
Abstract
During the course of development, molecular mechanisms underlying activity-dependent synaptic plasticity change considerably. At immature CA3–CA1 synapses in the hippocampus, PKA-driven synaptic insertion of GluA4 AMPA receptors is the predominant mechanism for synaptic strengthening. However, the physiological significance of the developmentally restricted GluA4-dependent plasticity mechanisms is poorly understood. Here we have used microelectrode array (MEA) recordings in GluA4 deficient slice cultures to study the role of GluA4 in early development of the hippocampal circuit function. We find that during the first week in culture (DIV2–6) when GluA4 expression is restricted to pyramidal neurons, loss of GluA4 has no effect on the overall excitability of the immature network, but significantly impairs synchronization of the CA3 and CA1 neuronal populations. In the absence of GluA4, the temporal correlation of the population spiking activity between CA3–CA1 neurons was significantly lower as compared to wild-types at DIV6. Our data show that synapse-level defects in transmission and plasticity mechanisms are efficiently compensated for to normalize population firing rate at the immature hippocampal network. However, lack of the plasticity mechanisms typical for the immature synapses may perturb functional coupling between neuronal sub-populations, a defect frequently implicated in the context of developmentally originating neuropsychiatric disorders.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Background and Introduction
Synaptic plasticity driven by synchronous activity of the network is a key mechanism guiding refinement of synaptic connectivity during development. At the immature synapses, mechanisms similar to long-term potentiation (LTP) and long-term depression (LTD) are thought to initiate processes that lead to stabilization or elimination of the latent synapse, and consequently fine-tune the functional network to the ongoing activity templates. To facilitate this process, immature synapses are equipped with signaling mechanisms that allow them to efficiently transform network activity-patterns into long-lasting changes in synaptic structures. The physiological significance of such developmentally restricted plasticity mechanisms is best understood in the sensory systems, where patterned activity during a critical period of development affects the physical structure of the brain as well as behavior [17, 21]. In the hippocampus, both pre- and postsynaptic plasticity mechanisms operating specifically during early postnatal development have been characterized (e.g. [18, 22, 23]). However, the significance of these mechanisms for the development of the hippocampal circuitry and the extent by which they can be compensated for is less well understood.
At CA3–CA1 glutamatergic synapses in the hippocampus, a number of key molecular components in both pre- and postsynaptic compartments undergo age dependent modifications (e.g. [11, 12, 22, 34]). Postsynaptically, GluA4 subunits of AMPA receptors are predominantly expressed during the first postnatal week, while the levels of the other subunits are low [37]. GluA4 expression is sufficient to alter the signaling mechanisms of LTP and support PKA—dependent LTP at CA3–CA1 synapses [6, 24, 37, 36]. Indeed, the loss of GluA4 expression at the CA1 pyramidal neurons during postnatal maturation of the circuitry can fully explain the developmental switch in LTP mechanisms from PKA dependent to CaMKII dependent [24, 36]. In addition to Hebbian LTP, GluA4 also contributes to induction requirements of homeostatic plasticity. At GluA4 expressing immature synapses, the threshold for homeostatic scaling of glutamatergic transmission is lower as compared to adult or GluA4 lacking synapses, which facilitates tuning of AMPA transmission to changes in the intrinsic activity of the immature circuits [14].
These data show that GluA4 has a central role in regulation of synaptic efficacy specifically during the early postnatal period of activity-dependent refinement of the CA3–CA1 circuitry. Accordingly, in the absence of GluA4, maturation of AMPA transmission at CA3–CA1 synapses is delayed, evidenced by significantly lower AMPA/NMDA ratio in the GluA4−/− slices as compared to WTs during postnatal development (P6-P18; [25]). Here, we have studied how the GluA4-dependent transmission and plasticity contributes to functional maturation of the hippocampal CA3–CA1 circuitry using microelectrode array (MEA) recordings in transgenic mice lacking GluA4.
Materials and Methods
Animals
Experiments were performed using wild type (WT) and GluA4−/− (C57Bl) mice [8] of either sex. All experiments with animals were done in accordance with the University of Helsinki Animal Welfare Guidelines.
Slice Culture
For preparation of slice cultures, hippocampi were dissected from P6-P7 day old WT or GluA4−/− mice in ice cold Gey’s balanced salt solution (GBSS; Sigma) supplemented with glucose (6.5 mg/ml). The hippocampi were mounted on 2% liquid agarose and 400 µm thick slices were cut using a McIlwain tissue chopper. After 1 h recovery in oxygenated (95% O2 and 5% CO2) GBSS, the slices were transferred to MED64-probe plates (MED-P515A, Alpha Med Scientific, Japan) coated with polyethylenimine solution (0.1% PEI in 25 mM Borate buffer). 250 µl of culture medium [Neurobasal A (Gibco), 2% B27-supplement (Gibco), 2 mM l-glutamine and chloramphenicol] was added and the plates were maintained in humidified CO2 incubator (+35 °C) on a rocking stage. The culture media was changed every second day.
Immunostaining
Immunostainings for slice cultures were done at DIV4 and DIV8. Slices were cultured as described [34], fixed with 4% paraformaldehyde overnight, washed with PBS-T (PBS with 0.3% Triton-X) and blocked with 1% DMSO and 4% normal goat serum in PBS-T overnight. Primary antibodies (Rabbit anti-GluA4 1:100 Millipore; Mouse anti-PV 1:5000 SWANT, Switzerland; mouse anti-GAD67 1:500 Millipore) were added to the blocking solution and slices were incubated 48 h at +4 °C. Slices were then washed with PBS before incubation with secondary antibodies overnight + 4 °C. (Goat anti-mouse Alexa405 1:1000; Goat anti-rabbit Alexa647 1:1000 Life Technologies). Slices were washed with PBS-T, 50% glycerol, 80% glycerol and mounted with 80% glycerol. Confocal images were acquired using a LSM Zeiss 710 confocal microscope (alpha Plan-Apochromat 63×/1.46 OilKorr M27 objective). Images were processed with ImageJ and Corel Paint Shop Pro ×8 and brightness and contrast were corrected.
Microelectrode Array Recordings
The spontaneous activity in slice cultures was recorded for a 10–15 min periods at DIV2-DIV8 using the MED 64-amplifier (MED-A64HE1, Alpha MED Scientific). The data were collected with the Mobius Software (Alpha MED Scientific) using a low cut frequency of 0.1 Hz, high cut frequency of 5000 Hz and a sampling rate of 20 kHz.
Data Analysis
For analysis, the raw data files were imported into NeuroExplorer (NE) software (Nex Technologies). For analysis the channels lying in the pyramidal layer in CA3 and CA1 hippocampal regions were selected (minimum three channels per region). Recordings were filtered using second order Butterworth band pass filter (frequency band 50–5000 Hz) to alleviate baseline fluctuations and slow frequency components. Spike detection was performed using the method for unsupervised spike detection [30] in the NE and the threshold was set to four times the median of the background noise of the signal. Detected spikes were verified visually. Bursts were detected using the firing rate based method in the NE, where a firing rate histogram with bin size of 5 ms was computed for each channel of interest and the bins that had values higher than 3.5 × (Mean + SD of firing rate) were considered as a burst.
For calculation of spike time tiling coefficient (STTC) and correlation coefficient, at least two CA3 and CA1 channel pairs separated by at least 300 µm were selected for each slice. The values for all the channel pairs in each slice were averaged and considered as n = 1. Cross correlograms were calculated using the Cross-correlogram tool in NE, with an interval t0 ± t (t = 20 ms) and bin size of 0.25 ms. Obtained values were used to calculate population Pearson correlation coefficient in excel.
For calculation of STTC, time stamps for detected spikes were exported to MatLab. STTC was calculated according to [5] as follows:
here P1 is the fraction of spikes from the 1st electrode that occur within time \(\pm \Delta{\text{t}}\) of any spike from the 2nd electrode. T1 is the proportion of total recording time that lies within time \(\pm \Delta{\text{t}}\) of any spike from the 1st electrode. P2 and T2 are calculated similarly for the 2nd electrode. \(\Delta{\text{t}}\) was set to 5 ms. The coefficient has a range [−1, 1] and equals + 1 in case of autocorrelation. To ensure that \(\Delta{\text{t}}\) does not introduce false correlations random shuffling procedure was used. Traces were divided into 20 bins, which then were randomly permuted. STTCs computed for randomly shuffled traces using the same value of \(\Delta{\text{t}}\) were close to 0 and significantly lower than STTCs for original traces.
Connectivity maps were created in MATLAB using biograph function. Nodes of graph correspond to the positions of channels in MEA layout and edges were plotted only for channels in CA1 and CA3 regions, based on STTC measure. The width of edge line indicates the strength of corresponding connection and is calculated as follows:
here \({W}_{ij}\) is a weight between channels i and j, calculated as STTC, a and b—scaling parameters, set to 10 and 0.1 respectively, and thresh—threshold, set to 0.5. Connections with weights below the threshold were not included in the graphs.
Statistical Analysis
All statistics were calculated on raw data using two-way ANOVA with Holm-Sidak post-hoc comparison in Sigma Plot software. All data are presented as mean ± standard error of the mean (SEM). P < 0.05 was considered statistically significant. In figures, the significance levels are indicated by asterisks as follows: *p < 0.05, **p < 0.01, ***p < 0.001.
Results
GluA4 Deficiency has no Significant Effect on the Excitability of the CA3–CA1 Circuit During Early Development
To study the role of GluA4 in functional development of the hippocampal circuitry, we cultured hippocampal slices from neonatal (P6-7) WT and GluA4−/− mice on top of microelectrode array (MEA) probes, allowing recording of the network activity patterns at different stages of development in vitro (DIV).
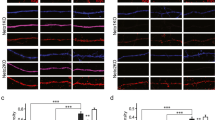
In the brain, GluA4 is expressed in the pyramidal neurons during the first postnatal week [37] Subsequently, its expression in the pyramidal neurons gradually decreases and expression starts to emerge in parvalbumin positive (PV) fast-spiking interneurons, reaching adult levels during the third postnatal week of development [29, 37]. Since this developmental profile might be different in vitro, we first validated the cell-type specific expression profile of GluA4 in our culture conditions. At DIV4, GluA4 immunostaining was clearly detected in the CA1 pyramidal cell layer. The intensity of GluA4 staining in the CA1 pyramidal cells diminished during development but was still visible at DIV8 (Fig. 1a). In our cultures, specific staining with anti-parvalbumin antibody was not detected either at DIV4 or DIV8 (not shown). Therefore, we chose to use GAD67 staining to investigate whether GluA4 localized to GABAergic neurons. GAD67 staining was observed in cells scattered in various layers of the cultured slice as expected (Fig. 1b). However, careful inspection of the double stained slice cultures failed to find any co-localization of GluA4 with GAD67 at DIV4. At DIV8, the staining pattern was very similar as in DIV4, and no apparent double positive cells were detected (Fig. 1c). These data support that GluA4 is predominantly expressed in the pyramidal neurons but not in GABAergic interneurons during the first week in culture.

GluA4 is not expressed in GAD67 positive cells during first week in vitro. a Example images illustrating the immunostaining against GluA4 in area CA1 of cultured hippocampal slices at DIV4 and DIV8. b Example images of the immunostaining against GAD67 in area CA1 of cultured hippocampal slices at DIV4 and DIV8. c Double immunostaining for GluA4 (red) and GAD67 (blue) in area CA1 of cultured hippocampal slices at DIV4 and DIV8. Scale bar: 50 µm. (Color figure online)
The population spiking activity in the cultured slices was recorded at DIV2, 4, 6 and 8, using channels located at the CA3 and CA1 pyramidal cell regions. In the WT slices, the mean frequency of detected spikes increased during the time in culture both in areas CA1 and CA3 (CA1: DIV2 5.64 ± 0.37 Hz, DIV8 10.20 ± 1.58 Hz, pDIV2,8 < 0.001; CA3: DIV2 5.41 ± 0.50 Hz, DIV8 9.64 ± 2.09 Hz, pDIV2,8 p < 0.001; Fig. 2a, b), while no significant differences in the population spike frequency were observed between CA3 and CA1 at any developmental stage. At DIV2–4, the spikes occurred asynchronously or in short bursts, while at DIV6 and DIV8, long hypersynchronous bursts of activity were detected with interleaved silent periods (Fig. 2a). In GluA4−/− slices, the overall activity patterns were not different from the WT during the first week in vitro (Fig. 2a). Thus, there were no significant differences between genotypes in mean population spike frequency, burst frequency, % occurrence of spikes in bursts or in the burst duration at DIV2–6 (Fig. 2b–e). At DIV8, however, the population spike frequency in GluA4−/− slices was significantly lower as compared to WT (CA3: WT 9.64 ± 2.09 Hz, GluA4−/− 6.21 ± 0.41 Hz, p = 0.005; CA1: WT 10.20 ± 1.58 Hz, GluA4−/− 6.18 ± 0.42 Hz, p < 0.001; Fig. 2b). These data show that loss of GluA4 has no major influence on network excitability during the first week in culture, when it is predominantly expressed at pyramidal neurons.

Overall network activity is similar in WT and GluA4−/− slices during the first week in culture. a Examples traces showing the spontaneous activity recorded with MEA electrodes located in CA3 and CA1 pyramidal regions from WT and GluA4−/− slice cultures at DIV 2, DIV4, DIV 6 and DIV 8. b Pooled data on the mean population spike frequency in CA3 and CA1 areas in WT (n = 24) and GluA4 deficient (n = 26) slice cultures, at different stages of development in vitro. c Average burst frequency for the same recordings as in (b). d The mean % of spikes in bursts for the same recordings as in (b). e The mean burst duration for the same recordings as in (b)
Absence of GluA4 Does not Influence Population Firing Rate Homeostasis
GluA4 promotes homeostatic scaling of glutamatergic transmission at immature CA3–CA1 synapses [14]. Therefore, we were next interested to see whether impaired synaptic scaling in the GluA4 deficient slices affects the stability of firing rates of the CA1 neuronal populations. To this end, recordings were made at DIV2 and DIV3 with 8–15 h intervals, corresponding to the time scale of the fast homeostatic response at the WT immature networks [13, 14]. The relative change in the mean population spike frequency was plotted (Fig. 3a, b), and the coefficient of variation (CV) in the frequency in the sequential recordings was calculated for each slice. Even if the mean population firing rate in the CA1 area varied slightly more in the GluA4−/− as compared to WTs, there were no significant difference in the coefficient of variation (CV) between the genotypes (WT 0.81 ± 0.35, n = 9; GluA4−/− 1.32 ± 0.25, n = 11; p = 0.26; Fig. 3c). These data show that impaired synaptic scaling in the GluA4−/− slices does not destabilize the excitability of the CA1 network and suggest that other mechanisms can efficiently compensate for perturbed synaptic scaling to maintain firing rate within physiological range.

GluA4 deficiency has no significant effect on population firing rate homeostasis. a Relative changes in population firing rate for WT (n = 9) slices in four consecutive recordings within 8–15 h intervals at DIV2-3. b Equivalent data as in (a) for GluA4−/− (n = 11). c Mean CV for the population firing rate for WT and GluA4−/− slices, calculated from the data shown in (a) and (b)
GluA4 Deficiency Perturbs Synchronization of the CA3–CA1 Neuronal Populations During Development
During the first two postnatal weeks (P6–P15), the AMPA/NMDA ratio of evoked EPSCs at CA3–CA1 synapses in GluA4 deficient mice is significantly lower as compared to WTs [25]. We presumed that synaptic inputs influence the CA1 excitability predominantly during the bursts, when CA1 pyramidal neurons receive high-frequency glutamatergic input from the CA3. Indeed, even if the population firing rate was not affected at GluA4−/− slices, we found that the spike interval within the bursts was longer and the number of spikes per burst was lower in the GluA4−/− slices as compared to WTs and this effect was most consistent in the CA1 area (Fig. 4a–c).

GluA4 deficiency affects the population firing frequency within the bursts. a Example traces of recordings, filtered with 100 Hz high pass Butterworth filter to illustrate the population spikes within bursts for CA3 and CA1 pyramidal regions from WT and GluA4−/− slice cultures at DIV 2 and DIV 6. b Pooled data on the inter-spike interval (ISI) within the bursts for CA3 and CA1 areas in WT (n = 24) and GluA4−/− (n = 26) slice cultures. c Equivalent data as in (b), for the number of spikes/burst
Firing frequency within the bursts reflects the strength of excitatory synaptic drive but is also critically influenced by GABAergic inhibition. To study the development of functional synaptic connectivity between CA3–CA1 pyramidal cells more directly, we went on to analyse temporal pairwise-correlation of the spiking activity between pyramidal cell populations. Connectivity maps between electrodes located in the pyramidal regions were build based on STTC, a measure for temporal correlation that is independent on the basal activity level [5]. These maps indicated that development from DIV2 to DIV6 was associated with reorganization of the functional connectivity both in the WT and GluA4−/− slices (Fig. 5a). For quantification, the STTC and correlation coefficient was calculated for CA3 and CA1 channel pairs separated by at least 300 µm in each slice. In the wild-type slices, CA3 and CA1 neuronal populations became increasingly synchronized during development in vitro (DIV6 vs. DIV2, correlation coefficient, 380 ± 56%, p < 0.001; STTCCA3,CA1 124 ± 6% p = 0.07; Fig. 5b, c). In GluA4−/− slices, however, the temporal correlation of the CA3 and CA1 population spiking activity was not increased to the same extent as in the WT during development (DIV6 vs. DIV2, correlation coefficient, 273 ± 41%, p < 0.001; STTCCA3,CA1 105 ± 4% p = 0.75) and at DIV6 the level of correlation was significantly lower as compared to WTs (between genotypes, p < 0.001 for both correlation coefficient and STTCCA3,CA1; Fig. 5b, c). The differences in synchronization of CA3–CA1 population activities between the genotypes persisted during development and were still significant at DIV10 (correlation coefficient: WT 0.59 ± 0.033, n = 13 ; GluA4−/−: 0.48 ± 0.031, n = 15, p < 0.001; STTCCA3,CA1: WT 0.58 ± 0.02, n = 13; GluA4−/− 0.51 ± 0.018, n = 15, p < 0.01). Together, these data show that in the absence of GluA4, developmental synchronization of the firing activities of the CA3 and CA1 neuronal populations is impaired.

Loss of GluA4 perturbs synchronization of the CA3-CA1 population firing activities during development in vitro. a Examples of connectivity maps for WT and GluA4−/− slices cultures, based on STTC of the spiking activity between all the channels located in the pyramidal regions. The thickness of the line indicates the strength of the connection. b Pooled data showing the STTCCA3,CA1 for WT (n = 13) and GluA4−/− (n = 15) slice cultures at DIV 2 and DIV 6. c Pooled data on the correlation coefficient for firing activity in CA3–CA1 channel pairs WT (n = 13) and GluA4−/− (n = 15) slice cultures at DIV 2 and DIV 6
Discussion
The precise role of developmentally restricted plasticity mechanisms in wiring of limbic circuits is poorly understood. To get insight on their physiological significance, we have here studied how the GluA4 dependent plasticity mechanisms operating specifically at immature synapses contribute to functional development of CA3–CA1 circuit. We find that the loss of GluA4 has no effect on the overall excitability of the immature hippocampal network, but significantly impairs developmental synchronization of the CA3 and CA1 population firing activities. These data show that even if synapse-level defects in transmission and plasticity are efficiently compensated for to maintain physiological firing rates, they may perturb functional coupling between neuronal populations, a defect frequently implicated in the context of neuropsychiatric disorders.
PKA-driven synaptic insertion of GluA4 is the predominant mechanism for synaptic strengthening at immature hippocampal synapses [14, 24, 25]. GluA4 is efficiently transported to synapses with initially low or silent AMPA-mediated transmission in response to Hebbian activity [6, 25, 37], which is provided by the spontaneously occurring bursts of synchronous activity in the neonatal hippocampus [14,15,16, 27]. Accordingly, in the absence of GluA4, AMPA transmission in immature CA1 is less responsive to changes in intrinsic activity of the network [14]. Studies using recombinant GFP tagged receptors have indicated that phosphorylation of GluA4 subunits at Ser862 is sufficient for its synaptic recruitment in contrast to GluA1, that requires at least two Ser/Thr sites to be phosphorylated for its synaptic incorporation (e.g. [3, 6, 19, 20]). These differences in trafficking rules are thought to facilitate plasticity at immature synapses expressing GluA4 AMPA receptors but lacking the molecular scaffolds that support efficient intracellular signaling at mature spines. From postnatal day 10 onwards, GluA4 is expressed in and contributes significantly to excitatory drive to fast-spiking PV interneurons [29]. However, before the onset of interneuronal expression, corresponding to the first week in culture in our conditions, GluA4−/− slices provide a model to study the physiological significance of the immature-type synaptic mechanisms on early development of the hippocampal circuitry.
Despite its central role in mediating AMPA transmission and plasticity at immature synapses, the population firing rate and burst frequency in GluA4 deficient slices were not significantly different from the WTs during the first week in culture. Moreover, GluA4−/− mice are grossly normal with no apparent morphological changes in the brain [8, 31], suggesting that many of the GluA4 functions at the immature circuitry are efficiently compensated for. Indeed, the GluA4−/− mice show apparently normal LTP at immature CA3–CA1 synapses that is mediated via GluA1 and CaMKII dependent mechanisms, in contrast to the PKA dependent LTP that is predominant at this developmental stage in the WTs [24]. In addition, even if immature synapses in GluA4 deficient mice are less responsive to alterations in network activity, they are capable of synaptic scaling with a higher threshold for induction as compared to WTs [14]. These data are consistent with previous studies indicating that individual AMPA-R subunits are dispensable and can be largely substituted by other types of glutamate receptors to maintain synaptic functionality [10]. Our data from GluA4 deficient mice extend this finding from individual synapses to immature neuronal networks, which were able to maintain stable population firing rates surprisingly similar to that in the WT despite the loss of GluA4-mediated transmission and plasticity.
The relative importance of excitatory inputs in determining the spontaneous firing rate varies between cell types, depending on the activity of the circuit elements projecting to the particular neuron as well as its intrinsic properties. Stable population firing rates in GluA4−/− slices implies that alternative mechanisms, such as changes in intrinsic excitability or GABAergic inhibition are sufficient to maintain excitability in the absence of GluA4 dependent synaptic scaling. Interestingly, in cultured hippocampal neurons, network firing rates were not compensated following long-term AMPAR blockade in contrast to accurate homeostatic control following increase of synaptic inhibition [32]. This suggests that the extent by which changes in excitatory drive can be compensated for to maintain firing rate might be limited, thus explaining the need for redundancy in the synapse-level plasticity mechanisms which enables functionality in the face of perturbations.
Connectivity maps based on the temporal coincidence of population spiking activity indicated that significant reorganization of functional connectivity occurred during the first week in culture, leading to strengthening of the CA3–CA1 connectivity in the WT slices. In the GluA4 deficient slices, changes in the connectivity were evident but the synchrony between CA3 and CA1 neuronal populations was not significantly increased during development in vitro. The lack of immature-type plasticity mechanisms at CA3–CA1 synapses in the GluA4 deficient slices could well explain the impaired developmental synchronization of the CA3–CA1 population firing activities. According to the data obtained from acute neonatal slices [14, 25], GluA4 lacking AMPA receptors at silent or initially weak connections are expected to respond belatedly to intermittent synchrony at the immature network, complicating stabilization and strengthening of the nascent connections. At DIV8, CA1 pyramidal neurons in GluA4−/− slices had significantly lower population firing rate as compared to WTs, which could be a consequence of impaired development of the CA3–CA1 connectivity. However, even if GluA4 expression was not detected at GAD positive interneurons during the first week in culture, we cannot completely rule out the possibility that low GluA4 expression levels, below our detection limit, existed in the interneurons and affected synchronization directly or indirectly, for example via altered development and migration of GABAergic interneurons [1].
Human GluA4 gene (Gria4) has been identified as susceptibility gene for schizophrenia [26] and changes in the expression of GluA4 mRNA in the post-mortem samples from schizophrenic patients have been reported [2]. More recently, GluA4 was identified as a target regulated by the brain-enriched microRNA miR-124 that may contribute to social behavioral deficits in frontotemporal dementia [9] and to major depression [28]. Patients with neuropsychiatric disorders such as schizophrenia frequently have deficits in neuronal synchrony, including deficits in local oscillations and long-range functional connectivity (e.g. [33]). GluA4−/− mice show disrupted network oscillations and are prone to epileptiform activity [8, 28]. In addition, schizophrenia-related phenotypes including drastically impaired prepulse inhibition of the acoustic startle response and enhanced sensitivity to the locomotor stimulatory effects of a NMDA receptor antagonist, MK-801, have been reported [31]. Many of the phenotypes in adult GluA4 deficient mice can be attributed to the defects in glutamatergic drive to PV interneurons, since ablation of GluA4 selectively in hippocampal PV IN’s disrupts hippocampal population gamma rhythms [4, 8]. However, a large amount of evidence points to neurodevelopmental model in the origin of schizophrenia (e.g. [7]) supporting the idea that the defects in developmental plasticity, resulting in disordered connectivity, may also contribute to the disease mechanism.
References
Akgül G, McBain CJ (2016) Diverse roles for ionotropic glutamate receptors on inhibitory interneurons in developing and adult brain. J Physiol 594(19):5471–5490
Beneyto M, Meador-Woodruff JH (2006) Lamina-specific abnormalities of AMPA receptor trafficking and signaling molecule transcripts in the prefrontal cortex in schizophrenia. Synapse 60:585–598
Boehm J, Kang M-G, Johnson RC, Esteban J, Huganir RL, Malinow R (2006) Synaptic incorporation of AMPA receptors during LTP is controlled by a PKC phosphorylation site on GluR1. Neuron 51:213–225
Caputi A, Fuchs EC, Allen K, Le Magueresse C, Monyer H (2012) Selective reduction of AMPA currents onto hippocampal interneurons impairs network oscillatory activity. PLoS ONE 7(6):e37318
Cutts CS, Eglen SJ (2014) Detecting pairwise correlations in spike trains: an objective comparison of methods and application to the study of retinal waves. J Neurosci 34(43):14288–14303
Esteban JA, Shi SH, Wilson C, Nuriya M, Huganir RL, Malinow R (2003) PKA phosphorylation of AMPA receptor subunits controls synaptic trafficking underlying plasticity. Nat Neurosci 6:136–143
Fatemi SH, Folsom TD (2009) The neurodevelopmental hypothesis of schizophrenia revisited. Schizophr Bull 35:528–548
Fuchs EC, Zivkovic AR, Cunningham MO, Middleton S, Lebeau FEN, Bannerman DM, Rozov A, Whittington MA, Traub RD, Rawlins JNP, Monyer H (2007) Recruitment of parvalbumin-positive interneurons determines hippocampal function and associated behavior. Neuron 53:591–604
Gascon E, Lynch K, Ruan H, Almeida S, Verheyden JM, Seeley WW, et al (2014) Alterations in microRNA-124 and AMPA receptors contribute to social behavioral deficits in frontotemporal dementia. Nat Med 20:1444–1451
Granger AJ, Shi Y, Lu W, Cerpas M, Nicoll RA (2013) LTP requires a reserve pool of glutamate receptors independent of subunit type. Nature 493:495–500
Groc L, Gustafsson B, Hanse E (2006) AMPA signalling in nascent glutamatergic synapses: there and not there! Trends Neurosci 29:132–139
Hanse E, Taira T, Lauri S, Groc L (2009) Glutamate synapse in developing brain: an integrative perspective beyond the silent state. Trends Neurosci 32:532–537
Huupponen J, Molchanova SM, Taira T, Lauri SE (2007) Susceptibility for homeostatic plasticity is down-regulated in parallel with maturation of the rat hippocampal synaptic circuitry. J Physiol 581(Pt 2):505–514
Huupponen J, Atanasova T, Taira T, Lauri SE (2016) GluA4 subunit of AMPA receptors mediates the early synaptic response to altered network activity in the developing hippocampus. J Neurophysiol 115(6):2989–2996
Huupponen J, Molchanova SM, Lauri SE, Taira T (2013) Ongoing intrinsic synchronous activity is required for the functional maturation of CA3–CA1 glutamatergic synapses. Cereb Cortex 23(11):2754–2764
Kasyanov AM, Safiulina VF, Voronin LL, Cherubini E (2004) GABA-mediated giant depolarizing potentials as coincidence detectors for enhancing synaptic efficacy in the developing hippocampus. Proc Natl Acad Sci USA 101:3967–3972
Katz LC, Shatz CJ (1996) Synaptic activity and the construction of cortical circuits. Science 274(5290):1133–1138
Lauri SE, Vesikansa A, Segerstrale M, Collingridge GL, Isaac JTR, Taira T (2006) Functional maturation of CA1 synapses involves activity-dependent loss of tonic kainate receptor mediated inhibition of glutamate release. Neuron 50:415–429
Lee HK, Takamiya K, He K, Song L, Huganir RL (2010) Specific roles of AMPA receptor subunit GluR1 (GluA1) phosphorylation sites in regulating synaptic plasticity in the CA1 region of hippocampus. J Neurophysiol 103:479–489
Lee HK, Takamiya K, Kameyama K, He K, Yu S, Rossetti L, Wilen D, Huganir RL (2007) Identification and characterization of a novel phosphorylation site on the GluR1 subunit of AMPA receptors. Mol Cell Neurosci 36:86–94
Levelt CN, Hübener M (2012) Critical-period plasticity in the visual cortex. Annu Rev Neurosci 35:309–330
Lohmann C, Kessels HW (2014) The developmental stages of synaptic plasticity. J Physiol 592:13–31
Luchkina NV, Sallert M, Clarke VR, Taira T, Lauri SE (2013) Mechanisms underlying induction of LTP-associated changes in short-term dynamics of transmission at immature synapses. Neuropharmacology 67:494–502
Luchkina NV, Huupponen J, Clarke VR, Coleman SK, Keinänen K, Taira T, Lauri SE (2014) Developmental switch in the kinase dependency of long-term potentiation depends on expression of GluA4 subunit-containing AMPA receptors. Proc Natl Acad Sci USA 111(11):4321–4326
Luchkina NV, Coleman SK, Huupponen J, Cai C, Kivistö A, Taira T, Keinänen K, Lauri SE (2017) Molecular mechanisms controlling synaptic recruitment of GluA4 subunit-containing AMPA-receptors critical for functional maturation of CA1 glutamatergic synapses. Neuropharmacology 112(Pt A):46–56
Makino C, Fujii Y, Kikuta R, Hirata N, Tani A, Shibata A, Ninomiya H, Tashiro N, Shibata H, Fukumaki Y (2003) Positive association of the AMPA receptor subunit GluR4 gene (GRIA4) haplotype with schizophrenia: linkage disequilibrium mapping using SNPs evenly distributed across the gene region. Am J Med Genet B Neuropsychiatr Genet 116B:17–22
Mohajerani MH, Sivakumaran S, Zacchi P, Aguilera P, Cherubini E (2007) Correlated network activity enhances synaptic efficacy via BDNF and the ERK pathway at immature CA3 CA1 connections in the hippocampus. Proc Natl Acad Sci USA 104:13176–13181
Paz JT, Bryant AS, Peng K, Fenno L, Yizhar O, Frankel WN, Deisseroth K, Huguenard JR (2011) A new mode of corticothalamic transmission revealed in the Gria4(-/-) model of absence epilepsy. Nat Neurosci 14(9):1167–1173
Pelkey KA, Barksdale E, Craig MT, Yuan X, Sukumaran M, Vargish GA, Mitchell RM, Wyeth MS, Petralia RS, Chittajallu R, Karlsson RM, Cameron HA, Murata Y, Colonnese MT, Worley PF, McBain CJ (2015) Pentraxins coordinate excitatory synapse maturation and circuit integration of parvalbumin interneurons. Neuron 85:1257–1272
Quiroga RQ, Nadasdy Z, Ben-Shaul Y (2004) Unsupervised spike detection and sorting with wavelets and superparamagnetic clustering. Neural Comput 16:1661–1687
Sagata N, Iwaki A, Aramaki T, Takao K, Kura S, Tsuzuki T, Kawakami R, Ito I, Kitamura T, Sugiyama H, Miyakawa T, Fukumaki Y (2010) Comprehensive behavioural study of GluR4 knockout mice: implication in cognitive function. Genes Brain Behav 9:899–909
Slomowitz E, Styr B, Vertkin I, Milshtein-Parush H, Nelken I, Slutsky M, Slutsky I (2015) Interplay between population firing stability and single neuron dynamics in hippocampal networks. Elife 4:e04378
Spellman TJ, Gordon JA (2015) Synchrony in schizophrenia: a window into circuit-level pathophysiology. Curr Opin Neurobiol 30:17–23
Vesikansa A, Sallert M, Taira T, Lauri SE (2007) Activation of kainate receptors controls the number of functional glutamatergic synapses in the area CA1 of rat hippocampus. J Physiol 583(Pt 1):145-157
Yashiro K, Philpot BD (2008) Regulation of NMDA receptor subunit expression and its implications for LTD, LTP, and metaplasticity. Neuropharmacology 55:1081–1094
Yasuda H, Barth AL, Stellwagen D, Malenka RC (2003) A developmental switch in the signaling cascades for LTP induction. Nat Neurosci 6:15–16
Zhu JJ, Esteban JA, Hayashi Y, Malinow R (2000) Postnatal synaptic potentiation: delivery of GluR4-containing AMPA receptors by spontaneous activity. Nat Neurosci 3:1098–1106
Funding
The funding was provided by Suomen Akatemia, Sigrid Juséliuksen Säätiö, Jane ja Aatos Erkon Säätiö and CIMO.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article

Cite this article
Atanasova, T., Kharybina, Z., Kaarela, T. et al. GluA4 Dependent Plasticity Mechanisms Contribute to Developmental Synchronization of the CA3–CA1 Circuitry in the Hippocampus. Neurochem Res 44, 562–571 (2019). https://doi.org/10.1007/s11064-017-2392-8
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s11064-017-2392-8