
Abstract
The present study has been designed to explore the molecular mechanism and signaling pathway targets of chlorogenic acid (CGA) and its main hydrolysates, caffeic (CA) and quinic acid in the protective effect against glutamate-excitotoxicity. For this purpose 8-DIV cortical neurons in primary culture were exposed to 50 μM l-glutamic acid plus 10 µM glycine, with or without 10–100 μM tested compounds. Chlorogenic acid and caffeic acid via their antioxidant properties inhibited cell death induced by glutamate in dose depended manner. However, quinic acid slightly protects neurons at a higher dose. DCF, JC-1 and Ca2+sensitive fluorescent dye fura-2, were used to measure intracellular ROS accumulation, mitochondrial membrane potential integration and intracellular calcium concentration [Ca2+] i . Results indicate that similarly, CGA acts as a protective agent against glutamate-induced cortical neurons injury by suppressing the accumulation of endogenous ROS and restore the mitochondrial membrane potential, activate the enzymatic antioxidant system by the increase levels of SOD activity and modulate the rise of intracellular calcium levels by increasing the rise of intracellular concentrations of Ca2+caused by glutamate overstimulation. PKC signaling cascade appear to be engaged in this protective mechanism. Interseling, CGA and CA also exhibit antiapoptotic properties against glutamate-induced cleaved activation of pro-caspases; caspase 1,8 and 9 and calpain (PD 150606,Calpeptin and MDL 28170).These data suggest that neuroprotective activity of CGA ester may occurs throught its hydrolysate,the caffeic acid and its interaction with intracellular molecules suggesting that CGA exert its neuroprotection via its caffeoly acid group that might potentially be used as a therapeutic agent in neurodegeneratives disorders associated with glutamate excitotoxicity.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
ROS generation is a normal physiological process, particularly for activation of numerous signal transduction pathways [1]. However, over production or dysregulation of ROS activity contributes to the development of some prevalent neurobiological diseases related to the central nervous system (CNS). Glutamate excitotoxicity play a key role in a number of neurodegenerative disorders, including Huntington’s disease and multiple sclerosis, Alzheimer’s disease, Parkinson’s disease [2, 3] as well as cerebral ischemic and traumatic brain injury and other neurological disorders [4].
Many studies have focused on bioactive compounds, in particular phenolic compounds and their antioxidant activities against oxidative stress, which could be directly associated with neuronal protection. Hydroxycinnamic acids are one of the most common groups of phenolic acids, which are present in many consumed fruit, vegetables and beverages. Among the most common and well-known hydroxycinnamic acids is chlorogenic CGA (5-O-caffeoylquinic acid). Chlorogenic acid, a caffeic acid ester linked to quinic acid, it is generally present in berries, cherries, apples, kiwis, artichokes, eggplants, plums and coffee and it is known to have several biological functions, including anti-inflammatory, antibacterial, anticarcinogenic and antioxidant activities [5]. It has been also found that chlorogenic acid has the ability to attenuate oxidative stress in numerous neurological disorders; it has been demonstrated that it acts as a neuroprotective agent against scopolamine-induced amnesia and significantly improved the impairment of short-term or working memory induced by scopolamine [6]. In addition, CGA has been reported to inhibit H2O2-induced apoptotic neuronal death through the up-regulation of the anti-apoptotic proteins Bcl-2 and Bcl-XL as well as the blockage of H2O2-induced pro-apoptotic cleavage of caspase-3 and pro-PARP in primary cortical neurons [7].
Chlorogenic acid is broken down into caffeic and quinic acids before absorption. The neuroprotective effects of caffeic acid (3, 4-dihydroxycinnamic acid), as a major metabolite of chlorogenic acid, abundant in carrots and various berries [8] have been investigated through in vitro studies. CA is already regarded as potent antioxidant, function dependent on its chemical structure [9]. It has been reported to have neuroprotective effects against Aβ-induced neurotoxicity in vitro and to inhibit peroxynitrite-induced neuronal injury [10, 11]. CA also acts as a selective 5-LOX inhibitor, protected mice from the aluminum induced neuronal damage through the down-regulation of overexpression of APP and amyloid beta protein [12]. The antioxidant-like effect of caffeic acid has been shown to be neuron-protective in vivo and ameliorates brain injuries after focal cerebral ischemia under pathological conditions [13] and attenuated the alterations induced by Quinolinic acid, an endogenous glutamate agonist with a relative selectivity toward N-methyl-d-aspartate (NMDA) receptor, induced oxidative damage, mitochondrial dysfunction and cellular alterations in rat striatum [14].
Quinic acid has been shown to exert neuroprotective and neurotrophic effect on Aβ-induced toxicity and potentiating activity in the neurite outgrowth of PC 12 cells in in vitro studies [15]. However, little is known about its mechanisms in the central nervous system.
More recently, chlorogenic acid and its metabolites exhibited neuroprotective activity against mechanistically distinct cell death-inducing agents in cultured cerebellar granule neurons [16]. Indeed, the role of CGA in protecting against neuronal death inin vitro study was reported and it has been shown that CGA inhibited glutamate-induced neuronal cell death and protects neurons against ischemia [17]. Chlorogenic acid is broken down into caffeic and quinic acids before gastrointestinal absorption and several data indicate their beneficial effects have been obtained under in vitro or in vivo conditions may be correlated with their parent compound [18] and its neuroprotective potential may result from its main metabolite(s). In support of this observations, in the present work, pharmacological approaches were used to investigate and compared the protective effects of chlorogenic acid and its major metabolites in the neuroprotective against l-glutamate induced excitotoxicity in cortical neuron cell culture. The underlying molecular mechanisms involved and signaling pathway transduction involved are investigated suggesting its potential role in the prevention in neurological disorders related to the glutamate excitotoxicity in the SNC.
Materials and Methods
Animals
Wistar rats were kept in a temperature-controlled room (21 ± 1 °C) under an established photoperiod (lights on from 7:00 am to 7:00 pm) with free access to food and water. All experiments were performed according to the recommendations of the ethical committee in Tunis and Spanish University for care and use of animals in conformity with NIH guidelines.
Chemicals
Neurobasal medium, B27 supplement, d(+)-glucose, l-glutamine, foetal bovine serum (FBS), trypsin-EDTA, and the antibiotic–antimycotic solution were obtained from Gibco (Invitrogen, Grand Island NY, USA), sodium pyruvate, calcein, chelerythrine, H89, U73122, 2,2-diphenyl-1-picrylhydrazyl (DPPH), and insulin were purchased from Sigma Aldrich (St. Louis, MO, USA). Caspases inhibitors (Ac-YVAD-H, Ac-IETD-H and Ac-LEHD-H) and Calpain inhibitors (PD150606, MDL28170, calpeptin) were purchased from international peptide. DCF: 2′, 7′-dichlorofluorescein (DCF), fura-2 AM and JC-1 were purchased from Molecular Probes Eugene, OR. Glutamate (Sigma, St. Louis, MO) and was first dissolved in an equimolar solution of NaOH. Chlorogenic acid, caffeic acid and quinic acid were purchased from Sigma Aldrich Spain; were dissolved in DMSO at a concentration of 100 mM as a stock solution.
Antioxidant Activity
Free radical scavenging activity was determined spectrophotometrically according to the method of DPPH assay. The DPPH· radical is reduced when reacting with an antioxidant compound, which can donate hydrogen. Serial concentrations of Trolox or stock solution compounds were diluted and then (25 μl) was placed in a cuvette with 975 μl of 0.04 mM methanolic solution of DPPH radical. Absorbance was measured at 517 nm 30 min in the dark later. All determinations were performed in triplicate. The DPPH inhibition percentage was calculated as the absorbance decrease of the antioxidant samples relative to the control. The antiradical activity was expressed as the DPPH inhibition percentage as calculated using the following formula:
Cortical Neurons
Cortical neurons were obtained from the cortical lobes of E18 Sprague-Dawley rat embryos, according to previously described procedures [19]. The cells were resuspended in B27 Neurobasal medium plus 10% FBS and then seeded onto poly-l-ornithinecoated glass coverslips (12 mm in diameter) or 6-well plates at 5 × 104 cells per coverslip or 2 × 106 cells per well. Culture dishes precoated with poly-d-lysine (50 mg/ml in sterile water) overnight.The cultures were essentially free of astrocytes and microglia. They were maintained at 37 °C and 5% CO2. Cultures were used 8–10 days after plating for excitotoxicity insult.
Induction of Excitotoxicity and Cell Viability Determination
To induce excitotoxicity insult, neurons were exposed to l-glutamic acid (50 µM) for 10 min in HBSS containing 2.6 mM CaCl2, 10 mM glucose, 10 mM glycine, pH 7.4, at 378 °C. Then, in order to evaluate the effects of tested compounds on excitotoxicity, cells were stimulated with l-glutamic acid in the presence of different concentrations of each compound (10, 25, 50 and 100 µM) and these compounds were maintained for 2 h after glutamate exposure. For cell survival experiments, neurons culture (5 × 104 cells/well) were incubated for 30 min with the fluorescent probe calcein acetoxymethyl ester (calcein-AM), then rinsed with phosphate-buffered saline (PBS). Fluorescence intensity was measured (λ excitation = 485 nm and λ emission = 530 nm) with a FL800TBI fluorescence microplate reader (Bio-Tek Instruments, Winooski, USA). The fluorescence in the control cells was taken as 100% viability. Data are mean percentages of viable cells versus the respective controls. Phenolic compounds stocks were dissolved in DMSO and were diluted in culture medium to the final desired concentration (maximal concentration of DMSO 0.001%). All experiments were performed in triplicate and the values provided here are the averages of at least three independent experiments.
Measurement of Cell Cytotoxicity
Measurement of LDH activity in culture medium was determined using a coloromertic method, the CytoTox96 ®non-radioactive Cytotoxicity assay. Neurons were seeded onto 96-well plates (2 × 104 cells/well). The CytoTox96 assay quantitatively measures lactate deshydrogenase (LDH), a stable cytosolic enzyme that is released upon cell lysis. Release LDH in culture supernatants is measeured with a 30-minute coupled enzymatic assay that results in the conversion of a tetrazoliumm salt (INT) into a red formazan product. The amount of color formed is proportional to the number of lysed cells. Visible wavelength absorbance at 490 nm data is collected using a standard 96-well plate reader. The results were expressed as a percentage of total LDH release from neurons. Data are expressed as the mean ± SEM from three independent experiments performed in quadruplicate. The results were expressed as a percentage of total LDH release from neurons.
Determination of Intracellular Reactive Oxygen Species (ROS)
ROS were detected by using a fluorescent probe, 2′, 7′-dichlorofluorescein (DCF) resulting from the deacetylation and oxidation of the non-fluorescent compound DCFH2-DA. After treatment, cells were incubated with 30 µM of cell permeant DCFH2-DA in Neurobasal serum-free loading medium at 37 °C for 30 min. At the end of the incubation, DCFH2-DA was removed and cells were washed twice with PBS. The intensity of fluorescence was measured (λ excitation = 485 nm and λ emission = 530 nm) with a FL800TBI fluorescence microplate reader (Bio-Tek Instruments, Winooski, USA). Results are expressed as the mean ± SEM from three independent experiments performed in quadruplicate.
Measurement of Mitochondrial Membrane Activity
Mitochondrial membrane potential was quantified using the JC-1 fluorescent probe. Neuronal cultures (5 × 104 cells/well) were exposed to l-glutamate alone or with phenolic compounds (100 µM) as described .At the end of treatments, neurons were incubated in the presence of the JC-1 probe (10 µg/ml) for 15 min at 37 °C and then washed twice with PBS. In living cells, the lipophilic dye JC-1 enter into the mitochondria through membrane potential and form aggregates producing an intense orange signal. Whereas, in dead cells, where the mitochondrial membrane potential collapses the monomeric JC-1 remains cytosolic and stains the cell in green. Fluorescence intensity was measured with a fluorescence microplate reader (Bio-Tek Instruments, Winooski, USA) and expressed as a ratio of the fluorescence emission at 590 nm (orange, intact mitochondrial membrane potential) versus 530 nm (green, collapsed mitochondrial membrane potential). Results are expressed as the mean ± SEM performed in quadruplicate from three independent experiments.
Measurement of Antioxidant Enzyme Activities
We next analyzed if CGA and its metabolites can modify the equilibrium of the endogenous antioxidant system. Cortical neurons (2 × 106 cells per well) were subjected to the excitotoxic stimulus in the absence or presence of CGA,CA and QA and at the end of the treatment, culture medium was removed; cells were washed twice with PBS and then homogenized in the same solution at 4 °C. Cells were harvested by centrifugation (350 g, 4 °C, 10 min) and the cell pellet were resuspended in 50 ml of ice-cold lysing buffer containing 50 mM Tris–HCl (pH 8), 10 mM EDTA, 100 mM phenylmethyl-sulfonylfluoride and 1% Triton X-100. Samples were then centrifuged (16,000×g, 20 min, 4 °C) and supernatants were finally stored at −20 °C until enzyme activity determinations.
The activity of SOD was measured using a spectrophotometric assay, which consists in measuring epinephrine autoxidation induced by superoxide anion. Samples were incubated for 3 min with a mixture containing bovine catalase (0.4 U/L), epinephrine (5 mg/mL) and Na2CO3/NaHCO3 buffer (62.5 mM, pH 10.2). The oxidation of epinephrine was measured at 480 nm.
Catalase activity was of H2O2. Samples were mixed with 30 mM H2O2 in PBS. The disappearance of H2O2 was measured at 240 nm for 180 s at 30-s intervals. Catalase activity was calculated using the extinction coefficient of 40/mM/cm for H2O2.
Determination of Calcium Release [Ca2+]i
Measurement of intracellular calcium of [Ca2+]i was determined by fluorescent probe fura-2 AM (Molecular Probes, Eugene, OR). Cells were incubated with 5 µM fura-2-AM in culture medium for 30 min at 37C°, and then washed with HBSS containing 20 mM HEPES [pH 7.4], 10 mM glucose, and 2 mM CaCl2. Experiments were carried out in a coverslip chamber, continuously perfused with incubation buffer at 2 ml/min. At the end of the assay, in situ calibration was performed with the successive addition of 10 mM ionomycin and 2 M Tris-50 mM EGTA, pH 8.5. The [Ca2+]i concentration was estimated by the 340/380 ratio method, using a Kd value of 224 nM. Data are expressed as the mean ± SEM from three independent experiments performed in quadruplicate.
Caspase Activity
The peptide caspase inhibitors are able to block caspase activity in vitro. We investigate the involvement of caspase 1, 8 and 9 in neuroprotective target of tested phenolic acid using synthetic caspase inhibitor peptides. For that purpose, neurons were seeded onto 96-well plates (2 × 104 cells/well), and incubated at day 8 in vitro with glutamate plus glycine in the presence or absence of polyphenols, as described above. Caspase inhibitors: Caspase 1 inhibitors (Ac-YVAD-H); Caspase 8 inhibitor (Ac-IETD-H) and Caspase 9 inhibitor (Ac-LEHD-H) were prepared in DMSO according to the instructions of the manufacturer (Peptides International, Louisville, KY) and added to cultures 30 min prior to glutamate induction. All experiments were performed in quadruplicate, and the values provided are the average of at least three independent experiments.
Calpain Activity
To evaluate the Calpain activity on live cells, calpain peptide inhibitor (International peptide) was used according to the manufacturer’s instructions. Neuronal cultures (5 × 104 cells/well) were exposed to l-glutamate alone or with phenolic acids (100 µM) in the presence of the calpain inhibitors 3-(4-iodophenyl)-2-mercapto-(Z)-2-propenoic acid (PD150606) (100 µM), calpeptin (20 µM), or N-[[(phenylmethoxy)carbonyl]-l-valyl]-phenylalaninal (MDL28170) (10 µM). Calpain inhibitors were added for 30 min before exposure to l-glutamate. Calcein-AM was used here to quantify cell viability and Fluorescence was measured using a Synergy-HT fluorimeter (Bio-Tek Instruments Incl., Beverly, MA, USA). All experiments were performed in quadruplicate, and the values provided are the average of at least three independent experiments.
Statistical Analysis
Data are presented as the mean ± SEM from three independent experiments performed in quadruplicate. Statistical analysis of the data was performed by using Student’s t-test and ANOVA, followed by Bonferroni’s test. **p < 0.01; ***p < 0.001 and NS not statistically different from control; # p < 0.05; ## p < 0.01; ###p < 0.001 versus glutamate-treated cells and ns not statistically different from glutamate alone.
Results
Antioxidant Potential: DPPH Assay
The antioxidant effects of chlorogenic acid (CGA) and its metabolites, caffeic acid (CA) and quinic acid (QA), were investigated. All three chemicals were separately used at a concentration range of 10–100 µM and tested by DPPH assay. The scavenging DPPH radical assay results indicated that examined compounds here could scavenge DPPH free radical at a dose dependent manner. CA and CGA exhibited potent antioxidant activity against the DPPH radical formation. At 100 µM, the highest DPPH activity was observed with CA followed by CGA; (101.10 and 97.5%, respectively) while QA revealed itself as a weak antioxidant activity (Fig. 1).

Antioxidant activities of chlorogenic, caffeic acid, and quinic acid determined by the DPPH assay. Values represent the mean ± SEM of triplicates from three different experiments (n = 3). ANOVA followed by the Bonferroni’s test. #p < 0.05; ##p < 0.01; ###p < 0.001 and ns, not statistically different when compounds were compared
Effect of Chlorogenic Acid and its Metabolites on Glutamate Mediated—Excitotoxicity
We evaluated the effectiveness of chlorogenic acid and its occurring metabolites against neuronal death following excitotoxic insults mediated by NMDA receptors(glutamate 50 µM plus glycine 10 µM; 10 min) as a model of neuronal excitotoxicity. As seen in Fig. 2, l-glutamate-induced decrease in cell viability by approximately 40% and was ameliorated dose-dependently by chlorogenic acid and caffeic acid; More there, both CGA and caffeic acid significantly enhanced cell survival to a similar degree and shows a complete reversal of the toxic effect glutamate at different concentrations. Although only a high concentration of quinic acid (100 µM) exhibited a significant increase of cell viability but the results did not reach statistical significance compared with cells treated with caffeic or chlorogenic acid. We used a concentration 100 µM in the following experiments since it had the maximal neuroprotection with the tested compounds.

Effects of graded concentrations (10–100 µM) of each compound chlorogenic acid caffeic acid and quinic acid on glutamate induced-excitotoxicity. Cells were incubated with l-glutamate/glycine (50/10 µM) for 10 min followed by tested compounds at the indicated concentrations were maintained for 2 h after glutamate exposure. After the treatments the medium was changed and cells were maintained at 37 °C in a humidified atmosphere of 5% CO2 for others 24 h. Each value is the mean (± SEM) calculated from at least six different wells from four independent cultures. ANOVA followed by the Bonferroni’s test. * < 0.05 ; **p < 0.01; ***p < 0.001 and NS not statistically different from control; #p < 0.05; ##p < 0.01; ###p < 0.001 versus.glutamate-treated cells and ns not statistically different from glutamate alone
ROS Scavenging Activity and Mitochondrial Membrane Potential
Glutamate excitotoxicity lead to excessive production of ROS causing oxidative damage and cell death. In order to evaluate the ability of tested phenolic compounds to directly scavenge free radicals and endogenous ROS generations, neurons were incubated with fluorescent probe CMH2DCFDA which forms the fluorescent DCF compound upon oxidation with ROS. Results show that ROS generation as the consequences of glutamate excitotoxic insults, are similarly attenuated by CA and CGA and increase above control levels, but quinic acid did not alter the glutamate-induced increase in ROS production and failed to protect neurons against the overproduction of ROS after excitotoxicity insult (Fig. 3).

Effect of CGA and its related compounds on intracellular accumulation of ROS produced by activation of NMDA glutamate receptors in cultured cortical neurons. Cortical neuronswere stimulated with glutamate (50 µM) in the presence of glycine (10 µM) and treated with CGA,CA andQA (100 µM) following stimulation at different times. Cellular ROS were detected by measuring the fluorescence of 2′, 7′-dichlorofluorescein (DCF), and the results are expressed as a percentage of fluorescence with respect to that of control cells. Each value represent the mean (± SEM) of triplicates from three different experiments (n = 3). ANOVA followed by the Bonferroni’s test. *p < 0.05 ; **p < 0.01; ***p < 0.001 and NS not statistically different from control; #p < 0.05; ##p < 0.01; ###p < 0.001 versus glutamate-treated cells and ns not statistically different from glutamate alone
Considering the temporal pattern of free radical production after glutamate exposure (50 µM; 10 min) ROS production rise up until reaching its maximum at 60 min after the excitotoxic insult by 2.41 fold compared with non treated cells. The reduction of ROS levels of caffeic acid was maintained at later stages until 90 min and had a statistically significant reduction levels, although chlorogenic acid attenuates oxidative stress caused by glutamate disappeared at 60 min after excitotoxic insult.
Since the generation of ROS in excitotoxic insults is associated with alterations and permeabilize the mitochondrial membrane, we next measured mitochondrial membrane potential using JC-1 as a fluorescent probe. Neurons exposed to l-glutamate /Glycine show decrease of the red signal (590 mm); indicating the alteration of mitochondrial membrane integrity. Incubation of cells with CGA and caffeic acid totally suppressed the deleterious effects of l-glutamate on mitochondrial membrane potential and kept the mitochondrial membrane potential around the value of the resting potential only for 30 min (96.34 ± 2.6 and 100. 2 ± 5.9%, respectively). However, lower intensity was observed with quinic acid with green fluorescent stains indicating altered membrane in dead cells and failed to decrease the depolarization of the mitochondrial membrane potential after excitotoxic insult (Fig. 4).

Effect of CGA and its metabolites on glutamate-induced alteration of mitochondrial membrane potential in cultured cortical neurons at differente time course. CGA and CA attenuate the depolarization of the mitochondrial membrane. The mitochondrial transmembran tential was assessed by using the JC-1 probe, and the ratio of fluorescence emissions 590 versus 530 nm. The results are expressed as percentage of control. Each value is the mean (± SEM) calculated from at least four different wells from three independent cultures (n = 3). ANOVA followed by the Bonferroni’s test. ANOVA followed by the Bonferroni’s test. *p < 0.05 ; **p < 0.01; ***p < 0.001 and NS not statistically different from control; #p < 0.05; ##p < 0.01; ###p < 0.001 versus glutamate-treated cells and ns not statistically different from glutamate alone
Modulation of the Antioxidant Enzyme System
The antioxidant defense system has been developed by the organism as a protective mechanism against ROS formation. Among the most reported endogenous antioxidant systems are the activity of the enzymes SOD and CAT. In order to know if the protective effects of tested compounds against l-glutamate-induced oxidative stress are related to the antioxidant enzyme system. The results in Fig. 5 show that l-glutamate significantly decreased the levels of the antioxidant enzymes CAT, and SOD when compared with the levels in the control cells. More specifically, catalase activity was half of the respective control values. However, levels of SOD was restored above control levels when neurons were treated with CGA and caffeic acid but not in the presence of quinic acid. Remarkably,QA has succeeded to restore catalase levels.

Effect of CGA and its metabolites on SOD and catalase activities in cultured rat cortical neurons 1 h post-excitotoxic stimulus in the absence or presence of 100 µM CGA,CA and QA. SOD activity was determined by measurement of epinephrine autoxidation induced by superoxide anion, and catalase activity by the decrease of rate H2O2. Data were normalized with respect to non cells represented as a percentage of U/mg protein with respect to its control (100%). Each value is the mean (± SEM) of at least four different dishes from three independent cultures (n = 3). ANOVA followed by the Bonferroni’s test. *p < 0.05;**p < 0.01, ***p < 0.001 and NS not statistically different from control. #p < 0.05; ##p < 0.01 ; ###p < 0.001 versus glutamate -treated cells. ns not statistically different when compounds were compared
CGA and CA were almost identically effective; CGA increased by 98.56 ± 3.98 and CA by 105.6 ± 4.7%).
Effect of CGA and its Metabolites on Intracellular Calcium
Excitotoxic insult mediates by glutamate receptors cause an elevation in the concentration of cytosolic Ca2+. To further investigate the potential mechanisms underlying CGA and its related compounds -mediated cell death, the cytoplasmic free Ca2+ concentration was determined in cell culture with free-2 specific probe. Firstly, the time course of [Ca2+]i (mean ± 4 SEM) with respect to basal values before incubation with glutamate was determined (Fig. 6a). We observed previously that NMDA receptor activation in cortical neurons caused a significant [Ca2+]i increases which are reduced by CGA and Caffeic acid but not by quinic acid. Histogram in Fig. 6b depicts the changes in the peak amplitude of [Ca2+]i. The intracellular calcium concentration data in Fig. 6b represents the time to peak [Ca2+]i response increases when agonist as added.

Effect of l-glutamate and phenolic compounds on intracellular calcium conecentration. Cells were stimulated by glutamate/glycine (50/10 µM) and followed by 100 µM of each compounds. [Ca2+]i was determined using fluorescent probe fura 2-AM. a Curves in A illustrate the time course of the [Ca21]i increase ± SEM (n = 3)with respect to basal values. b [Ca2+]i increases induced by L-glutamate 50 µM are reduced by CGA and caffeic acid but not by quinic acid. ANOVA followed by the Bonferroni’s test. *p < 0.05;**p < 0.01, ***p < 0.001 and NS not statistically different from control. #p < 0.05 ##p < 0.01; ###p < 0.001 versus l-glutamate-treated cells; ns not statistically different when compounds were compared
Signal Transduction Pathways Involved in the Neuroprotective Effect of Chlorogenic Acid and its Related Compounds
To investigate the signaling cascade involved in the neuroprotection action of each phenolic compounds ,the whole ester, the chlorogenic acid as its main related metabolites via glutamate overstimulation, neurons were pre-incubated with the PKA inhibitor H89 (2 × 10−5M), the PLC inhibitor U73122 (10−5 M), the PKC inhibitor chelerythrine (10−6 M) for 30 min, at 37 °C. The PLC inhibitor and the PKC inhibitor totally abrogated the neuroprotective action of CGA and CA against glutamate induced cell death. In contrast, the selective PKA inhibitor H89 did not modify the effect of tested compounds on glutamate-induced cell death; suggesting that PKC signaling pathways’ is involved in the neuroprotective mechanism of both CGA and CA. Meanwhile, for the quinic acid, its protective effects is totally abrogated on cell survival when neurons were pretreated with the PKA inhibitor suggesting that the neuroprotective activity of quinic acid can be accounted for by activation of the PKA (Fig. 7).

Differential intracellular signaling pathways involved in the neuroprotective effect of CGA and its related compounds on cortical neuron cells. Cultured neurons at 8 DIV were pre-incubated for 30 min in the absence or presence of with the PKA inhibitor H89 (2 × 10−5 M), the PLC inhibitor U73122 (10−5 M), the PKC inhibitor chelerythrine (10−6 M) and then stimulated for 10 min with l-glutamate 50 µM without or with tested compounds at the same dose (100 µM). Cell survival was quantified by measuring FDA fluorescence intensity, and the results are expressed as percentages of the control. Each value is the mean (± SEM) of at least of six different wells from three independent cultures. ANOVA followed by the Bonferroni’s test. * < 0.05 ; **p < 0.01; ***p < 0.001 and NS not statistically different from control; #p < 0.05; ##p < 0.01; ###p < 0.001 versus glutamate-treated cells and ns not statistically different from glutamate alone
Effects of Caspases and Calpain Inhibitor on Glutamate-Induced Cell Death
Caspases and calpain inhibitor were able to block caspases and calpain activity in neurons. In l-glutamate treated cells, cell survival was about 72.77% (P < 0.05) compared with that of the control group. Glutamate-induced cell death was fully blocked by caspases specific inhibitors on cortical neurons cell culture. Pretreatment with specific caspase inhibitor Ac-YVAD-H, Ac-IETD-H and Ac-LEHD-H (100 μM) 30 min before glutamate exposure had no effect on the neuroprotective effect of CGA and CA which suggested that caspase-dependent apoptosis accounted for the neurporotective effect of these phenolic compounds against glutamate excitotoxicity.
Calpain or Ca2+-dependent proteases play an important role in the execution of apoptosis and activation of calpain has been implicated in neuronal apoptosis. Pretreatment with specific calpain inhibitor also totally abrogated the deleterious effect of glutamate induced cell death and preserve the neuroprotective effect of CGA and CA (Fig. 8).

Effect of caspases and calpain Inhibition activities in cultured neuron cortical rat at 30 min post stimilus. a Cells were pre incubated for 30 min with peptide caspase inhibitors at 100 µM:YVAD (casp1 inhibitor) ; IETD (casp-8 inhibitor) or LEHD (casp-9 inhibitor) in the absence or presence of tested compounds before NMDA receptors activation. b Neurons were incubated for 30 min with calpain inhibitor PD150606 (100 µM);Calpeptin (20 µM) or MDL 28,170 (10 µM) before addition of agonists, and cell survival was measured at 30 min post stimilus. In all data ,values are not statistically different versus glutamate-treated cells. Values are illustrated as a percentage with respect to the corresponding control, untreated cells (100%), and data are means ± SEM., n = 3. In all instances, significance is given as **p < 0.01; ***p < 0.001; NS not statistically different versus control. ##p < 0.01; ###p < 0.001 and ns not statistically different versus glutamate-treated cells
Discussion
Genarally, “excitotoxicity” was referred to as a neuronal degeneration triggered by the over- or prolonged activation of glutamate receptors in the central nervous system (CNS) by excitatory amino acids [20]. Glutamate excitotoxicity appears to play an important role in a number of neurodegenerative diseases, including Alzeimer diseases, Parkinson diseases, Huntington disease and multiple sclerosis [2, 3].
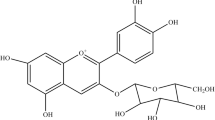
Chlorogenic acid (CGA) is a cinnamic acid, which possesses different isomeric forms, is the predominant phenolic compounds in coffee and berries. Pharmacological effects of chlorogenic acid have been demonstrated on the CNS, and appear to exert many beneficial effects against oxidative stress to reduce the risk of developing neurodegenerative disorders [7, 16, 17]. CGA is capable of mediating oxidative stress and attenuating cell apoptosis due to different oxidative stressors and free radicals by modulating the accumulation of reactive oxygen species and by regulating the expression of key proteins and enzymes involved in cell apoptosis [21].
Since, chlorogenic acid the whole compounds is known to be poorly absorbed, extensively metabolized in vivo, and in the small intestine, it is hydrolyzed to caffeic acid and quinic acid which have been identified in the plasma and urine of rats fed [22]. The neuroprotective effect of CGA and its hydrolysates, caffeic acid and quinic acid against glutamate excitotoxicity were investigated with differences between observations [16, 17] but molecular mechanisms and signaling pathways transduction targeted accounting for neuroprotective effect of CGA,the whole ester and its hydrolysates, separately requires further research.
In a first investigation, we demonstrated that CGA did not differ statistically to caffeic acid to protect neurons against l-glutamate induced neuronal death. In contrast, quinic acid showed a lower range of neuroprotective activity and levels of cell viability reach of statistical significance only with a higher dose 100 µM.
Our results were in accordance with some previous studies, which have investigated the neuroprotective effects of CGA and its metabolites in vitro, indicating that CGA protects neurons from glutamate-induced neuronal cell death [17]. However, in the study of Taram et al. chlorogenic acid did not protect Cerebellar granule neurons CGNs and primary cultures of rat cortical neurons and only caffeic acid provided significant protection from excitotoxicity-induced cell death [16]. Differences between these observations could be due to several factors such as cell type, primary cell cultures or stable cell line cultures, tissue culture conditions, source of cells (fetal, middle age or adult brains), age of cultures, purity of cultures concentration and duration (minutes to hours) of glutamate exposure, nature of the neurotoxin used, and extent of Ca2+ influx [19].
Excitotoxicity thought overstimulation of over activation of glutamate receptors has been reported as the most important trigger of neurotoxicity upon excitatory amino acids and neuronal death in some conditions mediated by the extracellular glutamate release and interaction with ionotropic receptos agonists receptors [23]. The ability of glutamic acid excitotoxicity is mediated, in most cases, by an interaction with NMDA receptors, leading to an uncontrollable rise of ROS and intracellular calcium concentrations. l-Glutamate also induces the partial depolarization of the mitochondrial membrane that triggers alterations in the membrane potential of mitochondria [20] and causes enhanced intracellular substrate oxidation and reactive oxygen species ROS production leading to apoptotic neuronal death. Measurement of mitochondria activity with the membrane potential-sensitive probe JC-1 revealed that treatment with glutamate resulted in a decrease of the proportion of active mitochondria, and that treatement with both caffeic and CGA prevented the deleterious effect of glutamate.Caffeic acid appears the most reactive compounds to neutralize the excess of ROS production. Its strongly suggest that the mechanism for antioxidant action of CA is associated with the formation of the iron complex, which inhibits the reaction between hydroxyl radical and the target molecule improving the capacity of CA to inhibit ROS formation.
In turn, the results obtained with chlorogenic acid and caffeic acid are consistent with previous reports showing that excitotoxic insults produce a rapid mitochondrial membrane depolarization which can be abolished by antioxidants [24]. CGA and CA may protect mitochondria through its direct free radicals scavenging ability and acting as a chain-breaking antioxidant though the the anti-radical properties of polyphenols lead to reduction of hidroxyl and superoxide radicals [25].
As for the antioxidant/scavenging activity of these compounds, was well documented [9, 26] and it was widely reported that chlorogenic acid and related compounds are well known to be antioxidants [27]. The scavenging of the DPPH radical is a simple model reaction providing relevant information about the ability of acids to scavenge free hydroxyl radicals. Antioxidant activity of phenolic acids and their derivatives depends on the number and position of hydroxyl groups bound to the aromatic ring, the binding site and mutual position of hydroxyl groups in the aromatic ring, and the type of substituents [28].
CGA have been the subject of scientific research due to their antioxidant capacity to scavenge free radicals, which is attributed to double bonds and hydroxyl groups in its structure. In contrast to these observations, the binding of quinic acid to caffeic acid causes an increase in the antioxidant activity of caffeic acid while decreasing DPPH radical scavenging activities [29]; some other reports indicate that chlorogenic and caffeic acids are equally effective antioxidants activity [30].
In agreement with our results, caffeic acid has potent antioxidant properties, a function dependent on its chemical structure and the presence of a catechol moiety [28] suggesting that this antioxidant activity might contribute to its neuroprotective properties. The antioxidant activity of CA was previously studied by Nardini et stronger al. and reported that CA inhibited, in a dose-dependent manner, human LDL lipoperoxidation induced by cupric ions [31]. CA is able to reduce lipoperoxyl radicals (ROO·)—by donating a hydrogen atom–to its corresponding hydroperoxide, which inhibits the lipid peroxidation chain reaction [25].
Another hypothesis to restore mitochondrial membrane dysfonction is chelating ability of phenolic compound. A previous study has suggested that CA has a chelating-antioxidant mechanism and it is strongly suggest that is works in vitro by efficiently chelating iron ions and, thus, minimizing the effects of Fenton-generated hydroxyl radicals. The antioxidant capacity of CA is due to the formation of complexes with metal ions, capable of preventing in vitro oxidative damage [32]. CA is able to complex Fe(II) ions and to inhibit in vitro hydroxyl radical formation and lipid peroxidation of rat liver membranes chain reaction which gives it the capability to act in the cell membrane microenvironment of different tissues preventing lipid peroxidation and other oxidative processes mediated by lipid peroxidation products [33].
Additionally, chlorogenic acid revealed the ability to form complexes with metal ions. This could be attributed to a more complex structure of others phenolic acid which is a cinnamate ester obtained by formal condensation of the carboxy group of trans-caffeic acid with the 3-hydroxy group of quinic acid. Thus, the presence of group carboxylic group in hydroxycinnamic acids family may have an influence on the chelation capacity, as well as on scavenging activity (iron-chelation properties of phenolic acids bearing catechol groups) [34].
All these reactions provoke inhibition or reduction in the formation of free radicals, they interrupt the propagation of free radical chain reactions, or they delay the start or reduce the reaction rate. Furthermore, metal chelation can result in prevention of metal redox cycling, occupation of all metal coordination sites, formation of insoluble metal complexes, steric hindrance of interactions between metals, and formation of lipid intermediates [35].
Scavenging or detoxification the excess ROS is achieved by an efficient antioxidative enzymatic system. Endogenous antioxidants play an important role in alleviating oxidative stress and its consequences [36]. Previous studies have suggested that anti-oxidant enzymes such as superoxide dismutase (SOD) and catalase (CAT) can function as a defense mechanism against ROS-mediated cellular damage, thereby proposing that these anti-oxidant enzymes may be useful as a therapeutic agent against ROS-related diseases [37]. Herein, we demonstrate that the CGA is able to stimulate both SOD and catalase activities in neurons. The mechanism involved in the polyphenols induced increase of SOD and catalase activity in a model of glutamate induced excitotoxicity is currently unknown, but it has been demonstrated that dephosphorylation of SOD and catalase in a calcium- and protein kinase C-dependent manner is associated with an increase in the activity of these two enzymes [38, 39]. Thus, the stimulatory effect of CGA and CA on SOD and catalase activities could be ascribed to the intracellular calcium increase inducing SOD and catalase activation through a dephosphorylation process. This finding supposes that chlorogenic acid via its caffeoyl groupement maintain the equilibrium of the endogenous enzymatic antioxidant to reinforce defense and remedy deleterious effects of exocitotoxic damage.
Another hypothesis were suggested and support the fact that ROS generation during glutamate overstimulation is likely may due to the Ca2 + influx into the cytosol leading to neuronal damage via activation of enzymatic system in association with ROS generation and lipid peroxidation [40]. Depolarization of mitochondrial membrane also can activate plasma membrane voltage Ca2 + channels leading to additional Ca2 + influx. In support of this hypothesis, it has been shown it have been reported that CGA in coffee protects neurons from glutamate neurotoxicity during brain ischemia by regulating Ca2 + entry into neurons [17]. Caffeic acid also protects primary cultures of rat cortical neurons from the excitotoxic effects of L-glutamate by attenuated excessive calcium influx caused by glutamate [41]. Apparently, caffeic acid potently modulates the intracellular calcium concentration neuron cells through the control calcium influx or activation of calcium sensitive molecules. Thus, the inhibition of Ca2 + elevation by CGA through caffeic acid moiety and this effect might be attributed to a direct reduction in the amount of calcium entering through voltage-dependent Ca2 + channels or to the secondary effects caused by other processes such as the alteration plasma membrane potential following modulation of K+ channels and support the finding of the capacity of some polyphenols to reduce calcium entry via glutamate receptors [42].
Collectively, these data suggest that the intrinsic radical scavenging activity of phenolic compounds has been linked to scavenge the excess of intracellular ROS generation and modulating mitochondrial membrane potential in a response to cytosolic Ca2 + dysregulation followed by alteration of membrane potential and permeability transition, suggesting that these events may be considered as an initial line of defense against glutamate induced cell death.
Activation of MAPK kinase cascade signaling pathway is proposed to be involved in the neuroprotective effect of chlorogenic acid and its related hydrolysates against glutamate excitotoxicity using specific potein kinases inhibitor peptides: U73122 PLC inhibitor, Chelerytrine, PKC inhibitor or H89, PKA inhibitor. PLC inhibitor U73122 and the PKC inhibitor chelerythrine abrogated similarly the neuroprotective effect of chlorogenic acid and caffeic acid. These observations suggest that chlorogenic acid and its related compound caffeic acid and quinic acid compounds separately, exert their protective effects trough different targeted transduction pathways. More there, the neuroprotective activity of chlorogenic acid the whole compounds can be can contributed by the caffeoyl groupement and ascribed specifically to activation of the PLC/PKC signaling pathway. Since, activation of PKC by polyphenols is a key event in neuroprotection, and already known as a potent regulator of apoptotic pathways and was shown to be the target of many polyphenols for providing survival signaling. PKC signaling pathways regulate important molecular events involved in associative memory storage, and signaling deficits of PKC signaling pathways play an important role in the pathophysiology of neurodegenerative disorders like Alzheimer disease [43]. Protein kinase C is a calcium regulated kinase that modulate NMDA receptors function. PKC, enhance voltage-dependent Ca2 + channel function and glutamate release [41]. However, quinic acid appears to exert its neuroprotective through the PKA signaling pathway.
It is widely accepted that permeabilization and mitochondrial dysfunction causes activation of caspases, the effectors of apoptotic cell death [42]. We have thus investigated the possible effect of phenolic tested compounds on l-glutamate–induced caspase activation in cultured cortical neurons. Therefore, active caspases can be controlled by of peptide inhibitors that directly interact with the protease; these peptide inhibitors are able to block caspases and inhibit their activities and per consequent inhibit cell death in vitro [44]. Caspases were divided into two important intracellular family group, upstream initiators and downstream effectors. The upstream initiators activate the effectors caspases in response to un-upstream stimuli to leads to the apoptotic phase execution by cleavage of various substrates as a pro-enzyme [45]. Caspase 1, 8 and 9 were considered as caspases initiator of Caspase 3 that has already been identified as a key mediator of neuronal cell death [46]. Caspase 8 and caspases 9 have been implicated as principal procaspases and thus critical to distingue cell death pathway: Caspases 8 activation is involved in the extrinsic or receptor-mediated pathways; the intrinsic or mitochondrial pathway involves activation of caspase 9 [47]. Herein, caspase1, caspase 8 and caspase 9 inhibitor was used to block caspases activities. Our results showed that similarly CGA and caffeic acid inhibited-l glutamate-induced cleaved activation of caspase-8 and -9 which further confirmed that CGA and CA almost, similarly could inhibit l-glutamate-induced apoptosis in cortical neurons indicating that the protective effect of CGA and CA can be accounted for by an inhibition of caspase 8 and 9 that in turn inhibit caspase 3. Both caspases 8 and 9 then activate caspase 3, the major effector and the key mediator of neuronal cell death.
Moreover, an increase in the intracellular Ca2+ level is thought to trigger a cascade of biochemical processes, including calpain activation. It has been demonstrated that calpains is implicated in an early involvement in glutamtae excitotoxicity induced cell death [48] and that caspases have the propriety to up-regulate calpain activities through modification of calpastatin (an endogenous calpain inhibitor) by proteolytic cleavage [49]. In addition, the rise levels of Ca2+ may be one of the causes of activations of proteolytic enzymes as calpains, which are considered mediators of both necrotic cell death and apoptosis [50]. Pretreatment of cortical neurons with calpain inhibitors, PD150606, calpeptin, and MDL28170 prior to tested compounds totally abrogated the protective effect of caffeic acid. Caffeic acid can inhibit glutamate induced calpain activity supporting its pronounced neuroprotective potential probably resulting from oxidative stress suppression.
Altogether, these data indicate that CGA acts as a protective agent against l-gluatamte induced cortical neurons injury through maintain antioxidant enzymatic system and inhibiting apoptosis, as well as regulating neurons MAPK signaling cascade. These finding suggest that activation of PKC by caffeoyl group of chlorogenic acid appears to be a key event in its neuroprotective effect via proposed following mechanisms: maintain mitochondrial membrane potential, scavenging endogenous ROS production and increasing the levels of SOD activity.
Conclusion
Recently, numerous bio-molecules such as phenolic compounds, having therapeutic abilities, have been developed as new drug candidates. However, their large molecular sizes and poor hydrophilicities reduce their bioavailabilities or delivery to cells or tissues. This data demonstrate that the neuroprotective effects of chlorogenic acid a cinnamate ester obtained by formal condensation of the carboxy group of trans-caffeic acid with the 3-hydroxy group of quinic acid, on glutamate-induced excitotoxicity and brain damage might be attributed to the caffeoyl group, a relation structure function that is associated with anti oxidative ,anti apoptotic activities leading to the anti-excitatory potential of these active compounds and to have clinical benefits and might potentially be used as a therapeutic agent for neurodegenerative diseases. The neuroprotective effects of these compounds in vivo are currently under investigation.
Abbreviations
- CNS:
-
Central nervous system
- DCF:
-
Dichlorofluorescein
- FDA:
-
Fluorescein diacetate
- H2O2 :
-
Hydrogen peroxide
- LDH:
-
Lactate dehydrogenase
- PKA:
-
Protein kinase A
- PKC:
-
Protein kinase C
- PLC:
-
Phospholipase C
- ROS:
-
Reactive oxygen species
- MAPK:
-
Mitogen apoptotic protein kinase
- DPPH:
-
2, 2-diphenyl-1-picrylhydrazyl
- DCF:
-
2’, 7’-dichlorofluorescein
- CGA:
-
Chlorogenic acid
- CA:
-
Caffeic
- QA:
-
Quinic acid
- PBS:
-
Phosphate-buffered saline
- LDH:
-
Lactate deshydrogenase
- CAT:
-
Catalase
- SOD:
-
Superoxide dismutase
- NMDA:
-
N-methyl-d-aspartate
- DMSO:
-
Dimethyl sulfoxide
- HBSS:
-
Hank’s Balanced Salt Solution
References
Martindale JL, Holbrook NJ (2002) Cellular response to oxidative stress: signaling for suicide and survival. J Cell Physiol 192:1–15
Blandini F, Greenamyre JT, Nappi G (1996) The role of glutamate in the pathophysiology of Parkinson’s disease. Funct Neurol 11:3–15
Zeron MM, Chen N, Moshaver A, Lee AT, Wellington CL, Raymond MR (2001) Mutant huntingtin enhances excitotoxic cell death. Mol Cell Neurosci 17:41–53
Choi DW (1988) Glutamate neurotoxicity and diseases of the nervous system. Neuron 1:623–634
Dos Santos MD, Almeida MC, Lopes NP, De Souza G (2006) Evaluation of the antiinflammatory, analgesic and antipyretic activities of the natural polyphenol chlorogenic acid. Biol Pharm Bull 29:2236–2240
Kwon SH, Lee HK, Kim JA, Hong SI, Kim HC, Park YI, Lee CK, Lee YB, Lee SY, Jang CG (2010) Neuroprotective effects of chlorogenic acid on scopolamine-induced amnesia via anti-acetylcholinesterase and anti-oxidative activities in mice. Eur J Pharmacol 649:210–217
Kim J, Lee S, Shim J, Kim HW, Kim J, Young JJ, Yang H, Park J, Choi SH, Yoon JH, Lee KW, Lee HJ (2012) Caffeinated coffee, decaffeinated coffee, and the phenolic phytochemical chlorogenic acid up-regulate NQO1 expression and prevent H2O2-induced apoptosis in primary cortical neurons. Neurochem Int 60:466–474
Mattila P, Pihlava JM, Hellstrom J (2005) Contents of phenolic acids, alkyl- and alkenylresorcinols, and avenanthramides in commercial grain products. J Agric Food Chem 53:8290–8295
Pavlica S, Gebhardt R (2005) Protective effects of ellagic and chlorogenic acids against oxidative stress in PC12 cells. Free Radic Res 39:1377–1390
Vauzour D, Corona G, Spencer JPE (2010) Caffeic acid, tyrosol and p-coumaric acid are potent inhibitors of 5-S-cysteinyl-dopamine induced neurotoxicity. Arch Biochem Biophys 501(1):106–111
Vauzour D, Vafeiadou K, Spencer J JPE (2007) Inhibition of the formation of the neurotoxin 5-S-cysteinyl-dopamine by polyphenols. Biochem Biophys Res Commun 362(2):340–346
Yang JQ, Zhou QX, Liu BZ, He BC (2008) Protection of mouse brain from aluminum-induced damage by caffeic acid. CNS Neurosci Ther 14(1):10–16
Zhou Y, Fang SH, Ye YL, Chu LS, Zhang WP, Wang ML, Wei EQ (2006) Caffeic acid ameliorates early and delayed brain injuries after focal cerebral ischemia in rats. Acta Pharmacol Sin 27:1103–1110
Kalonia H et al (2009) Effect of caffeic acid and rofecoxib and their combination against intrastriatal quinolinic acid induced oxidative damage, mitochondrial and histological alterations in rats. Inflammopharmacology 17:211–219
Hur JY, Soh Y, Kim BH, Suk K, Sohn NW, Kim HC, Kwon HC, Lee KR, Kim SY (2001) Neuroprotective and neurotrophic effects of quinic acids from Aster scaber in PC12 cells. Biol Pharm Bull 24:921–924
Taram F, Winter AN, Linseman DA (2016) Neuroprotection comparison of chlorogenic acid and its metabolites against mechanistically distinct cell death-inducing agent sin cultured cerebellar granule neurons. Brain Res 1648:69–80
Mikami Y, Yamazawa T (2015)Chlorogenic acid, a polyphenol in coffee, protects neurons against glutamate neurotoxicity. Life Sci 139:69–74
Azuma K, Ippoushi K, Nakayama M et al (2000) Absorption of chlorogenic acid and caffeic acid in rats after oral administration. J Agric Food Chem 48:5496–5500
Zhang YM, Bhavnani B (2005) Glutamate-induced apoptosis in primary cortical neurons is inhibited by equine estrogens via down-regulation of caspase-3 and prevention of mitochondrial cytochrome c release. BMC Neurosci 6:13. https://bmcneurosci.biomedcentral.com/articles/10.1186/1471-2202-6-13
Olney JW (1969) Glutamate-induced retinal degeneration in neonatal mice. Electron microscopy of the acutely evolving lesion. J Neuropathol Exp Neurol 28:455–474
Anggreani E, Lee CY (2017) Neuroprotective effect of chlorogenic acids against Alzheimer’s disease. Int J Food Sci Nutr Diet 6(1):330–337
Dupas C, Baglieri AM, Ordonaud C, Tom D, Maillard M (2006) Chlorogenic acid is poorly absorbed, independently of the food matrix: a Caco-2 cells and rat chronic absorption study. Mol Nutr Food Res 50:1053–1060
Campos-Esparza R, Sanchez-Gomez MV, Matute C (2009) Molecular mechanisms of neuroprotection by two natural antioxidant polyphenols. Cell Calcium 45:358–368
Ibarretxe G, Sánchez-Gómez MV, Campos-Esparza MR, Alberdi E, Matute C (2006) Differential oxidative stress in oligodendrocytes and neurons after excitotoxic insults and protection by natural polyphénols. Glia 53(2):201–211
RiceEvanc C (2001) Flavonoid-antioxidant. Curr Med Chem 8:797–807
Gottlieb M et al (2006) Neuroprotection by two polyphenols following excitotoxicity and experimental ischemia. Neurobiol Dis 23:374–386
Kono Y, Kobayashi K, Tagawa S, Adachi K, Ueda A, Sawa Y, Shibata H (1997) Antioxidant activity of polyphenolics in diets. Rate constants of reactions of chlorogenic acid and caffeic acid with reactive species of oxygen and nitrogen. Biochim Biophys Acta 1335(3):335–342
Rice-Evans CA, Miller JM, Paganga G (1996) Structure-antioxidant activity relationship of flavonoids and phenolic acids. Free Radic Biol Med 20:933–956
Sroka Z, Cisowski W (2003) Hydrogen peroxide scavenging, antioxidant and anti-radical activity of some phenolic acids. Food Chem Toxicol 41:753–758
Marinova EM, Toneva A, Yanishlieva N (2009) Comparison of the antioxidative properties of caffeic and chlorogenic acids. Food Chem 114:1498–1502
Nardini M, D’Aquino M, Tomassi G, Gentili V, Di Felice M, Scaccini C (1995) Inhibition of human low-density lipoprotein oxidation by caffeic acid and other hydroxycinnamic acid derivatives. Free Radic Biol Med 19:541–552
Hung TM, Na M, Thuong PT, Su ND, Sok D, Song KS, Seong YH, Bae K (2006) Antioxidant activity of caffeoyl quinic acid derivatives from the roots of Dipsacus asper Wall. J Ethnopharmacol 108:188–192
Genaro-Mattos TC, Maurício ÂQ, Rettori D, Alonso A, Hermes-Lima M (2015) Antioxidant activity of caffeic acid against iron-induced free radical generation—A chemical approach. PLoS ONE 10(11):14624
Andjelkovi M, Van Camp J, De Meulenaer B, Depaemelaere G, Socaciu C, Verloo M, Verhe R (2006) Iron-chelation properties of phenolic acids bearing catechol and galloyl groups. Food Chem 98:23–31
Heiss WD, Kessler J, Mielke R, Szelies B et al (1994) Long-term effects of phosphatidylserine, pyritinol, and cognitive training in Alzheimer’s disease: a neuropsychological EEG, PET investigation. Dementia 5:88–98
Noctor G, Foyer CH (1998) Ascorbate and glutathione: keeping active oxygen under control. Annu Rev Plant Biol 49:249–279
Matés JM (2000) Effects of antioxidant enzymes in the molecular control of reactive oxygen species toxicology. Toxicology 153:83–104
Hopper RK, Carroll S, Aponte AM, Johnson DT, French S, Shen RF, Witzmann FA, Harris RA, Balaban RS (2006) Mitochondrial matrix phosphoproteome: effect of extra mitochondrial calcium. Biochemistry 45:2524–2536
Kumar S, Suryanarayanan A, Boyd KN, Comerford CE, Lai MA, Ren Q, Morrow AL (2010) Ethanol reduces GABAA alpha1 subunit receptor surface expression by a protein kinase C gamma-dependent mechanism in cultured cerebral cortical neurons. Mol Pharmacol 77:793–803
Alkon DL, Sun MK, Nelson TJ (2007) PKC signaling deficits: a mechanistic hypothesis for the origins of Alzheimer’s disease. Trends Pharmacol Sci 28:51–60
Lin TY, Chung CY, Lu CW, Huang SK, Shieh JS, Wang SJ (2013) Local anesthetics inhibit glutamate release from rat cerebral cortex synaptosomes. Synapse 67:568–579
Orrenius S (2008) Reactive oxygen species in mitochondria-mediated cell death. Drug Metab Rev 39:443–455
White RJ, Reynolds IJ (1996) Mitochondrial depolarization in glutamate-stimulated neurons: an early signal specific to excitotoxin exposure. J Neurosci 16(18):5688–5697
Poręba M, Stróżyk A, Salvesen GS, Drąg M (2013) Caspase substrates and inhibitors. Cold Spring Harb Perspect Biol 5(8). doi:10.1101/cshperspect.a008680
Liu H, Radhakrishna B (2005) Endoplasmic reticulum stress–associated caspase 12 mediates cisplatin-induced LLC-PK1 cell apoptosis. JASN Express 16:1985–1992
Yuan B, Yankner A (2000) Apoptosis in the nervous system. Nature 407:802–809
Earnshaw WC, Martins LM, Kaufmann SH (1999) Mammalian caspases: structure, activation, substrates, and functions during apoptosis. Annu Rev Biochem 68:383–424
Leon R, Wu H, Jin Y, Wei J, Buddhala C, Prentice H, Wu JY (2009) Protective function of taurine in glutamate-induced apoptosis in cultured neurons. J Neurosci Res 87(5):1185–1194
Wang KW (2000) Calpain and caspases: can you tell the difference. Trends Neurosci 23:20–26
Wingrave JM, Schaecher KE, Sribnick EA, Wilford GG, Ray SK, Hazen-Martin DJ, Hogan EL, Banik L (2003) Early induction of secondary injury factors causing activation of calpain and mitochondria-mediated neuronal apoptosis following spinal cord injury in rats. J Neurosci Res 73(1):95–104
Acknowledgements
This work was supported by the Research Unit 00-UR-08-01 University of Sciences, Tunis and by a grant from the Tunisian, Ministry of Higher Education and Scientific Research Tunisia. The authors would like to thank Prof. Matute Carlos from the Instituto Del pays Vasco, Spain for their supportive advices. We acknowledge the financial support from the Ministry of Higher Education and Scientific Research (Tunisia).
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article

Cite this article
Rebai, O., Belkhir, M., Sanchez-Gomez, M.V. et al. Differential Molecular Targets for Neuroprotective Effect of Chlorogenic Acid and its Related Compounds Against Glutamate Induced Excitotoxicity and Oxidative Stress in Rat Cortical Neurons. Neurochem Res 42, 3559–3572 (2017). https://doi.org/10.1007/s11064-017-2403-9
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s11064-017-2403-9