
Abstract
Ischemic stroke is characterized by high morbidity, mortality and disability rate worldwide. Because of its complexity in pathogenesis and lack of effective therapeutic strategies and drugs, great breakthrough has not yet been made in the treatment of cerebral ischemic stroke. Therefore, to explore a more effective and safer therapeutic strategy for cerebral ischemic stroke has been the focus of numerous researchers. Neuroprotective effects of sonic hedgehog (Shh) signaling pathway in ischemic stroke have been reported in recent studies, but have not been fully elucidated. In our review, we elaborate the roles of Shh signaling in ischemic stroke from different aspects, including oxidative stress, excitotoxicity, neuroinflammation, apoptosis, angiogenesis, neuroplasticity, neurogenesis, astrogliosis and oligodendrogenesis. Meanwhile, Shh signaling based therapeutic approaches for cerebral ischemic stroke are also included in our review. We hope it will benefit the readers to better understand the roles of Shh signaling pathway in cerebral ischemic stroke and provide more comprehensive insights for basic research and novel strategies for the clinical treatment of cerebral ischemic stroke.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Ischemic stroke (also referred to as “cerebral ischemia”) exhibits one of the highest rates of morbidity and disability, which leads to a heavy economic burden on society [1]. To date, thrombolysis is the only Food and Drug Administration (FDA)-approved therapy for the treatment of acute ischemic stroke patients. However, only a small number of patients benefit from thrombolysis therapy because of its narrow therapeutic window, and various co-morbid medical conditions limit the use of thrombolysis [2]. Thus, a novel therapeutic strategy to restore the central nervous system (CNS) and promote subsequent functional recovery for patients with cerebral ischemia is urgently needed. Abundant evidence has demonstrated the role of the sonic hedgehog (Shh) signaling pathway in CNS repair and regeneration after brain injury, including ischemia [3]. Shh signaling is up-regulated following CNS injury, and it is finely regulated depending on the severity and type of injury [4]. Numerous studies have investigated the roles of Shh in physiological and pathological processes and have made substantial progress. The present review provides an overview of the protective roles and potential mechanisms of the Shh signaling pathway in cerebral ischemia to provide a more comprehensive reference for basic researchers and to aid in the development of better clinical therapeutic interventions for ischemic stroke.
Characteristics of Sonic Hedgehog Signaling Pathway
Researches into Hh family for the past three decades have revealed many fundamental components of the Hh pathway. In mammals, the Hh signaling pathway is triggered by one of three family members, including Sonic hedgehog (Shh), Desert hedgehog (Dhh) and Indian hedgehog (Ihh) [5] (Fig. 1). Among the Hh homologues, Shh is the most widely studied one, which plays an important role in neurogenesis and neural patterning during the development of CNS [6]. Shh in humans is identified as a homology to the Hh in Drosophila. Shh is able to be proteolytically cleaved to form two secreted proteins: a 25 kDa C-terminal domain of Shh (Shh-C) acts as protease and 19 kDa N-terminal domain (Shh-N) mediates the signaling activities [6] (Fig. 1).

Schematic of the canonical Shh signaling pathway. In Hh secreting cells, the N-terminal fragment (HhN) is generated by full-length Hh in the modification of cholesterol. Hh signaling reception is facilitated by Ihog/Boi in Drosophila and Cdo/Boc/Gas1 in mammals. Once secreted, the biologically active N-terminal fragment of Shh (Shh-N); exerts its effects by binding to patched1 (Ptch1) with high affinity. The combination of Shh and Ptch1 blocks the normal inhibition of smoothened (Smo). Within the nucleus of the responding cell, zinc-finger transcription factors of the Ci/Gli family (Gli1-3) act in the last known step of the Shh pathway
Dispatched (Disp) and Patched1 (Ptch1) are both transmembrane proteins and have similar sequence. Disp is required for the secretion of cholesterol modified Shh, while Ptch1is functioning as a receptor for the Shh [7] (Fig. 1). In mammals, the primary cilium is the main target site for the Shh binding to Ptch1 and Shh cannot exert signaling activity in cells without the combination to the primary cilium [8]. In the absence of Shh, Ptch1 inhibits the activity of G protein-coupled receptor Smoothened (Smo) [9]. Smo is a transmembrane protein that allows downstream nuclear translocation of glioma-associated oncogene homolog 1 (Gli1), a key regulator of the Shh pathway [4] (Fig. 1). The transcription factor Gli3 functions as a transcriptional inhibitor, while Gli2 acts as a transcriptional activator in the stimulation of Shh and can trigger the transcription of Gli1 [10] (Fig. 1). Inactivation of Gli1 results in the repression of Shh signaling pathway and its activation presents a high level of the pathway activity [11]. Gli1 up-regulates the expression of target genes downstream of Shh pathway, including Gli1 itself and Ptch1 [10]. Binding of Shh to Ptch1, leads to removal of its inhibitory effect on Smo followed by activation of downstream pathway and consequently initiates the expressions of target genes associated with cellular cycle progression [12]. Thus, Ptch1 and Gli1 are both regular components and transcriptional targets of the Shh pathway. Especially, nuclear translocation of Gli1 leads to the activation of its target genes, including cyclin E, cyclin D, N-Myc, Bcl-2, mammalian achaete scute homolog-1 (Mash1), and Bmi1 which are involved in cell proliferation, anti-apoptosis, adult neurogenesis, and self-renewal [13] (Fig. 1). The steroidal alkaloid cyclopamine, a natural Shh antagonist, induces conformational change of Smo and impedes the nuclear translocation of its downstream components [14].
The high affinity of Shh binding to Ptch1 (in mammals) requires the presence of at least one of three co-receptors: Boc (Boi, counterpart in drosophila), Cdo (Ihog, counterpart in drosophila) or Gas1 [9]. Boc (Boi) and Cdo (Ihog) are transmembrane proteins, but Gas1 (no homolog in drosophila) binds to cell membrane via a GPI anchor [15] (Fig. 1). Shh can affect cells 300 µm away from the secretion site in an autocrine and paracrine manner [12]. The three co-receptors are found to up-regulate Shh pathway and make cells sensitive to low level of Shh released from a remote site [16]. The definite mechanism mediates the indirect inhibitory effect of Ptch on Smo remains elusive [6]. The genetic details of the Shh pathway have been well characterized, however, more precise mechanisms underlying the multiple steps in the movement of Shh ligand and the nuclear transduction of Shh signaling need to be further elucidated.
There are three non-canonical Shh signaling pathways besides the aforementioned canonical one: Type I, core components of Shh signaling interacting with others in a " non-contiguous " or “atypical” way; Type II, the signaling components directly interact with constituents of other molecular pathways and opposed to the indirect regulation of Gli-mediated transcription cellular processes; Type III, signaling pathway which functions through Smo in an Gli- independent way or via Ptch1 in an Smo-independent way [6, 17, 18]. It is worth noting that, despite the presence of non-canonical pathways, the canonical Shh signaling pathway is the principal mechanism used for interpreting most relevant consequences of disturbed Shh signaling. The non-canonical signaling pathways serve as a modifier to the canonical pathway, which may play a role in moderating disruptions, fluctuations or play as a checkpoint in the pathway [17].
Potential Mechanisms of the Neuroprotective Effects of Shh in Cerebral Ischemia
Recent studies have suggested that Shh can not only play a positive role in neurogenesis of the CNS, but also exhibit neuroprotective effects in adults [3, 19]. Animal models of stroke have confirmed this hypothesis. Moreover, the activation of specific Shh pathway components protected brain function after ischemia [20].
Oxidative Stress
Oxidative stress is the primary mechanism of cerebral ischemia [21]. An overproduction of free radicals or dysfunction of the body’s anti-oxidation system creates an imbalance in oxygen free radical metabolism in vivo, which results in oxidative stress [22]. Brain tissues are particularly vulnerable to oxidative stress because of their high oxygen utilization as well as the high levels of polyunsaturated fatty acids and low levels of anti-oxidant enzymes in neural cell membranes. Cerebral ischemia increases the level of reactive oxygen species (ROS) in cerebral tissue. Excessive ROS generation activates diverse signaling pathways and regulates the expression of genes that encode various pro-inflammatory proteins [21, 22] (Fig. 2). Several beneficial factors are induced to combat ischemia-induced injury following stroke onset [20].

Simplified diagram of the role of the Shh signaling pathway in oxidative stress, excitotoxicity, neuroinflammation and apoptosis. The Shh signaling pathway can exert protective roles in the cerebral ischemia via the inhibition of oxidative stress, excitotoxicity, inflammation and apoptosis of neurons. The Shh/PI3K/Akt pathway may be the underlying mechanism. Shh signaling pathway activation exerts protective effects by increasing anti-oxidant enzymes (such as superoxide dismutase (SOD) and glutathione peroxidase (GSH-PX)), decreasing apoptotic genes (such as P53 and caspase-3), and increasing anti-apoptotic genes (such as Bcl-2) and BDNF. Shh is also involved in protecting neurons against NMDAR-dependent excitotoxicity
Shh attenuates protein oxidation and lipid peroxidation following middle cerebral artery occlusion (MCAO). MCAO is a classical and generally accepted model to induce cerebral ischemia in rats/mice [23]. The Shh signaling pathway plays an important role in protecting against oxidative stress by increasing the activities of the anti-oxidant enzymes glutathione peroxidase (GSH-PX) and superoxide dismutase (SOD) [24] (Fig. 2). Rats with permanent middle cerebral artery occlusion (pMCAO) exhibit a significant increase in Ptch1, Gli1 and SOD1 expression in ischemic cortex-at 6 h, 12 h, 24 h and 48 h after pMCAO, but not 72 h after pMCAO. Ptch1, Gli1 and SOD1 may play a role in pathological progression during the acute stage [25]. Inhibition of the Shh pathway exacerbates neurological deficits and increases the brain water content and infarct size during the acute phase of cerebral ischemia, which are caused by the down-regulation of Ptch1, Gli1 and SOD1 [20] (Fig. 2). Hydrogen peroxide (H2O2) is widely used to simulate oxidative stress because it is the primary type of ROS and exhibits membrane permeability [26]. A previous study used H2O2 to induce oxidative stress in rat primary cortical neurons and identified activation of the endogenous Shh pathway in cortical neurons under oxidative stress [24]. Brain-derived neurotrophic factor (BDNF) is an important neurotrophic factor that widely exists in the brain and is reported to have protective effects against oxidative stress [27]. H2O2 treatment induces a slight increase in BDNF expression in primary neurons, while exogenous Shh pretreatment significantly upregulates the expression of BDNF in primary neurons. This effect of exogenous Shh is partially reversed by cyclopamine (a specific antagonist of Shh signaling) treatment [24]. Therefore, activation of Shh signaling can exert neuroprotective effects against oxidative stress, partly via the regulation of BDNF (Fig. 2).
Oxidative stress also activates mitogen-activated protein kinases (MAPKs), which include several important members, such as extracellular signal-regulated kinase (ERK), p38 kinase, and phosphoinositol 3-kinase (PI3K) [28]. Increasing evidence has indicated that ERK inhibition protects neurons from death and apoptosis induced by oxidative stress [29]. The PI3K pathway exerts protective effects via activation of its direct downstream effector, Akt kinase, which controls multiple biological processes including cell growth, proliferation, survival, apoptosis and glycogen metabolism [30]. ERK, PI3K/Akt and the Shh pathway are involved in cell apoptosis and survival during oxidative stress, and further research examined the relationship between these factors. H2O2-induced oxidative stress in cultured cortical neurons did not influence the expression of Akt, ERK and p38 proteins; however, it decreased phosphorylated-Akt (p-Akt) and increased phosphorylated-ERK (p-ERK) and phosphorylated-p38 (p-38). Exogenous Shh treatment increased p-Akt expression and alleviated p-ERK expression, which was partially reversed following the addition of an inhibitor (cyclopamine) of the Shh pathway. However, Shh treatment did not impact the phosphorylation of p38 protein expression [31]. The neuroprotective effects of Shh against oxidative stress are independent of the p38 MAPK pathway; however, it is associated with PI3K/Akt pathway activation and ERK pathway inhibition (Fig. 2).
Excitotoxicity, Neuroinflammation and Apoptosis
Excitotoxicity is an important mechanism in cerebral ischemia, and it refers to the hyperactivation of glutamate receptors, in which the n-methyl-d-aspartate receptor (NMDAR) plays a key role due to its high Ca2+ permeability [32]. Overexpression of NMDAR leads to neuronal death via the induction of a cascade of downstream signaling molecules [33]. Superfluous Ca2+ influx through NMDARs activates the overexpression of nitric oxide (NO) via the activation of neuronal nitric oxide synthase (nNOS), which exacerbates neuronal injury [34, 35]. nNOS promotes the transcriptional activity of Shh, which is a compensatory mechanism to protect neurons from NMDAR-mediated excitotoxicity and cerebral ischemic stroke [36] (Fig. 2).
Neuroinflammation is destructive and protective in brain injury and is a trigger for Shh signaling [37,38,39]. Cytokine exposure to astrocytes activates the NF-κB pathway directly in vivo and induces Shh transcription in these cells. Increased neuroinflammation and the upregulation of Gli1 in the ipsilateral cortex suggest Shh pathway activation in the early stage after stroke [38] However, increased Gli1 levels and the lack of neuroinflammation on the contralateral side indicate that neuroinflammation is not the only trigger of Shh signaling after stroke. Neuroinflammation may be a neuroprotective mechanism in the early stages of stroke, during which cell proliferation is induced to repair the injured blood–brain barrier (BBB) [38, 40] A favorable argument for its positive role in neuroinflammatory signals during acute brain ischemia is that treatment with various anti-inflammatory agents is largely unsuccessful or detrimental; this finding indicates that neuroinflammation aids in the recovery from brain ischemia via the upregulation of Shh, at least in the acute stage. However, the failure to restrict neuroinflammation ultimately results in widespread neuronal death in the later stage of brain ischemia [38]. Neuronal death caused by cerebral ischemia can triggers local immune responses, which result in microglial activation and leukocyte infiltration to the injured brain tissue. This activation produces multiple inflammatory mediators and leads to BBB damage, cerebral edema, hemorrhage and neuronal death [41]. Activation of Shh decreases the mRNA levels of the microglia/ macrophage marker CD11b and the pro-inflammatory cytokines IL6, TNFα and IL1β in the ischemic cortex in the late stage after MCAO [4]. Shh also regulates the inflammation via a reduction of NF-κB activation, which is a well-established upstream target of iNOS [25]. Therefore, Shh provides a protective effect for stroke via the regulation of microglia/macrophages and a series of related inflammatory cytokines (Fig. 2).
Apoptosis is an active, programmed cell death process that activates by intrinsic and extrinsic signals [42]. Shh significantly upregulated the expression level of Bcl-2 (an anti-apoptotic protein) and downregulated the expression level of Bax (a pro-apoptotic protein) in the primary cultured neurons of rats with H2O2-induced oxidative stress compared to H2O2 treatment alone, and the addition of the PI3K/Akt pathway blocker LY294002 or the Shh blocker cyclopamine reversed these effects. The PI3K/Akt pathway is necessary for the anti-apoptotic effects of Shh and the Shh/PI3K/Bcl-2 pathway, which is the underlying mechanism to protect impaired neurons against oxidative stress-induced apoptosis [31]. Cerebral ischemia is characterized by high levels of oxidative stress in neurons, which leads to necrosis and apoptosis. Neuronal ischemic necrosis is difficult to reverse; however, apoptosis is able to be altered via the regulation of its upstream signals. The treatment strategy for ischemic stroke is aimed at apoptotic cells, which are also the target for drug action [43]. Shh is a necessary survival factor in various cell types, and the Shh signaling pathway inhibits apoptosis [44]. Gli1and Gli3 upregulate the anti-apoptotic Bcl2 gene, which is dependent on the canonical Shh signaling pathway [45, 46] (Fig. 2). Ptch1 regulates apoptosis via caspase activation in the neural tube without the participation of Shh in the non-canonical signaling pathway [47]. Shh signaling promotes cell survival via inhibition of the p53 pathway, which is essential for the acceleration of apoptosis [48] (Fig. 2). Shh also suppresses cell death by reducing the generation of active caspase-3, which is an executioner enzyme of apoptosis [49] (Fig. 2). Shh decreases the number of TUNEL+ cells in the cortices of rats in a stroke model, which is consistent with its anti-apoptotic role [13]. Therefore, the elucidation of the underlying mechanism by which the Shh signaling pathway exerts anti-apoptotic effects in cerebral ischemia may improve the therapeutic approach to cerebral ischemic stroke.
Angiogenesis
In general, stroke is caused by a rapid cerebrovascular stenosis or occlusion, and it is always accompanied by the necrosis of brain cells and deficits in neurological function [50]. Organisms possess multiple compensatory mechanisms to increase the oxygen supply in ischemic brain tissue, which promotes angiogenesis [51]. Angiogenesis is defined as the formation of new capillary networks via vascular endothelial cell proliferation, migration, sprouting, vascular division and branching of the original blood vessels [52]. Angiogenesis in ischemic brain tissue is an important factor in determining local blood flow, neuronal restoration and regeneration, and the recovery of neurological function. Angiogenesis is also the basis for the reconstruction and remodeling of synaptic links in neurons [53]. Early collateral circulation of ischemic stroke depends on the openness of the original vascular network, and it relies heavily on neovascularization in the later stage. New blood vessels begin to appear 3 days after cerebral ischemia and continue to increase until at least 21 days [54]. Stroke patients with a high cerebral vascular density exhibited a better prognosis than patients with a lower vascular density [55]. Various angiogenic factors, such as vascular endothelial growth factor (VEGF) and angiopoietin-1/angiopoietin-2 (Ang-1/Ang-2), promote angiogenic and vascular repair processes in an animal model of cerebral ischemia [56, 57].
Shh signaling is essential to the development of embryonic blood vessels and is able to be reactivated during vessel repair in adults [58]. However, the endogenous regulation of Shh signaling in ischemia is largely unknown; identification of the mechanism by which Shh promotes the angiogenesis of blood vessels may provide novel therapeutic targets for the treatment of ischemic diseases. Previous reports have demonstrated that Shh signaling partially rescued heart and skeletal muscle from ischemic injury via the promotion of angiogenesis [59, 60]. A recent study reported that Shh can also enhance post-ischemic angiogenesis in a pMCAO model [61]. VEGF is an important factor for angiogenesis in ischemic tissue, as it enhances vascular permeability [62]. Shh promotes angiogenesis and neuron survival by increasing VEGF expression (Fig. 3), and VEGF inhibition weakens the pro-angiogenic effects of Shh in models of ischemic stroke [61]. Shh promotes the formation of capillary-like tubes and the migration of rat brain micro-vessel endothelial cells (RBMECs), partially via VEGF. It also significantly promotes cell proliferation and angiogenesis via the upregulation of Ki-67 (a marker for cell proliferation) and CD31 (a marker for cell proliferation) after pMCAO. In contrast, administration of a VEGF antibody or cyclopamine decreases the pro-angiogenesis effect of Shh [61]. However, Shh-induced blood vessels exhibit more smooth muscle cells and greater lumens than VEGF-induced vessels [63], which suggests that Shh exerts stronger effects on the promotion of angiogenesis than VEGF, and the underlying mechanism must be further clarified.

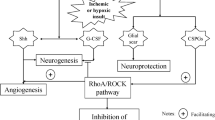
Simplified diagram of role of the Shh signaling pathway in angiogenesis, neurogenesis, astrogliosis and oligodendrogenesis. The Shh signaling pathway improves the recovery of neurological function after cerebral ischemic stroke by increasing angiogenesis, neurogenesis, and oligodendrogenesis and reducing astrogliosis. Granule neurons in the dentate gyrus of the hippocampus and olfactory bulb interneurons are continuously produced in adults. The Shh pathway in the SVZ is involved in generating specific neuronal progeny. Shh induces angiogenesis by increasing the expression of VEGF and Ang-1, which contribute to neurogenesis. Shh secreted by astrocytes interacts with cerebral endothelial cells or attenuates glial scar formation to facilitate the repair of BBB integrity
Shh increases Ang-1 expression, which is crucial for the sprouting of endothelial cells (Fig. 3). Ang-1 prevents vasogenic brain edema via the inhibition of VEGF-induced vascular leakage [64]. Shh decreases Ang-2 expression, which is a negative modulator of Ang-1. Cyclopamine significantly reverses this pattern of Shh-mediated Ang-1/Ang-2 gene expression, and it blocks the Shh signaling pathway through Smo. These results suggest that Ang1/Ang-2 are downstream of Gli1 [63]. Shh promotes cerebral angiogenesis via activation of the RhoA/ROCK pathway or the nuclear receptor NR2F2, which are part of the non-canonical pathway, in an in vitro model of cerebral ischemia [65, 66]. Thus, Shh promotes cerebral angiogenesis via both canonical and non-canonical pathways.
Zonula occludens-1 (ZO-1) is a well-studied tight junction protein that reflects the pathological changes of the BBB, and it is a valuable marker of the endothelial barrier [67]. Shh-triggered Ang-1 is primarily produced in astrocytes under ischemic injury, and secreted Ang-1 acts on brain microvascular endothelial cells to upregulate ZO-1, which repairs the tight junction and limits brain edema and BBB leakage (Fig. 3). Cyclopamine does not reverse this effect, which suggests that Shh upregulates Ang-1 in astrocytes via non-canonical signaling and in a Gli-1 independent manner [64].
Vessels with newly formed endothelial cells have been identified in the peri-infarct area following Shh agonist treatment in a mouse stroke model [4]. Capillary tube formation and migration are widely used to evaluate angiogenesis in in vitro experiments [68]. Shh increases the number of luminal formation and tubular branches of brain microvessels. Moreover, Shh increases the permeability, migration distance and vitality of cerebral microvascular endothelial cells [61]. However, the associated mechanisms of Shh promotion of angiogenesis and vascular maturation require further examination.
Neuroplasticity
The capacity of neural regeneration is limited; however, the CNS circuitry exhibits substantial neuroplasticity after injury, which is vital for the CNS to recover from functional deficits [69]. Neuroplasticity is characterized by anatomical remodeling and functional restoration of the CNS, and various factors interfere with neuroplasticity and its assessment [70]. The formation of new synapses, axonal sprouting and growth factors secreted from surviving neurons and glia play a role in neuroplasticity [69]. The following three mechanisms are involved in brain neuroplasticity: (1) activation of functional pathways that are silent prior to injury; (2) compensatory regulation from parallel brain circuits and pathways to restore impaired functions; and (3) new synapse formation and sprouting of survival neurons. The first two mechanisms are short-term mechanisms, and the third mechanism is typical in long-term plasticity [70]. Shh is a pivotal regulator of multiple pathways in the developing, postnatal and adult brain and is proved to promote brain remodeling process [71]. Shh promotes neurite outgrowth of primary cortical neurons partially by regulating the expression of BDNF which plays multiple roles in maintaining and promoting brain plasticity [72]. Activation of Shh signaling stimulates axonal elongation in hippocampal neurons and accelerates its interaction with target neurons and synaptic connections as part of the recovery process for the ischemic stroke [73]. Selective inhibition of Shh signaling impairs the ability of mesenchymal stem cells to promote synaptogenesis, neurite outgrowth and functional recovery after focal ischemic stroke in mice [74]. Therefore, interventions to activate the Shh signaling will contribute to the promotion of neural restoration and functional recovery.
Neurogenesis
Neurogenesis has been observed in animal ischemic stroke models and the adult human ischemic boundary particularly in the sub ventricular zone (SVZ) [75]. The SVZ is the largest germinal zone in the adult brain, and it generates various immature neurons of different types daily. Shh protein is present in cells positive for the neuronal marker protein NeuN in the apical and basal surfaces of the SVZ [3, 16] Pharmacological and genetic research has demonstrated that the Shh pathway is vital for the post-natal maintenance and self-renewal of SVZ neural stem cells, the generation of transit-amplifying progeny, and the migration of the neuroblast. Progenitors located in the ventral SVZ respond to high levels of Shh and primarily generate interneurons of the deep granule. Strong Shh pathway activation overrides the intrinsic programming of neural progenitors, and the reprogramming of neural stem cells induces progenitors within the SVZ to differentiate into neurons of certain types, depending on relevant molecular signals [16] (Fig. 3). Newly generated neurons in the SVZ exhibit properties of stem cells and play a critical role in the remodeling process after cerebral ischemia [3]. Cerebral infarction increases neuronal proliferation, which leads to an early expansion of the neural progenitor pool in the SVZ. The neural progenitor cells differentiate into neuroblasts, which migrate from the progenitor pool of the SVZ to the ischemic cortex and striatum [76]. Endogenous neural progenitors migrate from germinal zones to brain damaged areas and differentiate into mature neurons (and/or glial cells) that blend with the pre-existing neurons after cerebral ischemia [14, 69].
Increasing evidences suggests that Shh is involved in the regulation of adult neurogenesis. Shh expression and its transcription factor Gli1 in neural progenitor cells (NPCs) are up-regulated in the ischemic brain. Activation of the Shh pathway improved motor function in stroke mice, which indicates a protective role for Shh signaling in stroke [3, 24]. The Shh signaling pathway regulates neuronal proliferation, migration, and apoptosis [77]. Recent studies have demonstrated that the activation of Shh signaling pathway leads to high Gli1 levels in the ventral SVZ and the genesis of several specific progeny of neurons [16] (Fig. 3). The Shh pathway is critical in the generation of transit-amplifying progeny, the self-renewal of stem cells and neuroblasts migrations [78]. Shh organizes the development of neural tubes by establishing a different region of the homologous domain along the dorsal ventral axis [7]. Shh induces numerous transcription factors in ventral progenitor cells and determines the fates of different cell types using a gradient concentration and time-dependent mechanism [79]. Shh in the ventral forebrain is necessary for the generation of cells in the medial and lateral ganglionic eminences, and in the midbrain and hindbrain, it is necessary for the generation of dopaminergic and serotonergic neurons [7]. Shh controls the division of adult stem cells, which contribute to the maintenance of the neural stem cell niche in neurogenic regions such as SVZ, which is vital for adults [80] (Fig. 3). Therefore, the up-regulation of Shh expression may represent a primary mechanism of neurogenesis. Moreover, cyclopamine reverses this effect [81].
Astrogliosis and Oligodendrogenesis
It is widely accepted that neurons comprise less than 25% of the cells in the brain, and astrocytes are the most abundant cell type in the brain, accounting for 30–65% of glia [82]. Astrocytes exert multiple functions, including the maintenance of optimal ionic conditions and osmotic balance for neurons, the processing of information via neurotransmitter recycling and the maintenance of metabolic homeostasis [83]. Oligodendrocytes provide axonal support via the production of the myelin sheath, which is critical for rapid impulse conduction across white matter tracts [82]. Coordination between neurons and glial cells is vital to the functional and structural integrity of the CNS [84]. Injuries to neural tissues lead to a reestablishment of the microenvironment, which contains neurons, astrocytes, microglia cells, oligodendrocytes and other cell types [85]. Microglia secrete inflammatory cytokines and arrive at the injury site within a few hours, whereas proliferating oligodendrocytes and astrocytes are observed during 24–72 h [86]. However, the role of these cells in microenvironment reconstruction in the context of cerebral ischemia must be further clarified.
The expression of Shh receptors is up-regulated in the ischemic area of neurons, astrocytes, microglia and oligodendrocytes in the early stage of ischemic stroke [4] (Fig. 3). The Shh-Smo pathway reduces astrocyte apoptosis and results in astrocyte activation [72]. Reactive astrocytes exhibit high levels of glial fibrillary acidic protein (GFAP), and these cells are the primary components of the glial scar [38, 87]. The glial scar separates the injury tissue from healthy tissue, fills the cavity of the lesion area, reduces the infiltration of immune cells and promotes BBB repair [88]. The glial scar and inflammatory environment restrict axonal sprouting and post-injury tissue regeneration [72]. Upregulated Shh production is dependent on cellular sources of pro-inflammatory cytokines and inflammation and astrocytes, microglia, oligodendrocytes and endothelial cells in the ischemic area. Shh increases the neurite outgrowth of cortical neurons and attenuates the formation of the glial scar to play a protective role in cerebral ischemia (Fig. 3). Astrocyte apoptosis also contributes to astrogliosis, and Shh-Smo pathway activation reduces the apoptosis of cultured astrocytes [13, 38, 72].
Oligodendrocytes are the only myelin-generating cells in the CNS. Oligodendrocytes exhibit rapid-pulse electrical conduction and are particularly sensitive to ischemic injury [89]. The Shh pathway is essential for the survival, proliferation and migration of oligodendrocytes [90], and regulates oligodendrogenesis (Fig. 3). Myelinating oligodendrocytes play critical roles in the remodeling processes of white matter in brain ischemia. Cerebrolysin (a mixture of neurotrophic peptides) enhanced oligodendrogenesis and neurogenesis and improved the neurological outcome in animal models of cerebral ischemia. The Shh pathway has been proven to play vital roles in cerebrolysin-mediated neurorestoration in the ischemic brain. In vitro and in vivo data have demonstrated that inactivation of the Shh signaling pathway using the pharmacological blocker cyclopamine abolished the beneficial effects of cerebrolysin. Shh promoted the migration of oligodendrocyte progenitor cells (OPCs) that originated from the SVZ into white matter and promoted OPC differentiation into mature oligodendrocytes in the white matter of the peri-infarct region [71, 91]. Oligodendrocytes die after MCAO, which leads to demyelination and damage to axonal conduction [92]. The Shh pathway contributes to remyelination and improves the neurological functional recovery after stroke [93].
Pharmacological and Cell-Based Therapeutic Approaches for Cerebral Ischemic Stroke
Ischemic stroke always results in structural and functional damage. Thrombolytic therapy using tissue-type plasminogen activator (tPA) is the only FDA-approved treatment for stroke, however, the effectiveness and application are limited because of its narrow time window after ischemia [3]. Therefore, safer and more efficient therapeutic approaches are currently under investigation to promote brain remodeling and the subsequent functional recovery of stroke patients.
Shh Signaling in Pharmacological Therapy
Shh is upregulated in multiple cell types in ischemic brain, including neurons, activated astrocytes and neural stem cells (NSCs) in the SVZ niche [3]. Sims et al. demonstrated that Shh played a role in the regulation of ischemia-induced neural progenitor cells proliferation and it may participate in injury remodeling [81]. Direct application of Shh peptide produced an improvement in neurological outcome after cerebral ischemia [3]. Bambakidis et al. affirmed that intrathecal administration of Shh protein upregulated Gli1 transcription and increased the proliferation and migration of neural precursor cells in the injury site in the rat MCAO model, which improved behavioral recovery [94]. Delayed treatment (24 h after stroke) with Salvianolic Acids for Injection (SAFI) activated Shh signaling pathway, which significantly promoted neurogenesis and long-term functional recovery after MCAO in mice [95]. The area of ischemia and severity of behavioral deficits were exacerbated in Shh knockout mice with cortical stroke. Animals that received post-stroke treatment with a Shh signaling agonist exhibited fewer deficits in behavioral function than vehicle-treated mice [3]. Mice treated with the Shh signaling agonist exhibited higher values in many horizontal movement parameters 7 days after stroke than the control group, however, there were no significant differences in the pre-stroke measurements [3]. Afterwards, Luo and his colleagues further explored the effect of delayed treatment (3–8 days after stroke) of the Shh agonist on long-term functional recovery after stroke and found that delayed administration of the Shh agonist after stroke improved survival of adult NSCs in both SVZ and subgranular zone, enhanced angiogenesis and cerebral blood flow reperfusion of ischemic brain, as well as improved long-term functional recovery of stroke in mice [96].
Purmorphamine (PUR) is a small molecular agonist of the Shh co-receptor Smo that exerts protective effects in the MCAO model [97]. PUR treatment decreased BBB permeability, up-regulated tPA expression and reduced apoptosis in the ischemic cortex. Inflammation and astrogliosis were alleviated 14 days after cerebral ischemia in PUR-treated animals. It also promoted neuron regeneration and neovascularization in the ischemic zone [4]. Cerebrolysin is a mixture of neurotrophic peptides that plays an important role in the promotion of neuronal regeneration and functional recovery in rats with ischemic stroke in a Shh signaling pathway-dependent manner [91]. Resveratrol is a natural polyphenolic phytoalexin which significantly improves neurological function, reduces infarct volume, enhances vitality and decreases apoptosis of neurons after stroke in vivo and in vitro. Shh signaling pathway is proved to be one of the mechanisms by which resveratrol exerts the protective effects above [98]. Sirtuin l (Sirt1) is a critical member of sirtuins enzyme family which presents in the CNS. Upregulation of Sirt1 plays a protective role in neuronal plasticity after ischemic stroke [99]. Resveratrol has been demonstrated to enhance neurite outgrowth and synaptogenesis by upregulating the expression of Sirt1, which result from the activation Shh signaling [100]. Therefore, the identification of potential pharmacological activators of the Shh signaling pathway may advance the treatment of cerebral ischemic stroke via improvements in neurogenesis and functional recovery.
Shh Signaling in Cell-Based Therapy
Cell therapy is a method to simulate the neural restorative process that naturally occurs in the brain [70]. Bone marrow stromal cell (MSC) treatment promotes oligodendrogenesis and remyelination via Shh/Gli1 signaling pathway activation in rats subjected to MCAO [93]. The Shh signaling pathway also promotes brain plasticity by increasing tPA expression and achieves better improvements in neurological recovery after MSC treatment in stroke models [74]. Recombinant mouse Shh (rm-Shh) and co-culture with mesenchymal stromal cells decreased transforming growth factor-β1 (TGF-β1), which regulates the expression of PAI-1 in astrocytes. The Shh pathway inhibitor cyclopamine reversed these effects. These results indicate that mesenchymal stromal cells stimulate parenchymal cells to secrete Shh, which subsequently promotes cellular activation of tPA directly via an increase in tPA expression or indirectly via a decrease in TGF-β1/PAI-1 expression [101].
MSC treatment increases endogenous tPA and accelerates neurite outgrowth within the ischemic zone in mice subjected to MCAO, which is activated by the Shh pathway [102]. MSC mediates gene expression in parenchymal cells via the release of exosomes that contain microRNAs (miRNAs) [103]. miRNAs belong to the short non-coding RNA family, which exerts crucial effects on neural stem cells during brain development and is associated with physio-pathological progresses via the downregulation of gene expression through translational repression and/or mRNA destabilization [104]. The Shh pathway regulates the expression of the miR17-92 cluster in progenitor cells in the ischemic brain by mediating a downstream molecular of Shh, Myc. The miR17-92 cluster is a direct transcriptional target of c-Myc, and the promoter region of the miR17-92 cluster is bound to c-Myc to a higher degree in neural progenitor cells following ischemic stroke [76]. Therapy based on marrow stromal cells has exhibited remarkable potential for an improved neurological outcome when employed soon after stroke onset.
Umbilical cord blood cells exhibit a lower risk of rejection, immunogenicity, and a greater potential for differentiation than bone marrow stem cells [105]. Neural stem cells (NSCs) exist in brain tissues throughout the lifespan and are able to be activated in certain circumstances. Umbilical cord blood mononuclear cells (UCBMCs) secrete various cytokines, which regulate the immune inflammatory responses, alleviate brain damage, and promote NSCs proliferation via Shh signaling pathway activation [106]. UCBMCs also contain numerous immature stem cells, which secrete neurotrophic and nutritional factors and these factors are able to activate the Shh pathway [107]. Transplantation with UCBMCs promoted NSC proliferation by increasing the levels of Shh and Gli1 proteins in hypoxic ischemic rats within the hippocampus and cortex, and cyclopamine reversed the neuronal protective effects [106]. Proliferating NSCs differentiate into mature neurons, which are associated with the Shh signaling pathway via the up-regulation of Ngn1 (which activates neuronal transcription factors to promote neurogenesis and restrain glial differentiation) and the down-regulation of BMP4 (which is likely to inhibit neurogenesis) [108,109,110]. These two studies used neonatal rats; however, the adult brain substantially differs from the neonatal brain. Therefore, the analogous studies in the neonate must be replicated in the adult.
Conclusion and Future Perspectives
Cerebral ischemia leads to a series of pathological events and results in irreversible damage to neurons within a few minutes of stroke [82]. Various factors are involved in the damaged tissues after stroke, including oxidative stress, neuroinflammation and apoptosis [111]. This summary of the most recent research literature shows that the Shh signaling pathway exhibits positive neuroprotective effects in cerebral ischemic stroke via multiple pathway such as the inhibition of oxidative stress, mitigation of excitotoxicity, suppression of neuroinflammation, increase in angiogenesis, and promotion of neurogenesis.
Neuronal restorative strategies to promote brain remodeling and functional recovery after cerebral ischemia are under extensive investigation. Targeting the factors responsible for damage and the related cellular pathways may represent potential treatments for cerebral ischemia. The present review provides a better understanding of the neuroprotective mechanisms of the Shh pathway in cerebral ischemic stroke to promote new insights into novel therapeutics. The Shh pathway plays ongoing and vital roles in cerebral ischemia to improve the infarction volume and functional outcome after ischemic injury. Endogenous activation of the Shh pathway with more effectiveness in brain parenchymal cells is a promising restorative treatment for cerebral ischemia and a potential therapeutic strategy for other neurological diseases. This review further elucidates the neuroprotective mechanisms of the Shh signaling pathway and clarifies the specific interactions between its downstream and upstream components, which can aid in the discovery of specific targets for basic research and more effective strategies for clinical therapy.
References
Cassella CR, Jagoda A (2017) Ischemic stroke: advances in diagnosis and management. Emerg Med Clin N Am 35:911–930
Wechsler LR, Jovin TG (2012) Intravenous recombinant tissue-type plasminogen activator in the extended time window and the US Food and Drug Administration: confused about the time. Stroke 43:2517–2519
Jin Y, Raviv N, Barnett A, Bambakidis NC, Filichia E, Luo Y (2015) The shh signaling pathway is upregulated in multiple cell types in cortical ischemia and influences the outcome of stroke in an animal model. PLoS ONE 10:e0124657
Chechneva OV, Mayrhofer F, Daugherty DJ, Krishnamurty RG, Bannerman P, Pleasure DE, Deng W (2014) A smoothened receptor agonist is neuroprotective and promotes regeneration after ischemic brain injury. Cell Death Dis 5:e1481
Pereira J, Johnson WE, O’Brien SJ, Jarvis ED, Zhang GJ, Gilbert MTP, Vasconcelos V, Antunes A (2014) Evolutionary genomics and adaptive evolution of the hedgehog gene family (Shh, Ihh and Dhh) in vertebrates. PLoS ONE 9:e74132
Patel SS, Tomar S, Sharma D, Mahindroo N, Udayabanu M (2017) Targeting sonic hedgehog signaling in neurological disorders. Neurosci Biobehav Rev 74:76–97
Ho KS, Scott MP (2002) Sonic hedgehog in the nervous system: functions, modifications and mechanisms. Curr Opin Neurobiol 12:57–63
Nozawa YI, Lin CW, Chuang PT (2013) Hedgehog signaling from the primary cilium to the nucleus: an emerging picture of ciliary localization, trafficking and transduction. Curr Opin Genet Dev 23:429–437
Cleveland TE, McCabe JM, Leahy DJ (2014) Detergent-solubilized Patched purified from Sf9 cells fails to interact strongly with cognate Hedgehog or Ihog homologs. Protein Expr Purif 104:92–102
Traiffort E, Angot E, Ruat M (2010) Sonic Hedgehog signaling in the mammalian brain. J Neurochem 113:576–590
Ruiz i Altaba A, Mas C, Stecca B (2007) The Gli code: an information nexus regulating cell fate, stemness and cancer. Trends Cell Biol 17:438–447
Briscoe J, Therond PP (2013) The mechanisms of Hedgehog signalling and its roles in development and disease. Nat Rev Mol Cell Biol 14:416–429
Huang SS, Cheng H, Tang CM, Nien MW, Huang YS, Lee IH, Yin JH, Kuo TBJ, Yang CCH, Tsai SK, Yang DI (2013) Anti-oxidative, anti-apoptotic, and pro-angiogenic effects mediate functional improvement by sonic hedgehog against focal cerebral ischemia in rats. Exp Neurol 247:680–688
Spaccapelo L, Galantucci M, Neri L, Contri M, Pizzala R, D’Amico R, Ottani A, Sandrini M, Zaffe D, Giuliani D, Guarini S (2013) Up-regulation of the canonical Wnt-3A and Sonic hedgehog signaling underlies melanocortin-induced neurogenesis after cerebral ischemia. Eur J Pharmacol 707:78–86
Filmus J, Capurro M (2014) The role of glypicans in Hedgehog signaling. Matrix Biol 35:248–252
Ihrie RA, Shah JK, Harwell CC, Levine JH, Guinto CD, Lezameta M, Kriegstein AR, Alvarez-Buylla A (2011) Persistent sonic hedgehog signaling in adult brain determines neural stem cell positional identity. Neuron 71:250–262
Jenkins D (2009) Hedgehog signalling: emerging evidence for non-canonical pathways. Cell Signal 21:1023–1034
Brennan D, Chen XL, Cheng L, Mahoney M, Riobo NA (2012) Noncanonical Hedgehog signaling. Vitam Hormon 88:55–72
Wu CL, Chen SD, Hwang CS, Yang DI (2009) Sonic hedgehog mediates BDNF-induced neuroprotection against mitochondrial inhibitor 3-nitropropionic acid. Biochem Bioph Res Commun 385:112–117
Ji H, Miao JY, Zhang XJ, Du YY, Liu HC, Li SY, Li LT (2012) Inhibition of sonic hedgehog signaling aggravates brain damage associated with the down-regulation of Gli1, Ptch1 and SOD1 expression in acute ischemic stroke. Neurosci Lett 506:1–6
Zhao HP, Han ZP, Ji XM, Luo YM (2016) Epigenetic regulation of oxidative stress in ischemic stroke. Aging Dis 7:295–306
Gonzalez J, Valls N, Brito R, Rodrigo R (2014) Essential hypertension and oxidative stress: new insights. World J Cardiol 6:353–366
Ikeda K, Negishi H, Yamori Y (2003) Antioxidant nutrients and hypoxia/ischemia brain injury in rodents. Toxicology 189:55–61
Dai RL, Zhu SY, Xia YP, Mao L, Mei YW, Yao YF, Xue YM, Hu B (2011) Sonic Hedgehog protects cortical neurons against oxidative stress. Neurochem Res 36:67–75
Ji H, Zhang X, Du Y, Liu H, Li S, Li L (2012) Polydatin modulates inflammation by decreasing NF-kappaB activation and oxidative stress by increasing Gli1, Ptch1, SOD1 expression and ameliorates blood-brain barrier permeability for its neuroprotective effect in pMCAO rat brain. Brain Res Bull 87:50–59
Ohno Y, Gallin JI (1985) Diffusion of extracellular hydrogen-peroxide into intracellular compartments of human-neutrophils—studies utilizing the inactivation of myeloperoxidase by hydrogen-peroxide and azide. J Biol Chem 260:8438–8446
Kwon DH, Kim BS, Chang H, Kim YI, Jo SA, Leem YH (2013) Exercise ameliorates cognition impairment due to restraint stress-induced oxidative insult and reduced BDNF level. Biochem Biophys Res Commun 434:245–251
Yan F, Yang Y, Yu L, Zheng X (2017) Effects of C-glycosides from Apios americana leaves against oxidative stress during hyperglycemia through regulating mitogen-activated protein kinases and nuclear factor erythroid 2-related factor 2. J Agric Food Chem 65:7457–7466
Yu J, Lin JJ, Yu R, He S, Wang QW, Cui W, Zhang JR (2017) Fucoxanthin prevents H2O2-induced neuronal apoptosis via concurrently activating the PI3-K/Akt cascade and inhibiting the ERK pathway. Food Nutr Res 61:1304678
Davis SM, Pennypacker KR (2017) Targeting antioxidant enzyme expression as a therapeutic strategy for ischemic stroke. Neurochem Int 107:23–32
Dai RL, Xia YP, Mao L, Mei YW, Xue YM, Hu B (2012) Involvement of PI3K/Akt pathway in the neuroprotective effect of sonic hedgehog on cortical neurons under oxidative stress. J Huazhong Univ Sci Technol 32:856–860
Huang M, Cheng G, Tan H, Qin R, Zou Y, Wang Y, Zhang Y (2017) Capsaicin protects cortical neurons against ischemia/reperfusion injury via down-regulating NMDA receptors. Exp Neurol 295:66–76
Chi OZ, Hunter C, Liu X, Weiss HR (2009) Effects of exogenous excitatory amino acid neurotransmitters on blood-brain barrier disruption in focal cerebral ischemia. Neurochem Res 34:1249–1254
Courtney MJ, Li LL, Lai YY (2014) Mechanisms of NOS1AP action on NMDA receptor-nNOS signaling. Front Cell Neurosci 8:252
Mukherjee P, Cinelli MA, Kang S, Silverman RB (2014) Development of nitric oxide synthase inhibitors for neurodegeneration and neuropathic pain. Chem Soc Rev 43:6814–6838
Zhang D, Wang H, Liu H, Tao T, Wang N, Shen A (2016) nNOS translocates into the nucleus and interacts with Sox2 to protect neurons against early excitotoxicity via promotion of Shh transcription. Mol Neurobiol 53:6444–6458
Gu L, Jian Z, Stary C, Xiong X (2015) T cells and cerebral ischemic stroke. Neurochem Res 40:1786–1791
Amankulor NM, Hambardzumyan D, Pyonteck SM, Becher OJ, Joyce JA, Holland EC (2009) Sonic Hedgehog pathway activation is induced by acute brain injury and regulated by injury-related inflammation. J Neurosci 29:10299–10308
Lalancette-Hebert M, Gowing G, Simard A, Weng YC, Kriz J (2007) Selective ablation of proliferating microglial cells exacerbates ischemic injury in the brain. J Neurosci 27:2596–2605
Das S, Basu A (2008) Inflammation: a new candidate in modulating adult neurogenesis. J Neurosci Res 86:1199–1208
Xiong XX, White RE, Xu LJ, Yang LY, Sun XY, Zou BD, Pascual C, Sakurai T, Giffard RG, Xie XM (2013) Mitigation of murine focal cerebral ischemia by the hypocretin/orexin system is associated with reduced inflammation. Stroke 44:764–770
Lo ACY, Woo TTY, Wonsg RLM, Wong D (2011) Apoptosis and other cell death mechanisms after retinal detachment: implications for photoreceptor rescue. Ophthalmologica 226:10–17
Ferrer I, Planas AM (2003) Signaling of cell death and cell survival following focal cerebral ischemia: life and death struggle in the penumbra. J Neuropathol Exp Neurol 62:329–339
Charrier JB, Lapointe F, Le Douarin NM, Teillet MA (2001) Anti-apoptotic role of Sonic hedgehog protein at the early stages of nervous system organogenesis. Development 128:4011–4020
Bigelow RLH, Chari NS, Unden AB, Spurgers KB, Lee SJ, Roop DR, Toftgard R, McDonnell TJ (2004) Transcriptional regulation of bcl-2 mediated by the sonic hedgehog signaling pathway through Gli-1. J Biol Chem 279:1197–1205
Cayuso J, Ulloa F, Cox B, Briscoe J, Marti E (2006) The Sonic hedgehog pathway independently controls the patterning, proliferation and survival of neuroepithelial cells by regulating Gli activity. Development 133:517–528
Thibert C, Teillet MA, Lapointe F, Mazelin L, Le Douarin NM, Mehlen P (2003) Inhibition of neuroepithelial patched-induced apoptosis by Sonic hedgehog. Science 301:843–846
Prykhozhij SV (2010) In the absence of Sonic Hedgehog, p53 induces apoptosis and inhibits retinal cell proliferation, cell-cycle exit and differentiation in zebrafish. PLoS ONE 5:e13549
Kumari S, Chaurasia SN, Kumar K, Dash D (2014) Anti-apoptotic role of sonic hedgehog on blood platelets. Thromb Res 134:1311–1315
Alves HC, Pacheco FT, Rocha AJ (2016) Collateral blood vessels in acute ischemic stroke: a physiological window to predict future outcomes. Arquivos de neuro-psiquiatria 74:662–670
Marti HJ, Bernaudin M, Bellail A, Schoch H, Euler M, Petit E, Risau W (2000) Hypoxia-induced vascular endothelial growth factor expression precedes neovascularization after cerebral ischemia. Am J Pathol 156:965–976
Chertok VM, Zakharchuk NV, Chertok AG (2017) [The cellular and molecular mechanisms of angiogenesis regulation in the brain]. Zhurnal nevrologii i psikhiatrii imeni. SS Korsakova 117:43–55
Li Y, Zhang X, Cui L, Chen R, Zhang Y, Zhang C, Zhu X, He T, Shen Z, Dong L, Zhao J, Wen Y, Zheng X, Li P (2017) Salvianolic acids enhance cerebral angiogenesis and neurological recovery by activating JAK2/STAT3 signaling pathway after ischemic stroke in mice. J Neurochem 43:87–99
Pan JR, Li Y, Pei Z, Li XP, Peng Y, Wang YD (2010) Hypoxic tissues are associated with microvessel density following brain ischemia-reperfusion. Neurol Sci 31:765–771
Chen J, Chopp M (2006) Neurorestorative treatment of stroke: cell and pharmacological approaches. NeuroRx 3:466–473
Shimamura M, Sato N, Aoki M, Kaneda Y, Ogihara T, Morishita R (2004) A novel therapeutic strategy to treat brain ischemia: overexpression of hepatocyte growth factor gene reduced ischemic injury without brain edema in rat model. J Hypertens 22:S84–S85
Hui Z, Sha DJ, Wang SL, Li CS, Qian J, Wang JQ, Zhao Y, Zhang JH, Cheng HY, Yang H, Yu LJ, Xu Y (2017) Panaxatriol saponins promotes angiogenesis and enhances cerebral perfusion after ischemic stroke in rats. BMC Complement Altern Med 17:70
Wang JM, Isenberg JS, Billiar TR, Chen AF (2013) Thrombospondin-1/CD36 pathway contributes to bone marrow-derived angiogenic cell dysfunction in type 1 diabetes via Sonic hedgehog pathway suppression. Am J Physiol Endocrinol Metab 305:E1464–E1472
Kusano KF, Allendoerfer KL, Munger W, Pola R, Bosch-Marce M, Kirchmair R, Yoon Y, Curry C, Silver M, Kearney M, Asahara T, Losordo DW (2004) Sonic hedgehog induces arteriogenesis in diabetic vasa nervorum and restores function in diabetic neuropathy. Arterioscler Thromb Vasc Biol 24:2102–2107
Pola R, Ling LE, Aprahamian TR, Barban E, Bosch-Marce M, Curry C, Corbley M, Kearney M, Isner JM, Losordo DW (2003) Postnatal recapitulation of embryonic hedgehog pathway in response to skeletal muscle ischemia. Circulation 108:479–485
Chen SC, Huang M, He QW, Zhang Y, Opoku EN, Yang H, Jin HJ, Xia YP, Hu B (2017) Administration of Sonic Hedgehog protein induces angiogenesis and has therapeutic effects after stroke in rats. Neuroscience 352:285–295
Greenberg DA, Jin KL (2013) Vascular endothelial growth factors (VEGFs) and stroke. Cell Mol Life Sci 70:1753–1761
Lee SW, Moskowitz MA, Sims JR (2007) Sonic hedgehog inversely regulates the expression of angiopoietin-1 and angiopoietin-2 in fibroblasts. Int J Mol Med 19:445–451
Xia YP, He QW, Li YN, Chen SC, Huang M, Wang Y, Gao Y, Huang Y, Wang MD, Mao L, Hu B (2013) Recombinant human Sonic Hedgehog protein regulates the expression of ZO-1 and occludin by activating angiopoietin-1 in stroke damage. PLoS One 8:e68891
He QW, Xia YP, Chen SC, Wang Y, Huang M, Huang Y, Li JY, Li YN, Gao Y, Mao L, Mei YW, Hu B (2013) Astrocyte-derived sonic hedgehog contributes to angiogenesis in brain microvascular endothelial cells via RhoA/ROCK pathway after oxygen-glucose deprivation. Mol Neurobiol 47:976–987
Li YN, Xia YP, Wang Y, Mao L, Gao Y, He QW, Huang M, Chen SC, Hu B (2013) Sonic Hedgehog (Shh) regulates the expression of angiogenic growth factors in oxygen-glucose-deprived astrocytes by mediating the nuclear receptor NR2F2. Mol Neurobiol 47:967–975
Papadopoulos D, Scheiner-Bobis G (2017) Dehydroepiandrosterone sulfate augments blood-brain barrier and tight junction protein expression in brain endothelial cells. BBA-Mol Cell Res 1864:1382–1392
Li YA, Mao L, Gao Y, Baral S, Zhou YF, Hu B (2015) MicroRNA-107 contributes to post-stroke angiogenesis by targeting Dicer-1. Sci Rep-UK 5:13316
Lee BH, Lim TH, Yoon YW, Yenari MA, Jeong Y (2015) Postinjury neuroplasticity in central neural networks. Neural Plast 2015:857085
Font MA, Arboix A, Krupinski J (2010) Angiogenesis, neurogenesis and neuroplasticity in ischemic stroke. Curr Cardiol Rev 6:238–244
Wang L, Zhang ZG, Gregg SR, Zhang RL, Jiao ZX, LeTourneau Y, Liu XS, Feng YF, Gerwien J, Torup L, Leist M, Noguchi CT, Chen ZY, Chopp M (2007) The sonic hedgehog pathway mediates carbamylated erythropoietin-enhanced proliferation and differentiation of adult neural progenitor cells. J Biol Chem 282:32462–32470
Berretta A, Gowing EK, Jasoni CL, Clarkson AN (2016) Sonic hedgehog stimulates neurite outgrowth in a mechanical stretch model of reactive-astrogliosis. Sci Rep 6:21896
Yao PJ, Petralia RS, Mattson MP (2016) Sonic Hedgehog signaling and hippocampal neuroplasticity. Trends Neurosci 39:840–850
Ding XS, Li Y, Liu ZW, Zhang J, Cui YS, Chen XG, Chopp M (2013) The sonic hedgehog pathway mediates brain plasticity and subsequent functional recovery after bone marrow stromal cell treatment of stroke in mice. J Cereb Blood Flow Metab 33:1015–1024
Bambakidis NC, Petrullis M, Xu K, Rothstein B, Karampelas I, Kuang Y, Selman WR, LaManna JC, Miller RH (2013) Improvement of neurological recovery and stimulation of neural progenitor cell proliferation by intrathecal administration of Sonic hedgehog (vol 116, pg 1114, 2012). J Neurosurg 118:488–488
Liu XS, Chopp M, Zhang RL, Zhang ZG (2013) MicroRNAs in cerebral ischemia-induced neurogenesis. J Neuropathol Exp Neurol 72:717–721
Wang GS, Zhiyuan ZY, Xu Z, Yin HJ, Bai L, Ma ZA, DeCoster MA, Qian GS, Wu GY (2010) Activation of the sonic hedgehog signaling controls human pulmonary arterial smooth muscle cell proliferation in response to hypoxia. BBA-Mol Cell Res 1803:1359–1367
Balordi F, Fishell G (2007) Hedgehog signaling in the subventricular zone is required for both the maintenance of stem cells and the migration of newborn neurons. J Neurosci 27:5936–5947
Ribes V, Briscoe J (2009) Establishing and interpreting graded Sonic Hedgehog signaling during vertebrate neural tube patterning: the role of negative feedback. CSH Perspect Biol 1:a002014
Akazawa C, Tsuzuki H, Nakamura Y, Sasaki Y, Ohsaki K, Nakamura S, Arakawa Y, Kohsaka S (2004) The upregulated expression of sonic hedgehog in motor neurons after rat facial nerve axotomy. J Neurosci 24:7923–7930
Sims JR, Lee SW, Topalkara K, Qiu JH, Xu J, Zhou ZP, Moskowitz MA (2009) Sonic Hedgehog regulates ischemia/hypoxia-induced neural progenitor proliferation. Stroke 40:3618–3626
Sajja VSSS, Hlavac N, VandeVord PJ (2016) Role of glia in memory deficits following traumatic brain injury: biomarkers of glia dysfunction. Front Integr Neurosci 10:7
Anderson MF, Blomstrand F, Blomstrand C, Eriksson PS, Nilsson M (2003) Astrocytes and stroke: networking for survival? Neurochem Res 28:293–305
Lopez-Leal R, Court FA (2016) Schwann cell exosomes mediate neuron-glia communication and enhance axonal regeneration. Cell Mol Neurobiol 36:429–436
Raghupathi R, Graham DI, McIntosh TK (2000) Apoptosis after traumatic brain injury. J Neurotraum 17:927–938
Hampton DW, Rhodes KE, Zhao C, Franklin RJM, Fawcett JW (2004) The responses of oligodendrocyte precursor cells, astrocytes and microglia to a cortical stab injury, in the brain. Neuroscience 127:813–820
Sofroniew MV (2009) Molecular dissection of reactive astrogliosis and glial scar formation. Trends Neurosci 32:638–647
Li LZ, Lundkvist A, Andersson D, Wilhelmsson U, Nagai N, Pardo AC, Nodin C, Stahlberg A, Aprico K, Larsson K, Yabe T, Moons L, Fotheringham A, Davies I, Carmeliet P, Schwartz JP, Pekna M, Kubista M, Blomstrand F, Maragakis N, Nilsson M, Pekny M (2008) Protective role of reactive astrocytes in brain ischemia. J Cereb Blood Flow Metab 28:468–481
Fruhbeis C, Frohlich D, Kuo WP, Amphornrat J, Thilemann S, Saab AS, Kirchhoff F, Mobius W, Goebbels S, Nave KA, Schneider A, Simons M, Klugmann M, Trotter J, Kramer-Albers EM (2013) Neurotransmitter-triggered transfer of exosomes mediates oligodendrocyte-neuron communication. PLoS Biol 11:e1001604
Seifert T, Bauer J, Weissert R, Fazekas F, Storch MK (2005) Differential expression of sonic hedgehog immunoreactivity during lesion evolution in autoimmune encephalomyelitis. J Neuropathol Exp Neurol 64:404–411
Zhang L, Chopp M, Meier DH, Winter S, Wang L, Szalad A, Lu M, Wei M, Cui Y, Zhang ZG (2013) Sonic hedgehog signaling pathway mediates cerebrolysin-improved neurological function after stroke. Stroke 44:1965–1972
McTigue DM, Tripathi RB (2008) The life, death, and replacement of oligodendrocytes in the adult CNS. J Neurochem 107:1–19
Zhang J, Li Y, Zhang ZG, Noffsinger L, Elias SB, Chopp M (2009) Bone marrow stromal cells increase oligodendrogenesis after stroke. Stroke 40:E171–E171
Bambakidis NC, Wang XK, Lukas RJ, Spetzler RF, Sonntag VKH, Preul MC (2010) Intravenous Hedgehog agonist induces proliferation of neural and oligodendrocyte precursors in rodent spinal cord injury. Neurosurgery 67:1709–1715
Zhang Y, Zhang X, Cui L, Chen R, Zhang C, Li Y, He T, Zhu X, Shen Z, Dong L, Zhao J, Wen Y, Zheng X, Li P (2017) Salvianolic acids for injection (SAFI) promotes functional recovery and neurogenesis via sonic hedgehog pathway after stroke in mice. Neurochem Int 110:38–48
Jin Y, Barnett A, Zhang Y, Yu X, Luo Y (2017) Poststroke Sonic Hedgehog agonist treatment improves functional recovery by enhancing neurogenesis and angiogenesis. Stroke 48:1636–1645
Sinha S, Chen JK (2006) Purmorphamine activates the Hedgehog pathway by targeting smoothened. Nat Chem Biol 2:29–30
Yu P, Wang L, Tang F, Zeng L, Zhou L, Song X, Jia W, Chen J, Yang Q (2017) Resveratrol pretreatment decreases ischemic injury and improves neurological function via Sonic Hedgehog signaling after stroke in rats. Mol Neurobiol 54:212–226
Jeong JK, Moon MH, Lee YJ, Seol JW, Park SY (2013) Autophagy induced by the class III histone deacetylase Sirt1 prevents prion peptide neurotoxicity. Neurobiol Aging 34:146–156
Tang F, Guo S, Liao H, Yu P, Wang L, Song X, Chen J, Yang Q (2017) Resveratrol enhances neurite outgrowth and synaptogenesis via Sonic Hedgehog signaling following oxygen-glucose deprivation/reoxygenation injury. Cell Physiol Biochem 43:852–869
Xin HQ, Li Y, Shen LH, Liu XS, Hozeska-Solgot A, Zhang RL, Zhang ZG, Chopp M (2011) Multipotent mesenchymal stromal cells increase tPA expression and concomitantly decrease PAI-1 expression in astrocytes through the sonic hedgehog signaling pathway after stroke (in vitro study). J Cereb Blood Flow Metab 31:2181–2188
Shen LH, Xin HQ, Li Y, Zhang RL, Cui YS, Zhang L, Lu M, Zhang ZG, Chopp M (2011) Endogenous tissue plasminogen activator mediates bone marrow stromal cell-induced neurite remodeling after stroke in mice. Stroke 42:459–464
Xin HQ, Li Y, Buller B, Katakowski M, Zhang Y, Wang XL, Shang X, Zhang ZG, Chopp M (2012) Exosome-mediated transfer of miR-133b from multipotent mesenchymal stromal cells to neural cells contributes to neurite outgrowth. Stem Cells 30:1556–1564
Hammond SM, Bernstein E, Beach D, Hannon GJ (2000) An RNA-directed nuclease mediates post-transcriptional gene silencing in Drosophila cells. Nature 404:293–296
Verina T, Fatemi A, Johnston MV, Comi AM (2013) Pluripotent possibilities: human umbilical cord blood cell treatment after neonatal brain injury. Pediatr Neurol 48:346–354
Wang XL, Zhao YS, Hu MY, Sun YQ, Chen YX, Bi XH (2013) Umbilical cord blood cells regulate endogenous neural stem cell proliferation via hedgehog signaling in hypoxic ischemic neonatal rats. Brain Res 1518:26–35
Shi W, Nie DK, Jin GH, Chen WW, Xia L, Wu XJ, Su X, Xu XD, Ni LC, Zhang XA, Zhang XH, Chen J (2012) BDNF blended chitosan scaffolds for human umbilical cord MSC transplants in traumatic brain injury therapy. Biomaterials 33:3119–3126
Wang XL, Zhao YS, Wang X (2014) Umbilical cord blood cells regulate the differentiation of endogenous neural stem cells in hypoxic ischemic neonatal rats via the hedgehog signaling pathway. Brain Res 1560:18–26
Lundell TG, Zhou Q, Doughty ML (2009) Neurogenin1 expression in cell lineages of the cerebellar cortex in embryonic and postnatal mice. Dev Dyn 238:3310–3325
Chalazonitis A, D’Autreaux F, Pham TD, Kessler JA, Gershon MD (2011) Bone morphogenetic proteins regulate enteric gliogenesis by modulating ErbB3 signaling. Dev Biol 350:64–79
Zhao JJ, Song JQ, Pan SY, Wang K (2016) Treatment with isorhamnetin protects the brain against ischemic injury in mice. Neurochem Res 41:1939–1948
Acknowledgements
This study was supported by National Natural Science Foundation of China (No. 81671891) to Zhongyuan Xia, National Natural Science Foundation of China (No. 81571147) to Xiaoxing Xiong and by National Natural Science Foundation of Hubei (No. 2016CFB167) to Bo Zhao.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
The authors declare that they have no conflict of interest.
Rights and permissions
About this article

Cite this article
Liu, L., Zhao, B., Xiong, X. et al. The Neuroprotective Roles of Sonic Hedgehog Signaling Pathway in Ischemic Stroke. Neurochem Res 43, 2199–2211 (2018). https://doi.org/10.1007/s11064-018-2645-1
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s11064-018-2645-1