenin overexpression in EBSS-treated N2a cells. f Proposed 14-3-3γ-mediated autophagic signaling in ischemic/starved neurons. All experiments were repeated three times
Further, we examined whether 14-3-3γ may activate autophagic signaling via β-catenin or not. Western blotting analysis demonstrated that β-catenin overexpression evidently reduced Beclin-1 and LC3-II levels in starved N2a cells (Fig. 5c, Supplemental Figure 7). Co-overexpression of 14-3-3γ rescued β-catenin-suppressed LC3 activation (Fig. 5d). Finally, Beclin-1 overexpression effectively reversed β-catenin-suppressed LC3 activation in starved N2a cells (Fig. 5e). These data together demonstrated that 14-3-3γ promoted Beclin-1-LC3 autophagic signaling in neuronal cells via binding to β-catenin upon starvation.
Discussion
In the present study, we found that starvation selectively up-regulated the γ isoform of 14-3-3 proteins, which correlated to autophagy activation in neurons in vitro and in vivo. 14-3-3γ up-regulated Beclin-1 and promoted LC3 cleavage in neuronal cells upon starvation, while β-catenin functioned oppositely. 14-3-3γ bound more p-β-catenin but less Beclin-1, suggesting that 14-3-3 proteins control neuronal autophagy via binding to β-catenin and then altering its signaling.
We are the first to clarify the functional roles of 14-3-3 isoforms in starvation-induced neuronal autophagy. Overexpression of DFP, which specifically blocks the interactions of all 14-3-3 proteins with their ligands, prominently inhibited Beclin-1 expression and suppressed LC3 activation in starved N2a cells, supporting that the overall effect of 14-3-3 proteins in neuronal cells was to promote induction (representing marker Beclin-1) and expansion (representing marker LC3) of autophagy. Overexpression of 14-3-3γ alone effectively abolished DFP-inhibited Beclin-1 and LC3 activation (Fig. 4e), suggesting an essential role of 14-3-3γ in regulating neuronal autophagy. Indeed, knocking-out endogenous 14-3-3γ alone completely suppressed Beclin-1 induction as well as LC3 activation in starved N2a cells (Fig. 4c). Further, overexpression 14-3-3γ alone confirmed its positive regulatory role in neuronal autophagy. However, overexpression of other 14-3-3 isoforms did not increase neuronal autophagosome (representing by LC3 puncta) (Fig. 4a). Combining with selective 14-3-3γ induction in starved N2a cells and neurons, our data strongly supported that the γ isoform of 14-3-3 was a key positive regulatory factor for neuronal autophagy upon starvation.
We elucidated a key pathological mechanism by which 14-3-3 regulates neuronal autophagic signaling. As a specific marker at the initial stage of autophagy, Beclin-1 was prominently upregulated in neurons upon ischemic insults [21, 22], suggesting a major role of Beclin-1-mediated signaling in pathological neuronal autophagy. β-catenin is a critical transcriptional factor highly associated with ischemic stroke [23]. Overexpressing β-catenin evidently reduced Beclin-1, suggesting that β-catenin is an important negative regulator of Beclin-1. Upon ischemia, p-β-catenin is prominently increased and translocated into the nuclei together with 14-3-3γ [5]. We speculate that the increased 14-3-3γ-p-β-catenin complex in the nuclei may antagonized unphosphorylated β-catenin function and thus upregulates Beclin-1. In addition, 14-3-3γ-p-β-catenin may function as a transcriptional factor to upregulate Beclin-1 directly. Other factors such as TNF-α and NF-κB are also potential Beclin-1 regulators in ischemic brains, which deserves for further investigations [24, 25]. In addition to controlling Beclin-1 expression, 14-3-3 also bind Beclin-1 directly. It is reported that 14-3-3 suppresses autophagy in cancer cells via binding to p-Beclin-1 [14]. We detected less 14-3-3γ-Beclin-1 in starved N2a cells, suggesting that a sift of 14-3-3-Beclin-1 to 14-3-3-β-catenin interaction is also a mechanism by which 14-3-3 promotes neuronal autophagy upon energy stress.
In summary, 14-3-3γ upregulates Beclin-1 and activated LC3-autophagic influx in neuronal cells upon starvation or ischemia via binding to p-β-catenin (Fig. 5f). Ischemia-induced 14-3-3γ-p-β-catenin-Beclin-1-LC3-autophagy is likely a major event in ischemic brains, which provides novel therapeutic strategies for neurological diseases such as stroke.
Abbreviations
- DFP:
-
Dimeric 14-3-3 peptide inhibitor (difopein)
- EBSS:
-
Earle’s balanced salt solution
- GFP:
-
Green fluorescent protein
- GST:
-
Glutathione S-transferase
- sgRNA:
-
Single guide RNA
- Ipsi:
-
Ipsilateral cortex
- Contra:
-
Contralateral cortex
- YFP:
-
Yellow fluorescent protein.
References
Jia H, Liang Z, Zhang X, Wang J, Xu W, Qian H (2017) 14-3-3 proteins: an important regulator of autophagy in diseases. Am J Transl Res 9:4738–4746
Xu J, Huai Y, Meng N, Dong Y, Liu Z, Qi Q, Hu M, Fan M, Jin W, Lv P (2017) L-3-n-butylphthalide activates Akt/mTOR signaling, inhibits neuronal apoptosis and autophagy and improves cognitive impairment in mice with repeated cerebral ischemia-reperfusion injury. Neurochem Res 42:2968–2981
Huang JR, Tan GM, Li Y, Shi Z (2017) The emerging role of cables1 in cancer and other diseases. Molecular pharmacology 92:240–245
Chen XQ, Fung YW, Yu AC (2005) Association of 14-3-3gamma and phosphorylated bad attenuates injury in ischemic astrocytes. J Cereb Blood Flow Metab 25:338–347
Lai XJ, Ye SQ, Zheng L, Li L, Liu QR, Yu SB, Pang Y, Jin S, Li Q, Yu AC, Chen XQ (2014) Selective 14-3-3gamma induction quenches p-beta-catenin Ser37/Bax-enhanced cell death in cerebral cortical neurons during ischemia. Cell Death Dis 5:e1184
Chen XQ, Liu S, Qin LY, Wang CR, Fung YW, Yu AC (2005) Selective regulation of 14-3-3eta in primary culture of cerebral cortical neurons and astrocytes during development. J Neurosci Res 79:114–118
Zhou XY, Hu DX, Chen RQ, Chen XQ, Dong WL, Yi CL (2017) 14-3-3 isoforms differentially regulate NFkappaB signaling in the brain after ischemia-reperfusion. Neurochem Res 42:2354–2362
Li H, Guo Y, Teng J, Ding M, Yu AC, Chen J (2006) 14-3-3gamma affects dynamics and integrity of glial filaments by binding to phosphorylated GFAP. J Cell Sci 119:4452–4461
Cavallucci V, Bisicchia E, Cencioni MT, Ferri A, Latini L, Nobili A, Biamonte F, Nazio F, Fanelli F, Moreno S, Molinari M, Viscomi MT, D’Amelio M (2014) Acute focal brain damage alters mitochondrial dynamics and autophagy in axotomized neurons. Cell Death Dis 5:e1545
Khan MW, Layden BT, Chakrabarti P (2018) Inhibition of mTOR complexes protects cancer cells from glutamine starvation induced cell death by restoring Akt stability. Biochim Biophys Acta 1864:2040–2052
Kang R, Zeh HJ, Lotze MT, Tang D (2011) The Beclin 1 network regulates autophagy and apoptosis. Cell Death Differ 18:571–580
Wild P, McEwan DG, Dikic I (2014) The LC3 interactome at a glance. J Cell Sci 127:3–9
Clapp C, Portt L, Khoury C, Sheibani S, Norman G, Ebner P, Eid R, Vali H, Mandato CA, Madeo F, Greenwood MT (2012) 14-3-3 protects against stress-induced apoptosis. Cell Death Dis 3:e348
Wang RC, Wei Y, An Z, Zou Z, Xiao G, Bhagat G, White M, Reichelt J, Levine B (2012) Akt-mediated regulation of autophagy and tumorigenesis through Beclin 1 phosphorylation. Science 338:956–959
Qiu XY, Hu DX, Chen WQ, Chen RQ, Qian SR, Li CY, Li YJ, Xiong XX, Liu D, Pan F, Yu SB, Chen XQ (2018) PD-L1 confers glioblastoma multiforme malignancy via Ras binding and Ras/Erk/EMT activation. Biochim Biophys Acta 1864:1754–1769
Yang Y, Qiu JG, Li Y, Di JM, Zhang WJ, Jiang QW, Zheng DW, Chen Y, Wei MN, Huang JR, Wang K, Shi Z (2016) Targeting ABCB1-mediated tumor multidrug resistance by CRISPR/Cas9-based genome editing. Am J Transl Res 8:3986–3994
Xie XQ, Zhang P, Tian B, Chen XQ (2017) Downregulation of NAD-Dependent Deacetylase SIRT2 Protects Mouse Brain Against Ischemic Stroke. Mol Neurobiol 54:7251–7261
Xiong XX, Pan F, Chen RQ, Hu DX, Qiu XY, Li CY, Xie XQ, Tian B, Chen XQ (2018) Neuroglobin boosts axon regeneration during ischemic reperfusion via p38 binding and activation depending on oxygen signal. Cell Death Dis 9:163
Mizushima N, Yoshimori T, Levine B (2010) Methods in mammalian autophagy research. Cell 140:313–326
Xing SS, Yang XY, Zheng T, Li WJ, Wu D, Chi JY, Bian F, Bai XL, Wu GJ, Zhang YZ, Zhang CT, Zhang YH, Li YS, Jin S (2015) Salidroside improves endothelial function and alleviates atherosclerosis by activating a mitochondria-related AMPK/PI3K/Akt/eNOS pathway. Vascul Pharmacol 72:141–152
Wang P, Liang J, Li Y, Li J, Yang X, Zhang X, Han S, Li S, Li J (2014) Down-regulation of miRNA-30a alleviates cerebral ischemic injury through enhancing beclin 1-mediated autophagy. Neurochem Res 39:1279–1291
Song DD, Zhang TT, Chen JL, Xia YF, Qin ZH, Waeber C, Sheng R (2017) Sphingosine kinase 2 activates autophagy and protects neurons against ischemic injury through interaction with Bcl-2 via its putative BH3 domain. Cell Death Dis 8:e2912
Wang W, Li M, Wang Y, Li Q, Deng G, Wan J, Yang Q, Chen Q, Wang J (2016) GSK-3beta inhibitor TWS119 attenuates rtPA-induced hemorrhagic transformation and activates the Wnt/beta-catenin signaling pathway after acute ischemic stroke in rats. Mol Neurobiol 53:7028–7036
Li W, Yang X, Zheng T, Xing S, Wu Y, Bian F, Wu G, Li Y, Li J, Bai X, Wu D, Jia X, Wang L, Zhu L, Jin S (2017) TNF-alpha stimulates endothelial palmitic acid transcytosis and promotes insulin resistance. Sci Rep 7:44659
Bai XL, Yang XY, Li JY, Ye L, Jia X, Xiong ZF, Wang YM, Jin S (2017) Cavin-1 regulates caveolae-mediated LDL transcytosis: crosstalk in an AMPK/eNOS/ NF-kappaB/Sp1 loop. Oncotarget 8:103985–103995
Acknowledgements
This work was supported by grants from the National Nature Science Foundation of China (Grant Nos. 81471386, 81672504), the Integrated Innovative Team for Major Human Diseases Program of Tongji Medical College, HUST (Grant No. 5001530026), the Fundamental Research Funds for the Central Universities, HUST (Grant No. 2017KFYXJJ048), China Postdoctoral Scientific Foundation (Grant No. 2018M633237).
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
The authors declare no conflict of interest.
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
About this article

Cite this article
Xiong, X.X., Hu, D.X., Xu, L. et al. Selective 14-3-3γ Upregulation Promotes Beclin-1-LC3-Autophagic Influx via β-Catenin Interaction in Starved Neurons In Vitro and In Vivo. Neurochem Res 44, 849–858 (2019). https://doi.org/10.1007/s11064-019-02717-4
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s11064-019-02717-4