
Abstract
Brief, non-harmful seizures (preconditioning) can temporarily protect the brain against prolonged, otherwise injurious seizures. Following focal-onset status epilepticus (SE) in preconditioned (tolerance) and sham-preconditioned (injury) mice, we screened for protein changes using a proteomic approach and identified several putative candidates of epileptic tolerance. Among SE-induced changes to both proteomic screens, proteins clustered in key regulatory pathways, including protein trafficking and cytoskeletal regulation. Downregulation of one such protein, ubiquitin carboxyl-terminal hydrolase isozyme L1 (UCHL1), was unique to injury and not evident in tolerance. UCHL1 inhibition decreased hippocampal ubiquitin, disrupted UPS function, interfered with seizure termination and exacerbated seizure-induced cell death. Though UCHL1 transcription was maintained after SE, we observed downregulation of the pro-translational antisense Uchl1 (AsUchl1) and confirmed that both AsUchl1 and rapamycin can increase UCHL1 expression in vivo. These data indicate that the post-transcriptional loss of UCHL1 following SE is deleterious to neuronal survival and may contribute to hyperexcitability, and are suggestive of a novel modality of rapamycin therapy.
Similar content being viewed by others
Avoid common mistakes on your manuscript.
Introduction
Epilepsy is a common neurological disorder characterised by unprovoked, recurrent seizures. Although numerous anti-epileptic drugs (AEDs) are available to patients, pharmacoresistance remains a major problem and no existing AED is efficacious in altering disease course, mandating indefinite prescriptions [1]. The identification of key contributors to the molecular pathogenesis of defined epilepsies therefore remains a research and health priority [2, 3]. One strategy is to exploit endogenous neuroprotective pathways activated in the brain in response to non-damaging stressors, a phenomenon termed tolerance [4]. Brief, non-harmful seizures (seizure preconditioning) can evoke a state of tolerance against the otherwise harmful effects of prolonged seizures (status epilepticus, SE [5]), where seizure-induced cell death is greatly reduced after periods of comparable seizure severity [6, 7]. Large-scale gene profiling work contrasting tolerance and non-tolerant (‘injury’) states has yielded important insights into factors contributing to epileptogenesis. These include changes to transcription of calcium signalling and excitotoxicity pathways [8], altered expression of various microRNAs and histone-modifying Polycomb Group proteins [9, 10], and differences in DNA methylation [11]. Functional work in animal models has shown targeting these processes can alter neuronal death and the emergence of spontaneous seizures [8, 9, 12]. Analysis of tolerance can reveal active neuroprotective mechanisms evoked by preconditioning, or can be indicative of important normative processes that become maladaptive or collapse during SE.
What remains absent from work to date on epileptic tolerance is an assessment of the proteome. Similar proteomic work revealed a novel role for epigenetic gene silencing in the phenotype of ischemic tolerance [13], a phenomenon that shares certain features with epileptic tolerance [5]. An equivalent comparison focusing on events unique to injury (in contrast to tolerance) complements prior transcript-based profiling work and may yield novel factors contributing to epileptogenesis. Indeed, new therapeutic target pathways recently emerged including those involved in cell growth and mechanistic target of rapamycin (mTOR) pathways, protein trafficking and post-translational regulation. Ubiquitination-dependent degradation of actin-binding proteins and the consequent synaptic remodeling may underlie rapid ischemic tolerance [14]. CRL4ACRBN, an E3 ligase, modulates surface expression of Ca2+ and voltage-activated K+ (BK) channels, thereby lowering seizure thresholds [15]. USP9X, a deubiquitinase, reduces proteasomal degradation of synaptic Prickle2, an evolutionarily conserved seizure-suppressive protein [16]. In each case, these enzymes displayed substrate-specificity and tractability in altering seizure activity. Widespread UPS inhibition and accumulation of ubiquitinated peptides follows SE, but surprisingly, it may be neuroprotective in epilepsy [17]; however, the effect of targeted ubiquitination and deubiquitination on seizure thresholds is substrate-dependent. USP9X, for example, is an important component in the endogenous anti-convulsant effect of Prickle signalling, but mediates the pro-convulsant activity of Prickle mutants when resulting in familial epilepsies [16].
Whether similar processes contribute to epileptic tolerance is unclear. Here, we searched for putative protein candidates using a proteomic screening approach in both injury and tolerance, highlighting potential differences between their proteomic signatures that may underlie the damage response to SE. Our screen was suggestive of disruption to regulatory cell processes in protein localisation, trafficking and ubiquitination. We focused on ubiquitin carboxyl-terminal hydrolase isozyme L1 (UCHL1), an abundant deubiquitinating enzyme which was reduced in injury compared to tolerance. Loss of UCHL1 was confirmed to be unique to the injury state and experimental inhibition in vivo disrupted ubiquitin homeostasis, depleted post-synaptic density protein 95 (PSD-95) expression, inhibited downstream ubiquitin–proteasome system (UPS) function and exacerbated seizure-induced neuronal cell death. These data shed light on a novel contributor to epilepsy pathology and validate proteomic approaches in identifying potential seizure-therapy targets.
Materials and Methods
Animal Procedures
Animal experiments were carried out as mandated by the European Communities Council Directive (86/609/EEC) and the European Union Parliament Directive (2010/63/EU). Experiments were prior approved by the Research Ethics Committee of the Royal College of Surgeons in Ireland or by the Centre for Laboratory Animal Resources in the Morehouse School of Medicine, under the supervision of the relevant Institutional Animal Care and Use Committee. Adult male C57BL/6 mice, aged 8–10 weeks (20–30 g), were obtained from Harlan or Charles River. Mice were housed in climate-controlled biomedical facilities on a 12 h light/dark cycle. Food and water were provided ad libitum.
Focal-onset SE was induced by intraamygdala (i.a.) microinjection of kainic acid (KA), as previously described [18]. For investigations involving tethered EEG recordings, three partial craniectomies were carried out and three cranial-mounted electrodes were set, as illustrated in Fig. 6a, prior to assembly of the microinjection assembly. KA microinjection (Sigma-Aldrich, in 0.2 µL phosphate-buffered saline [PBS]) was carried out in awake mice while non-seizure control mice received injection of vehicle alone. Dosages of both 0.3 and 1 µg KA were utilised to elicit moderate or severe SE, respectively [18]. For experiments involving epileptic tolerance, all mice undergoing SE received 1 µg KA, to ensure robust protection by preconditioning was evident. Subsequent studies on UCHL1 depletion following SE and not involving tolerance utilised 0.3 µg KA, to reduce morbidity. All mice undergoing SE received lorazepam (6 mg/kg, intraperitoneal [i.p.] in PBS) 40 min following i.a. KA injection to curtail seizures and reduce mortality. Seizure preconditioning was accomplished by i.p. injection of KA (15 mg/kg) 24 h prior to status epilepticus induced by i.a. KA [8].
Mice were killed at different time points following KA injection as outlined in the text. Paraformaldehyde (PFA, Sigma-Aldrich, 4%) perfused (transcardial) whole brains or hemispheres were dissected and sectioned on a vibratome (30 µm) prior to immunofluorescence. Saline perfused whole brains or hemispheres were fresh-frozen in 2-methylbutane at −30 °C and sectioned on a cryostat (12 µm) prior to histopathology.
Transgenic Mice Constitutively Expressing Ubiquitinated GFP
For investigations on the effect of inhibition of UCHL1, a deubiquitinating enzyme, transgenic mice constitutively expressing green fluorescent protein with an N-terminal ubiquitin (ubiquitinG76V-GFP) were employed [19]. The founding transgenic offspring were bred to C57BL/6N and backcrossed to C57BL/6N for approximately 30 generations before breeding with C57BL/6NJ mice to establish the commercially available colony. Breeding pairs were generously gifted by J. J. Lucas, Universidad Autónoma de Madrid.
EEG
Tethered EEG was recorded in free-moving, awake mice prior to and throughout SE and analysed using TWin software and Labchart Pro v7 (AD Instruments Ltd.). Electrographic data was binned into 30 s and 5 min allotments prior to analysis. Total power (µV2, a function of EEG amplitude over time) and spike frequency were calculated for whole electrographic traces as well as individual spectral bands (delta, 0–4 Hz; theta, 4–8 Hz; alpha, 8–16 Hz; beta, 16–32 Hz; and gamma, 32–50 Hz).
Drugs
Animals were randomised between treatment groups. Inhibition of UCHL1 was achieved through i.p. injection of LDN-57444 (0.5–2.5 mg/kg in 20% DMSO/PBS, Sigma-Aldrich) at 4 and 1 h prior to intraamygdala KA, unless otherwise stated. Rapamycin (Enzo Life Sciences) was administered i.p. (1 or 10 mg/kg) in a vehicle of ethanol (Sigma-Aldrich, 4%), polyethylene glycol-400 (Sigma-Aldrich, 4%) and Tween-80 (Sigma-Aldrich, 5%) in PBS, as described previously elsewhere [20]. Oligonucleotides targeting antisense Uchl1 (AsUchl1) were administered by intracerebroventricular (i.c.v.) injection into the right lateral ventricle (coordinates from bregma: AP = −0.3 mm, L = − 1.1 mm, with an injection depth of 2.0 mm). Mice received 2 µl of AsUchl1-targeting antagoNATs (Exiqon) or non-targeting scrambled controls (range 1 pmol to 100 pmol) in invivofectamine (Invivofectamine Reagent 2.0, Invitrogen). Anti-AsUchl1 antagoNATs were hybrid RNA-LNA unmodified oligonucleotide sequences of 16 nucleotide length. Two individual sequences, targeting two different sites in the first exon of AsUchl1 were administered simultaneously (as were two scrambled controls). These sequences each bind AsUchl1 at sites where it overlaps the sense Uchl1 mRNA, including the region of the translation start codon (see Fig. 9a). This overlap interaction is critical for AsUchl1-mediated translational effects on Uchl1 [21]. Anti-AsUchl1 AntagoNATs were injected in synthetic liposomes generated using invivofectamine as per manufacturer’s suggestions.
Mass Spectrometry
For proteomic analysis, individual ipsilateral hippocampus was dissected and homogenised in RIPA (radioimmunoprecipitation assay) buffer (50 mM tris [pH 8.0], 150 mM NaCl, 0.5% deoxycholate, 0.1% SDS, and a cocktail of protease inhibitors). Homogenates were fully lysed by three freeze–thaw cycles, centrifuged (10,000×g, 10 min at 4 °C) and the protein concentration quantified by the micro BCA Protein Assay (Pierce). For each treatment group and timepoint, lysates containing equal protein amounts from 6 animals were pooled (to a total of 400 µg) and analysed in triplicate using mass spectrometry (MS). Protein lysates were precipitated in methanol/chloroform [22] and resuspended in 50 mM ammonium bicarbonate. They were then denatured (0.06% RapiGest, 60 °C for 15 min), reduced (5 mM dithiothreitol, 60 °C for 30 min), and alkylated (10 mM iodoacetamide, room temperature for 30 min in darkness). Samples were digested in sequencing grade trypsin (Promega, 0.05 µg/µl, 37 °C overnight) and digestion was terminated by 0.5% trifluoroacetic acid, which also destroyed RapiGest in the sample. Yeast alcohol dehydrogenase (50 fmol/µl) was added to the digests as an internal standard for absolute quantification [23]. Flow separation of tryptic peptides was achieved using ultra performance liquid chromatography (nanoACQUITY UPLC, Waters) in preparation for MS. MS analysis of nanoUPLC elutes was carried out using a Q-Tof Synapt G2S mass spectrometer (Waters) using HDMSE acquisition mode. Analysis of MS data was completed using ProteinLynx Global Server (PLGS) software v2.3 (Waters) and a mouse protein database downloaded from the Universal Protein Resource (UniProt, http://www.uniprot.org) including Swiss-Prot entries only, aided by a previously described algorithm [24] and previously established criteria [13]. Using these assessments, a ‘femtomoles per microgram’ value was generated and used to calculate protein ratios between experimental conditions.
Histopathology and Immunofluorescence
To assess neuronal death, coronal cryosections (12 µm thickness, sectioned on a Leica CM1900 cryostat) were assayed using Fluoro-Jade B (FJB) staining and terminal deoxynucleotidyl transferase dUTP nick end labelling (TUNEL). For FJB staining, fresh-frozen tissue sections from three stereological levels [25] were air-dried, post-fixed in PFA solution (4%, 30 min), hydrated (100% ethanol into 70% ethanol into dH2O, 1 min each) and then immersed in 0.006% potassium permanganate solution (15 min, with agitation, Sigma-Aldrich). Sections were rinsed (dH2O, 2 × 1 min) and placed in 0.001% FJB solution in 0.1% acetic acid (Chemicon Europe Ltd.) for 30 min. Sections were rinsed again (dH2O, 9 × 1 min), dried, cleared in Histoclear II (National Diagnostics) and mounted in DPX (Sigma-Aldrich). DNA damage was analysed using a fluorescein-based TUNEL technique, according to the manufacturer’s instructions (Promega). Briefly, 12 µm cryosections were fixed (4% PFA, 30 min), washed (0.1% Triton-X in PBS, 3 × 5 min), permeabilised (3% Triton-X, 20 min) and washed again. Slides were then equilibrated in kit-specific equilibration buffer prior to reaction with recombinant Terminal Deoxynucleotidyl Transferase and fluorescein-12-dUTP (90 min, 37 °C, in darkness). Slides were immersed in saline sodium citrate, washed in PBS, mounted (Fluorsave, Merck Millipore) and imaged. Sections were examined using a Nikon 2000s epifluorescence microscope (Micron Optical) under Ex/Em wavelengths of 472/520 nm and imaged using a Hamamatsu Orca 285 camera (Micron Optical). Cell counts were performed for the primary hippocampal subfields, including the CA3, CA1, CA2, the granular layer of the DG and the hilar layer within the DG. Counts were carried out in duplicate, were the average of 2–4 adjacent sections and were assessed by an observer blinded to the experimental group or condition.
Immunofluorescence was performed on thicker coronal sections (Leica VT1000S vibratome, 30 µm) of the hippocampus. Sections were prepared from transcardially PFA-perfused whole brain and stored at -20 °C in cryoprotective medium (30% glycerol and 30% ethylene glycol in 1X PBS). Immunostaining was achieved following permeabilisation (0.1% Triton-X in PBS, 15 min), washing in glycine (1 M in PBS, 30 min) and blocking (1% bovine serum albumin in PBS, 60 min). Sections were incubated overnight at 4 °C with the following primary antibodies; α-NeuN (Millipore), mouse-raised, 1:400; α-UCHL1 (Cell Signalling Technology), rabbit-raised, 1:200. Immunoreactivity was visualised using AlexaFluor 488 and 568 secondary antibodies (1:400 dilution in blocking buffer, incubated for 2 h at room temperature, from Molecular Probes, OR, USA). Sections were labelled with Hoescht nuclear stain to visualise nuclei (1:10,000, 10 min). Tissue was mounted in FluorSave (Sigma-Aldrich). Images were captured using confocal microscopy (Zeiss Laser Scanning Microscope 710 Meta). For these exposures, 1 µm optical sections were taken at the midpoint of the 30 µm vibratome-sectioned samples, as determined by the limits of fluorescence. Mean fluorescence intensity of UCHL1 immunoreactivity was calculated in ImageJ for the entire pictograph and used as the basis of comparison between control and injury.
Gene Expression Analysis
Reverse-transcription (RT) quantitative PCR (qPCR) was carried out on microdissected hippocampal tissue samples. RNA (including mRNA and lncRNA) was isolated using a standard trizol extraction method. Tissue was mechanically homogenised in Trizol (Qiagen), after which chloroform-mediated phase separation was performed and isopropanol-mediated precipitation carried out. RNA was washed in 70% ethanol and reconstituted in RNase free H2O. Prior to RT, concentration of nucleic acid extracts were determined using a NanoDrop 2000 (Thermo Scientific), treated with DNAse1 (Invitrogen) to eliminate contaminating genomic DNA, and normalised to between 500 ng and 1 µg. RT of mRNA was performed using random hexamer primers (Fermentas) and Superscript II reverse transcriptase (Invitrogen). For lncRNA, RNA homogenates were reverse transcribed using the iScript Select cDNA synthesis kit, with kit-supplied random hexamer primers (Bio-Rad). The relative abundance of derived cDNA was determined by qPCR, carried out on a Lightcycler 2.0 (Roche) using Quantitect SYBR Green PCR kits (Qiagen). The following primer sequences were used; Uchl1, forward AGAGCCAAGTGTTTCGAGAAGA and reverse ATTCTCTGCAGACCTTGGCA; GAPDH, forward AACTTTGGCATTGTGGAAGG and reverse ACACATTGGGGGTAGGAACA; β-actin, forward GGGTGTGATGGTGGGAATGG and reverse GGTTGGCCTTAGGGTTCAGG. For antisense Uchl1, three sets of primers targeting distinct sequence domains were used, as used previously [21]; in the 3′ region, forward ACCTGCTTGCCAATAGACGA and reverse TGTATGGGAAGGTTGGCTGT; in the sense overlap region, forward GCACCTGCAGACACAAACC and reverse TCTCTCAGCTGCTGGAATCA; and in the SINEB2 repeat region, forward TCTCTCAGCTGCTGGAATCA and reverse TTCTCCTTATATCTCCCAGTCAGG. β-actin and GAPDH were used for the normalisation of RNA expression levels. Non-reverse transcribed extracts and non-template reactions were used as negative controls. The comparative cycle threshold method (2−ΔΔCT) was employed to determine the relative fold change in target transcript levels. Primer specificity was investigated using Taq Polymerase PCR in a Veriti Thermocycler (Applied Biosystems). Amplification products were run on a 2% agarose gel (100 V, 15 min, with 1:10,000 ethidium bromide) and imaged in a FujiFilm LAS-3000 (Fuji) under UV (312 nm, 0.125 s exposure).
Western Blotting
Homogenisation of whole hippocampus was carried out in lysis buffer (150 mM NaCl, 50 mM Tris–HCl, 1 mM EDTA, 1% NP-40, ph 8.0), where buffer included protease/phosphatase inhibitors PMSF (1:500), aprotinin (1:1000), leupeptin (1:1000) and vanadate (1:1000). Homogenates were centrifuged (14,000×g, 10 min at 4 °C) and the supernatant was retained. The pellet was also retained and later resuspended in SDS-lysis buffer for analysis of protein retained within the nuclear compartment. Protein concentration was determined using the micro BCA Protein Assay (Pierce). Lysates were boiled at 95 °C in gel-loading buffer and separated by SDS-PAGE (4%/6–15%, 90–130 V), before transfer by electroporation onto nitrocellulose or PVDF membranes (18 V, 60–90 min). Membranes were incubated overnight at 4 °C with primary antibodies raised against; ARP2 (Abcam); α-Tubulin (Sigma-Aldrich); Cdc42 (Cell Signalling Technology) GFP; GSK3β (Biosource); MDM2 (Santa Cruz Biotechnology); p53 (Santa Cruz Biotechnology); PSD-95 (Cell Signalling Technology); Sept7 (Novus Biologicals); Synj1 (Synaptic Systems); Ubiquitin (Enzo Life Sciences); UCHL1 (Cell Signalling Technology). Membranes were subsequently incubated with secondary horseradish peroxidase-conjugated antibodies (2 h, 20 °C, Jackson Immuno-Research), and visualised through Super Signal West Pico chemiluminescent substrate (Pierce). Images were captured with a FujiFilm LAS-4000 (Fuji) and densitometry performed using ImageJ. Chemiluminescent density plots for each sample were generated and the area of the region of interest (corresponding to the expected molecular weight) was normalised to the sample-matched loading control from the same immunoblot.
Statistics and Bioinformatics
All data and statistical analyses were carried out using Microsoft Excel, Graphpad Prism and Stata. Significance was accepted at P < 0.05. All data are presented as mean ± S.E.M. Protein expression, cell death counts and RNA transcription were assessed using unpaired two-tailed Student’s t tests and Mann–Whitney U tests (for two-group comparisons), or one-way ANOVA and Kruskal–Wallis tests (for multiple comparisons). Time-dependent EEG comparisons of power and frequency (for complete traces as well as individual frequency bands) were made using two-way analysis of variance, followed by the Bonferroni post-hoc test. Direct comparisons of fold changes over baseline between periods before and after LZ injection were made using unpaired two-tailed Student’s t test.
Following identification of regulated proteins in the proteomes of injury and tolerance, follow up bioinformatic analyses were performed using Excel (2007, Microsoft), GOstat (online resource, [26]) the Database for Annotation, Visualization and Integrated Discovery (DAVID v6.7, online resource, [27]) and Cytoscape (version 3.0, [28]) with the Enrichment Map plugin [29] to determine significant quantity differences, resolve functional ontological enrichments and visualise the proteome. Fisher’s exact test was performed independently for the three primary gene ontology hierarchies (molecular function, cellular component and biological process) and corrected using the Benjamini and Hochberg test for false discovery rate (FDR). For network visualisation, GO terms and the genes associated with each were mapped using the Enrichment Map plugin for Cytoscape. Functional clusters were manually annotated.
Results
Comparative Mass Spectrometry of the Global Proteome Following SE Reveals Divergences in the Proteome of Injury and Tolerance
We began by performing MS to assess the global proteome of the hippocampus following SE, in paradigms of injury and tolerance. Mice were injected with KA or vehicle control (i.a.), while the tolerance group also received systemic KA (i.p.) 24 h beforehand as a preconditioning stimulus. Mice receiving a preconditioning stimulus typically undergo a period of similar seizure severity and duration upon induction of SE, but with a reduction in seizure-induced cell death of approximately 50% [6, 7]. Mice were sacrificed at 3 and 24 h and hippocampal protein lysates from 6 animals per group were pooled (Fig. 1a) and run in triplicate. Upon identification of eluted peptides, inclusion criteria and quality control were applied. For those passing these criteria, protein quantities were obtained relative to ADH as an internal standard as previously described [13, 23] and normalised to femtomoles per microgram of total protein eluted. An average of 352 proteins were detected per sample. Using this finalised list of proteins, breakdown analysis across groups revealed that the majority of proteins were detected across all treatments (265/417, 3h post SE; 277/469, 24h post-SE) with subpopulations seen uniquely in certain treatments. A summary of all inclusion criteria, quality control and protein counts per sample are given in Supplementary File 1. A ratio cutoff was applied to proteins detected in all conditions, where those exceeding changes of ±30% compared to control were defined as upregulated or downregulated. Using this threshold, 17.95% of all proteins detected were considered to be changed after SE. In events where proteins were not uniformally detected, proteins were defined as +100% (not detected in control) or −100% (detected in control only). Comparing the extent of unique upregulation and downregulation events between injury and tolerance (compared to control) revealed that upregulation is the dominant response to SE, but particularly so for tolerance at 3 h, with 77.5% (69/89) of all unique changes versus control representing upregulation of proteins (Fig. 1b, c).

Proteome of SE, comparing injury and tolerance, reveals differences in protein regulation and expression. a Overview of the experimental design. b Protein upregulation and downregulation events as identified by MS in injury and tolerance. c Proportion of up- versus down-regulation events across regulated proteins identified by MS in injury and tolerance. I.p. intraperitoneal, KA kainic acid. Annotated protein lists for each experimental group (totalling 8 tables) can be found in the electronic supplementary material (File 1)
Gene Ontological Analysis Identifies Functional Clusters of Proteins that are Dysregulated Following SE
Having established criteria for defining functional differences in the proteome, we looked closer at such differences in both injury and tolerance. A finalised list of proteins altered following SE is given Supplementary File 1, segregated on the basis of treatment, timepoint and the direction of dysregulation (upregulation or downregulation). Several proteins of interest are immediately notable, with many pertaining to regulation of the cytoskeleton, plasticity and synaptic function, mitochondrial function and metabolism, apoptosis, ion transport and calcium binding, DNA binding and stress response. Some putative candidates are listed in Table 1. To give a more systematic evaluation of the proteome, we subjected our segregated lists to gene ontological (GO) analysis using GOstat (see Supplementary File 2). All listed p values were corrected for multiple testing using the Benjamini–Hochberg test for false discovery (P < 0.05). GO analyses point to a number of perturbed pathways pertinent to SE and epileptic tolerance. Proteins mediating protein trafficking and localisation, vesicle dynamics, endocytosis and clathrin assembly are enriched among upregulated proteins seen in injury at 3 h, but are enriched among proteins downregulated in tolerance at 3 h, indicating a key functional divergence between them. A similar situation is seen for proteins involved in regulation of the cytoskeleton and ion transport, where enrichment for proteins involved in these processes is seen among upregulated proteins in tolerance, but among proteins downregulated in injury, again at 3 h following SE. Processes such as metabolic processes, synaptic plasticity, neurotransmission and the regulation of mitochondria are also evident. At the later timepoint, 24 h following SE, we observe an enrichment of proteins involved in neurogenesis and axoneogenesis specifically in injury among upregulated proteins. Processes such as protein localisation and vesicle transport, initially pertinent to proteins upregulated in injury at 3 h, were observed as important to upregulation in tolerance at 24 h, suggesting that stunted cellular responses to SE may be a characteristic of tolerance. Using the Enrichment Map plugin for Cytoscape [28, 29], we grouped functionally related GO terms based on the overlap of proteins associated with distinct GO terms (Fig. 2). This visualisation analysis revealed a number of network clusters that represented major functional groups that were enriched in both injury and tolerance datasets.

Enrichment of major functional groups define injury and tolerance datasets. Functionally related GO terms were grouped based on the overlap of proteins associated with distinct GO terms using the Enrichment Map plugin for Cytoscape, for both injury (above) and tolerance (below). Inset UCHL1 is involved in several key, non-redundant functional groups implicated in proteomic profiling. Complete gene ontology lists for each experimental group can be found in the electronic supplementary material (File 2)
We next sought to validate observations on the proteome using western blotting. We used the same lysates as those used for MS and immunoblotted for Septin 7 (Sept7), Glycogen Synthase Kinase 3β (GSK3β), Cell Division Control Protein 42 (Cdc42), Actin-related Protein 2 (ARP2), Synaptojanin1 (Synj1) and UCHL1. Little is known of these proteins concerning epilepsy pathology, but they maintain important roles in neuronal function [30–33]. All six were predicted to be altered beyond the 30% cutoff established in the proteomic analysis. Sept7, GSK3β, Cdc42 and ARP2 were predicted to be dysregulated at 3 h, while Synj1 and UCHL1 were predicted to be altered at 24 h post-SE (Fig. 3a). We validated the dysregulation of four proteins predicted to change in injury (Sept7 and GSK3β, upregulated at 3 h; Synj1 and UCHL1, downregulated at 24 h, Fig. 3a, b). Tolerance-specific downregulation of Cdc42 at 3 h following SE was also confirmed. Western blotting indicated no SE-dependent change in ARP2, predicted to be upregulated specifically in tolerance at 3 h.

Follow-up biochemical analysis validates observations on the proteome. a In order to validate changes in protein expression as indicated by MS, western blotting was performed on ipsilateral hippocampal lysates from sham-preconditioned (injury) and seizure tolerant (tolerance) mice at 3 h following SE for Sept7, GSK3B and Cdc42, and at 24 h following SE for Synj1 and UCHL1. n = 5 or 6, *P < 0.05, **P < 0.01, ANOVA with Tukey post-hoc test. b Representative blots of data depicted in (a), as well as that of ARP2, are shown, n = 1 per lane. There was no significant change in ARP2 expression between treatments. Depicted blots for Sept7 and Cdc42 are each derived from the same gel. Depicted blots for Synj1 and UCHL1 are each derived from the same gel. The non-continuity of the depicted blot for Cdc42 is due to removal of interleaved lanes representing a different treatment group, for sake of clarity. CON control, INJ injury, TOL tolerance, TUB tubulin
UCHL1 Expression Levels are Decreased in the Hippocampus Following SE
Using GO and network analyses, we sought to select a candidate protein that showed divergent expression between injury and tolerance, and was a likely contributor to the major implicated functional groups, such as intracellular protein trafficking. UCHL1, an abundant DUB enzyme, is a key regulator of protein trafficking, cytoskeleton regulation and post-translational ubiquitination. In contrast to tolerance, in which both seizure-induced cell death and spontaneous seizures are reduced, UCHL1 was selectively downregulated in injury paradigms. Recent studies have implicated ubiquitination-mediated processes in regulating surface expression of ion channels [15] and degradation of synaptic proteins [16]. Targeting these processes altered seizure activity in these cases, while rescue of Ube3a, an E3 ligase, also alleviated other comorbidities in a model of Angelman syndrome [34]. GO and network analyses suggested that UCHL1 is involved in several key, non-redundant functional groups implicated in our proteomic profiling (Fig. 2, inset) and it was thus selected as a candidate protein for further study.
Western blot validation confirmed that UCHL1 is downregulated at 24 h following SE, as indicated by MS (one-way ANOVA, P = 0.0216; Tukey post-hoc, P < 0.05 compared to control). Confocal imaging revealed a patterned distribution of UCHL1 expression in the hippocampus of control mice, with somal expression evident throughout the pyramidal layer of the CA1, CA2 and CA3, as well as expression in unspecified neurons of the hilus (Fig. 4a). There was little UCHL1 expression in the granule layer. Immunoreactivity was almost entirely neuronal, as based on co-immunoreactivity with NeuN. Furthermore, UCHL1 immunoreactivity was observed in the dendritic field of CA3 and CA1 regions. Confocal imaging of staining in KA-treated mice suggested that UCHL1 immunoreactivity is reduced at 24 h following SE, in keeping with results from MS and Western blotting (Fig. 4b, c). Loss of CA3 immunoreactivity was profound, (Mann–Whitney U, P = 0.0286, Fig. 4b), while immunoreactivity was also reduced in regions of less-damaged CA1 fields (P = 0.0571, Fig. 4b), but not in the DG. Reduction of UCHL1 expression after KA was observed to impact both somal expression and staining in the stratum radiatum (Fig. 4c).

UCHL1 is downregulated in the ipsilateral hippocampus at 24 h after SE. a Confocal imaging showing UCHL1-immunoreactive cells in the mouse hippocampus. NeuN was used as a counterstain. Scale 50 μm. b Relative anti-UCHL1 immunofluorescence intensity was calculated for sections depicted in c where analysis of UCHL1 staining was carried out using confocal microscopy, in control animals and in animals that underwent SE 24 h previously (n = 4, Mann–Whitney U test, two-tailed). Scale bar 100 μm. Data expressed as mean ± SEM. *P < 0.05. CA cornu ammonis, CON control, DG dentate gyrus, INJ injury
Inhibition of UCHL1 Using LDN-57444 Depletes the Hippocampus of Monomeric Ubiquitin and Postsynaptic Density Protein PSD-95
To assess whether downregulation of UCHL1 contributes to seizure pathology or neuronal death, we explored the use of an inhibitor of UCHL1, LDN-57444 (termed LDN) as a means of manipulating UCHL1 activity in vivo. LDN is a reversible, competitive, isatin derivative that selectively inhibits UCHL1 hydrolase activity (IC50 = 0.88 µM) over a closely related DUB, UCHL3 (IC50 = 25 µM) [35]. Although no data is available on the pharmacokinetics of LDN in mice, LDN and other isatins readily cross the blood–brain barrier [30, 36, 37]. Previous work suggests that LDN is capable of exerting an inhibitory effect on UCHL1 in the hippocampus at systemic doses of 0.5 mg/kg [30, 36]; we used this reported effective dose to assess the inhibitory effect of LDN.
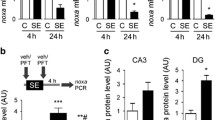
Mice were given 0.5 mg/kg i.p. LDN at 1, 4, 8 and 24 h prior to sacrifice, and levels of free monomeric ubiquitin were assessed by Western blotting, as a measure of UCHL1 inhibition [36]. These data suggested that the LDN-induced downregulation of free ubiquitin was maximal at 4 h following UCHL1 inhibition, though the effect was not significant (P = 0.1419, Fig. 5a, c). Increasing the dose of LDN to 1 and 2.5 mg/kg revealed a significant effect on free ubiquitin levels when assessed at 4 h (one-way ANOVA, P = 0.0011; Tukey post-hoc, P < 0.05 for 1 mg/kg dose and P < 0.01 for 2.5 mg/kg dose, when compared to vehicle control; Fig. 5b, c). High dose LDN (2.5 mg/kg) was observed to induce an increase in UCHL1 expression (one-way ANOVA, P = 0.0439; Tukey post-hoc, P < 0.05, compared to vehicle control; Fig. 5d, f), possibly as a compensatory mechanism. Both 1 mg/kg and 2.5 mg/kg doses of LDN were observed to deplete the hippocampus of postsynaptic density protein PSD-95 (one-way ANOVA, P = 0.0053; Tukey post-hoc, P < 0.05, compared to vehicle control; Fig. 5e, f). Given the efficacy of 1 mg/kg LDN in depleting the hippocampus of ubiquitin and PSD-95 with no reactive changes in UCHL1 expression, subsequent experiments using LDN were carried out with 1 mg/kg i.p. doses of LDN. In order to investigate possible effects of UCHL1 inhibition on the UPS, we employed a genetic reporter mouse strain (with C57Bl6/J background) constitutively expressing ubiquitinG76V-GFP to directly assess LDN-induced effects of ubiquitination-mediated proteasomal degradation. UCHL1 inhibition resulted in an accumulation of ubiquitinG76V-GFP at 4h (Mann–Whitney U, P = 0.0159, Fig. 5g, h), suggesting that loss of UCHL1 activity leads to downstream inhibition of the UPS.

UCHL1 inhibition depletes the hippocampus of monomeric ubiquitin and PSD-95, and inhibits the ubiquitin proteasome system. a Mice received 0.5 mg/kg LDN-57444 (i.p.) and hippocampal lysates were sampled at various times after injection. Monomeric ubiquitin levels were then assessed using western blotting. Semi-quantification was performed against endogenous tubulin (n = 4, one-way ANOVA with Tukey post-hoc test). b Mice received various doses of LDN-57444 before lysates were taken at 4 h. Ubiquitin levels were assessed as in a. c Representative blot used for the semi-quantification of ubiquitin levels depicted in a and b, n = 1 per lane. Observed molecular weights and experimental conditions are shown. d, e Western blotting was performed on hippocampal lysates from mice 4 h after receiving LDN-57444 at various doses. Semi-quantification was performed against endogenous tubulin (n = 4, one-way ANOVA with Tukey post-hoc test) for d UCHL1 and e PSD-95. f Representative blots used for the semi-quantification of proteins levels depicted in d and e, n = 1 per lane, with observed molecular weights and dose of LDN-57444 annotated. g Immunoblot showing ubiquitin-GFP accumulation in a reporter mouse for UPS inhibition. Analysis was carried out on hippocampal lysates from mice that received 1 mg/kg LDN-57444 or vehicle control 4 h previously (n = 5, Mann–Whitney U test, two-tailed). h Representative blots used for the semi-quantification of proteins levels depicted in f, n = 1 per lane, with observed molecular weights and experimental conditions depicted. i TUNEL and FJB staining was carried out on mice receiving 0.5 mg/kg LDN-57444 at various time-points previously, as noted. Scale bar 50 μm. All data expressed as mean ± SEM. **P < 0.01, *P < 0.05. CA cornu ammonis, DG dentate gyrus, LDN LDN-57444, TUB tubulin, UBQ ubiquitin, VEH vehicle
Previous work suggests UCHL1 inhibition is not cytotoxic at effective doses, as shown by MTT assays, LDH viability assays, and measurements of neuronal density in the CA1 of LDN-treated mice [36, 38]. We verified that LDN administration did not cause observable cell death. Staining sections for either FJB or TUNEL revealed no appreciable cell death in the hippocampus at 1, 4, 8 or 24 h after LDN injection (Fig. 5i).
UCHL1 Inhibition Prior to SE Attenuates the Electrographic Response to Anticonvulsant Lorazepam
As iterated previously, evidence is accumulating that substrate-specific activity of ubiquitin ligases and deubiquitinases can alter epileptogenesis [34] and the threshold for seizures [15, 16]. We hypothesised that UCHL1 inhibition would exacerbate seizure activity during SE. Seizures were evoked 4 h after UCHL1 inhibition (the point of maximal depletion of free ubiquitin), with the emergence of SE at approximately 10–15 min after injection of KA (Fig. 6b). Lorazepam (LZ) was injected (6 mg/kg i.p.) at 40 min following KA to curtail morbidity and mortality. EEG activity in LDN and vehicle treated controls was recorded with three skull mounted electrodes, with tissue sampling at 4 h and cell death assays at 24 h following SE (Fig. 6a). A second injection of LDN at 1 h after KA was included in order to prolong inhibition, as UCHL1 activity was previously reported to recover at 8 h following LDN injection [30].

UCHL1 inhibition prolongs seizures after status epilepticus. a Experimental design for assessment of effects of UCHL1 inhibitor on SE. Mice received LDN-57444 and then 4 h later were injected (i.a.) with KA to trigger SE, followed after 40 min by lorazepam. EEG recordings were made for 2 h after SE, during which time a second injection of LDN-57444 was performed. Cell death was assessed at 4 and 24 h after SE. b Representative power density analysis of EEG during SE as heatmaps for mice receiving vehicle or LDN-57444. c Power and spike frequency traces of 5 min time-bins were generated from EEG data for LDN- and vehicle-treated mice (n = 8 to 10, two-way ANOVA with Bonferroni post-hoc test). d The ratio against baseline for both power and frequency was calculated for periods before and after lorazepam treatment, in both groups of mice (n = 8–10, paired Student’s t test, two-tailed). P value is overlaid in yellow. e Representative trace of EEG power fractionation into five frequency bands using Labchart, displaying EEG evolution before and after KA injection (i.a.). f EEG power was fractionated as in e for both LDN- and vehicle-treated mice (n = 8–10). g Two-way ANOVA comparisons (with Bonferroni post-hoc test) were performed for alpha and delta frequency patterns between LDN- and vehicle-treated mice. All data expressed as mean ± SEM. *P < 0.05; # or **P < 0.01; ¤ or ***P < 0.001. KA kainic acid, LDN LDN-57444, LZ lorazepam, VEH vehicle
EEG recordings revealed that UCHL1 inhibition had no effect on the onset of SE based on measures of power and spike frequency for both ipsilateral and contralateral montage channels (two-way ANOVA, Fig. 6c). LZ injection strongly suppressed SE with a decrease in power and an increase in frequency for both vehicle and LDN treated mice, consistent with a transition from high-amplitude synchronous firing to a fast ictal pattern (Fig. 6c). To assess if UCHL1 inhibition had an effect on LZ efficacy, we directly compared the average fold change in power and frequency (relative to baseline EEG) of the SE period and the post-LZ period, defining the SE period as between KA + 15 min and KA + 40 min and the post-LZ period as between KA + 40 min and KA + 80 min. As per EEG traces (Fig. 6c), LZ induced a significant reduction in ipsilateral power (P = 0.0180, Fig. 6d) and significantly increased ipsilateral and contralateral frequency measures (P = 0.0006, Fig. 6d) in vehicle treated mice. In LDN treated mice, LZ was associated with a significant increase in ipsilateral frequency only (P = 0.0108, Fig. 6d), suggesting that the normalising effect of LZ is attenuated following UCHL1 inhibition. To more specifically delineate this effect, we separated the EEG power signal into five frequency-defined spectral bands and defined the fractional contribution of each band to the total power. As per the baseline signal (i.e., before KA injection) delta frequencies (0–4 Hz) represent approximately 40% of the power signal and alpha frequencies (8–16 Hz) account for approximately 15% of the total power signal (Fig. 6e). The onset of SE is typified by a disproportionately large delta fraction with an associated reduction in all other fractions, while LZ-induced normalisation of the EEG is characterised by a transient increase of the alpha fraction in particular before a return to baseline distribution within 60 min of LZ (Fig. 6e, f). However, UCHL1 inhibition was associated with an attenuation in the return to baseline distribution following LZ, both ipsilaterally and contralaterally (Fig. 6f). These animals experienced a prolonged period of time under the increased delta signal associated with initial SE, with little concomitant increase in the alpha signal (Fig. 6f). Direct comparisons of vehicle and LDN treated mice revealed a significant divergence in the ipsilateral alpha (two-way ANOVA, P < 0.0001/F(1,330) = 52.99 for drug; Bonferroni post-hoc test results are indicated, Fig. 6g) and ipsilateral delta fractions (two-way ANOVA, P < 0.0001/F(1,330) = 36.00 for drug; Bonferroni post-hoc test results are indicated, Fig. 6g). Similar results were seen for contralateral EEG traces (Fig. 6g). These divergences were confined to the initial 20 min period after LZ injection, and the EEG signal became indistinct within 100 min of KA injection (Fig. 6g).
UCHL1 Inhibition Prior to SE Exacerbates Seizure-Induced Neuronal Cell Death
We next appraised potential changes in seizure-induced neuronal cell death following UCHL1 inhibition, given data suggesting a refractoriness to LZ effects and previous reports that UCHL1 is neuroprotective in models of neurodegeneration [30, 36]. We performed FJB histopathology on cryostat-sectioned tissue at 24 h following KA, and performed counts of degenerating CA3, CA1, CA2, granule layer and hilar neurons in vehicle and LDN treated mice. Three stereological levels were assessed, corresponding to rostral, medial and ventral (or temporal) hippocampus (Fig. 7a). Average counts across all 3 stereological levels revealed that UCHL1 inhibition prior to SE exacerbates seizure-induced neuronal cell death in the CA3 of the hippocampus (one-way ANOVA for all treatments, P < 0.0001; Tukey post-hoc, P < 0.05, compared to vehicle-treated animals undergoing SE, Fig. 7b, c). The significant increase in cell death was most evident in the ventral hippocampus (two-way ANOVA, P = 0.0006/F(1,51) = 13.57 for drug; P < 0.05, Bonferroni post-hoc at ventral levels, Fig. 7d). This was supported by subsequent TUNEL labelling of ventral hippocampal slices from the same animals, revealing an LDN-induced increase in cell death following SE (P = 0.0080, Fig. 7e). There was no observed increase in SE-induced neuronal cell death in other subfields (data not shown). Further, UCHL1 inhibition did not affect cell death in the contralateral CA3 or when LDN was administered after SE (at 1 and 5 h after KA, data not shown). Western blotting of cell death signalling components at 4 h following SE revealed that inhibition of UCHL1 (prior to SE) was associated with increased expression of nuclear p53 (Kruskal–Wallis test, P = 0.0004; Dunn’s multiple comparisons test, P < 0.01 compared to non-SE control, Fig. 7f, g) and cleaved MDM2 (Mann–Whitney U, P = 0.0303, compared to vehicle SE, Fig. 7f, g). There was no observed difference in expression of either signal for vehicle treated mice following SE when compared to non-SE controls (data not shown). There was also no observed difference in Bax expression at 4 h following SE between vehicle and LDN treated mice (data not shown).

UCHL1 inhibition prior to SE is associated with increased hippocampal cell death. a Hippocampal neuropathology was assessed at rostral, medial and ventral stereological levels following SE. b Representative examples of neurodegeneration (FJB staining) in the ipsilateral CA3 of the hippocampus in vehicle and UCHL1 inhibitor-treated mice at 24 h. c Semi-quantitative analysis of FJB counts 24 h after SE or receiving sham saline injection, on cryosections from LDN- and vehicle-treated mice (n = 6–10, one-way ANOVA with Tukey’s post-hoc test). d Analysis of damage for each stereotaxic level, confirming strongest effect of LDN-7544 at ventral level following SE (n = 8–10, two-way ANOVA with Bonferroni post-hoc test). e TUNEL staining on ventral hippocampal sections confirming enhanced cell death in mice treated with UCHL1 inhibitor (n = 10, unpaired Student’s t test, two-tailed). f Semi-quantitative analysis of MDM2 and p53 levels detected by immunoblotting, on samples 4 h after SE or sham injection, in mice receiving LDN-57444 or vehicle (n = 4 to 6, Mann–Whitney U or Kruskal–Wallis test with Dunn’s multiple comparison test). g Representative images of MDM2 and p53 blots. Data expressed as mean ± SEM. *P < 0.05, **P < 0.01, ***P < 0.001. CA cornu ammonis, DG dentate gyrus, LDN LDN-57444, VEH vehicle
UCHL1 Depletion is Associated with a Downregulation of the lncRNA Antisense Uchl1
Next, we sought to determine if UCHL1 levels were altered following SE due to downregulation of mRNA levels. We employed RT-qPCR to assess Uchl1 transcript levels in microdissected subfields of the hippocampus following i.a. injection of KA. Optimal product amplification was confirmed via agarose DNA gel (Fig. 8b). RT-qPCR of reverse transcribed mRNA revealed that SE did not alter the levels of Uchl1 transcripts at 8 or 24 h following SE (Fig. 8d), suggesting that the observed downregulation of UCHL1 arises through post-transcriptional means. Given that Uchl1 translation is reported to be induced by an antisense long non-coding RNA, AsUchl1 [21], we next sought to determine if SE altered its levels in the hippocampus. Primers taken from the literature [21] and designed using Primer 3.0 software yielded three candidate primer pairs for amplifying AsUchl1 (see Fig. 8a), where optimal amplification was verified for a single pair, referred to as AsUchl1 SINEB2 primers (Fig. 8b). RT-qPCR of AsUchl1 in the CA3, CA1 and DG at 8 and 24 h following KA revealed a significant downregulation of AsUchl1 in the CA3 of the hippocampus at 8 h after KA (Mann–Whitney U, P = 0.0286, Fig. 8e, left). There was no other significant reduction in AsUchl1 following KA (8 or 24 h). AsUchl1 expression appeared to return to control levels after 24 h (Fig. 8e, right), while there was no observed downregulation in the contralateral CA3 at either timepoint (data not shown).

UCHL1 downregulation in the hippocampus is post-transcriptional and is preceded by depletion of AsUchl1. a Schematic depiction of the genetic landscape of Uchl1, in which a partially overlapping antisense non-coding RNA (AsUchl1) also resides. Points of primer pair binding are shown (arrows). The regulatory SINEB2 region is also highlighted. b DNA agarose gels were run to ensure successful, specific amplification with primer pairs targeting AsUchl1 and Uchl1. Primers targeting the SINEB2 region were used in subsequent investigations. c Hippocampal subfield microdissection was carried out. d, e RT-qPCR was carried out for each subfield at 8 h (left) and 24 h (right) for d Uchl1 mRNA following 1 µg KA and e AsUchl1 lncRNA following 0.3 µg KA (n = 4–6, Mann–Whitney U test, two-tailed). Data expressed as mean ± SEM, *P < 0.05. CA cornu ammonis, CON control, DG dentate gyrus, INJ injury
UCHL1 Protein Levels are Regulated by Antisense Uchl1 and mTOR In Vivo
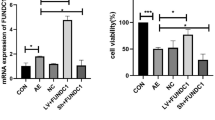
As downregulation of a pro-translational lncRNA targeting Uchl1 precedes UCHL1 downregulation following SE, we attempted to functionally implicate AsUchl1 in UCHL1 regulation in vivo. To this end, we employed hydrid RNA-LNA 16-mer oligonucleotides designed to bind AsUchl1 in sites overlapping the sense Uchl1 mRNA. We conceived an experimental setup involving two such anti-AsUchl1 oligonucleotides (termed ANTs) targeting the binding of AsUchl1 to 5′ Uchl1 as well as the sequence region encompassing the start codon of Uchl1 (Fig. 9a). Time-matched non-targeting scrambled sequences were used as experimental controls. I.c.v injection of 1 pmol, 10 pmol and 100 pmol doses in invivofectamine vehicle was carried out on adult mice and FJB histopathology of contralateral hippocampus was assessed at 24 h. FJB staining revealed that targeting AsUchl1 with LNA-RNA oligonucleotides is not potently cytotoxic, as no FJB-positive neurons were detected (Fig. 9b). Pilot studies (n = 2, data not shown) suggested that 10 pmol ANTs may be effacious in depleting UCHL1 expression in the hippocampus. Follow-up analysis by UCHL1 immunoblotting of hippocampal lysates 24 h after injection of 10pmol ANTs revealed a significant decrease in UCHL1 following targeting of AsUchl1 (Mann–Whitney U, P = 0.0286, compared to scrambled controls, Fig. 9c, d), suggesting a modest contribution of AsUchl1 to constitutive UCHL1 expression.

AsUchl1 positively regulates the expression of UCHL1 and may be induced by mTOR inhibition. a Synthetic oligonucleotides targeting the overlap region (red box) of AsUchl1 were employed to knockdown AsUchl1-mediated translation of Uchl1. b FJB staining of mice 24 h after receiving i.c.v. injection of anti-AsUchl1 or scrambled control (1–100 pmol) was carried out to assess potential RNA toxicity. c Immunoblotting was performed for UCHL1 expression 24 h after 10 pmol i.c.v. injection of anti-AsUchl1 or scrambled control (n = 4, Mann–Whitney U test, two-tailed). d Representative blots used for the semi-quantification of UCHL1 expression in c, n = 1 per lane, with observed molecular weights and experimental conditions annotated. e I.p. injection of rapamycin (1 mg/kg) was carried out in naive mice at 6 h prior to the sampling of hippocampal lysates. Western blotting was carried out for UCHL1 as above (n = 4, Mann–Whitney U test, two-tailed). f Representative blots used for the semi-quantification of UCHL1 expression in e, n = 1 per lane, with observed molecular weights and experimental conditions annotated. Data expressed as mean ± SEM. *P < 0.05. ANT anti-AsUchl1, RAPA rapamycin, SCR scrambled control, TUB tubulin
Previous reports suggested that AsUchl1-mediated translation of UCHL1 is a stress response signal following inhibition of mammalian target of rapamycin (mTOR) [21]. We investigated if rapamycin treatment might therefore boost UCHL1 expression in the hippocampus. When compared to vehicle control, UCHL1 expression was increased at 6 h following rapamycin (Mann–Whitney U, P = 0.0286) following 1 mg/kg i.p. injection.
Discussion
Models of epileptic tolerance and genome-wide profiling techniques are a powerful tool to identify genes and pathways that contribute to seizure-induced cell death and epileptogenesis [2, 5]. A proteomics screen here looking among proteins that were uniquely downregulated in injury mice but not in tolerance identified a number of ubiquitous regulatory proteins including UCHL1 that are strongly implicated in significantly enriched pathways. The present dataset includes other potentially interesting targets; here, we focused on UCHL1 as a demonstration of the validity of the approach. Investigating further, we confirmed that SE induces post-translational loss of UCHL1 through depletion of a lncRNA, and highlighted the contribution of UCHL1 to the regulation of excitability. Finally, we demonstrated that mTOR inhibition increases UCHL1 expression and proffer that the upregulation of UCHL1 is a hitherto unappreciated aspect of rapamycin efficacy.
The transcriptome of tolerance revealed that preconditioned animals display marked suppression of transcription in key functional groups governing excitability [8]. However, analysis of the miRNAome suggested that translation may be increased as a whole, as miRNA transcription was particularly suppressed in comparison to injury [9]. Here, we established for the first time the proteome of tolerance. As we did not specifically record EEG at this stage, we cannot exclude small differences in seizure severity or duration affecting results. However, no differences have been observed in any previous studies using this model [6, 7]. These data indicate rapid, active translation at 3 h after SE in preconditioned animals, with 77.5% of all changes in tolerance involving upregulation (totalling 69 proteins uniquely upregulated in tolerance). Upregulation remains dominant at 24 h after SE, though somewhat diminished, with 66.7% of changes involving upregulation. These data are suggestive of active neuroprotective mechanisms that are evoked during epileptic tolerance. However, transcriptional suppression of key pathways (as seen in the transcriptome) and maintenance of homeostatic processes (as seen here with UCHL1 expression) is demonstrative of distinct, coordinated responses that define epileptic tolerance. A similar principle seems evident with ischemic tolerance, where tolerance depends on protein synthesis, but is also associated with decreased transcription [39]. While ischemic tolerance may rely on the increased abundance of transcriptional repressors [13], our data highlight different strategies that may be involved in protection during epileptic tolerance. Regarding injury, SE was associated with dysregulation of a number of proteins previously implicated in proteomic profiling studies that may be fruitful in future investigations. These include those of the dihydropyrimidinase related protein family (DRP1, −2, −3 and −5 [40, 41]), dynamin-1 and -2 [42], synaptic proteins including various septins and T-complex proteins, α-internexin, NIPSNAP and SynGAP [43, 44], various heat-shock proteins [40, 42] and regulators of ubiquitin-conjugation such as UCHL1, UCHL5 and Ubiquitin-conjugating enzyme E2 [41, 42].
Here we show that SE is associated with a precipitous loss of UCHL1 in the hippocampus, while experimental depletion of UCHL1 leads to altered ubiquitin homeostasis and inhibition of the UPS. Importantly, UCHL1 inhibition leads to prolonged periods of synchronous discharge and increased seizure-induced cell death. UCHL1 is a dual DUB enzyme and ubiquitin ligase that has been implicated in Alzheimer’s disease, Parkinsonisms and neurodegeneration [30, 45–48], with functional roles in maintenance of synaptic structure, ubiquitin stabilisation and turnover, and regulation of proteasomal and lysosomal degradation [30, 36, 49–51]. Loss of UCHL1 expression is seen in retinal ischemia, where prior preconditioning rescues UCHL1 depletion [52]. Here, we observed a striking EEG phenotype when SE is preceded by UCHL1 inhibition, where animals receiving LDN underwent extended periods of high-amplitude, low-frequency activity uniquely responsible for seizure-induced cell death in this model, even after administration of the anticonvulsant lorazepam. It remains unclear whether UCHL1 inhibition directly impacts cell viability or whether LDN indirectly promotes cell death during SE via prolonged periods of high-amplitude discharge and inhibition of lorazepam efficacy. Nevertheless, our data point to destabilisation of ubiquitin homeostasis, alteration of key synaptic proteins, and dysregulation of the UPS as concomitant to the exacerbation of both SE and subsequent seizure-induced cell death, suggesting UCHL1 may be a key factor in the response to numerous insults, including SE and ischemia, where maintenance of expression is aligned with superior outcomes. Our data do not directly rule out a possible off-target effect of LDN on neurotransmission that may explain some of our observations, however. Though selective over other UCH isoforms [35], data is lacking regarding other potential neuronal targets of LDN, beside UCHL1. It is encouraging that the effect of LDN on synaptic morphology is rescued by exogenously applied ubiquitin [51], while the effect of LDN on contextual fear retention is mirrored by overexpression of a dominant-negative UCHL1 mutant [30], suggesting that the action of LDN is UCHL1-dependent, at least in these contexts.
The in vivo substrates of UCHL1 remain unclear. The primary function of UCHL1 is likely the deubiquitination of degraded peptide fragments, in tandem with stabilisation of unbound ubiquitin [49]. UCHL1 inhibition both in vivo [36] and in vitro [51] leads to a transient reduction in the level of monomeric ubiquitin by ~20% within 4 h. This is reflected in strains lacking functional UCHL1 expression [50, 53]. UCHL1 substrates may also include functionally mature proteins that are pertinent to epilepsy and SE, where ubiquitination regulates protein expression or localisation. Surface expression of certain transporter and signalling proteins, including NCAM [54], Glutamate Transporter GLT-1 [55] and Glycine Transporter GlyT2 [56], is regulated by UCHL1. Mechanistically, UCHL1 mediates rapid recycling of NCAM and GLT-1 within early endosomes through its DUB activity, reducing downstream degradation within the lysosome [54, 55]. Although ubiquitination status may depend on the ubiquitin pool (as regulated by UCHL1), these studies proffer that UCHL1 directly modulates ubiquitination of these proteins. For example, GLT-1 dynamics were not affected by UCHL3 inhibition [55], even though it too can regulate monomeric ubiquitin levels [56]. UCHL1 inhibition does not affect ubiquitination status or distribution of another glycine transporter, GlyT1 [55], and intracellular NCAM is colocalised with UCHL1 [54], suggesting that there are specific UCHL1 substrates in the brain.
Here, UCHL1 inhibition was coupled to an alteration in a key synaptic protein, PSD-95. It is likely that the effect of LDN on synaptic structures is specific to UCHL1 inhibition, as such changes are not observed following inhibition of UCHL3 [51]. Studies in vitro have noted that LDN treatment leads to an increase in the size of PSD-95-positive puncta alongside a decrease in puncta density [36, 51]. Although total PSD-95 immunoblot density is increased [51], at odds with our own data, neither study assessed PSD-95 levels from hippocampal lysates following LDN treatment. While total PSD-95 levels are increased in nm3419 mice lacking functional UCHL1 [51], these mice display redundant DUB activity through increased expression of Usp14, another DUB enzyme [50]. No previous report has assessed PSD-95 changes following UCHL1 inhibition in vivo in wildtype mice or addressed the ultrastructural effects of UCHL1 inhibition in vivo. LDN increased pathological synchronous activity during SE but did not induce cell death in itself, suggesting that LDN-induced loss of PSD-95 is associated with a change in network excitability without ablation of postsynaptic structures. This is reflected in PSD-95-null mice that display normal synaptic morphology but pronounced increases in long-term potentiation [57]. Redistribution of postsynaptic proteins including PSD-95 and NR2B-containing NMDA receptors has been observed following SE and contributes to excitotoxicity and epileptogenesis [58]. Therefore, PSD-95 depletion may contribute to the exacerbated EEG phenotype noted here.
UCHL1 inhibition was also observed to lead to UPS impairment. Accumulation of ubiquitinG76V-GFP appeared at 4 h after LDN administration. Data from other reports have been conflicting on this issue. Cartier et al. reported that in lysates derived from primary neuronal cultures, LDN treatment did not alter the cleavage of Suc-LLVY-AMC, a UPS substrate, and there was also no apparent change in the level of polyubiquitinated proteins [51]. However, that same study noted that LDN reduced clearance of exogenous ubiquitin-GFP [51]. Another study indirectly assessed UPS function in nm3419 mice (lacking functional UCHL1) and axj mice (lacking functional Usp14), asserting that UCHL1 contributes to lysosome-mediated degradation but not proteasomal degradation [50]. However, data here unequivocally demonstrate that acute disruption of UCHL1 function leads to an impairment in ubiquitin-dependent protein degradation in vivo. Redundant DUB activity by Usp14 in nm3419 mice may underlie these discrepancies [50]. The UPS is a vital component of synaptic plasticity mechanisms and dendritic structure, where activity-dependent alterations of UPS function and protein turnover represent a key regulatory mechanism in neuronal function [59, 60]. UPS function is altered in certain neurological disorders such as Parkinson’s, Alzheimer’s and Huntington’s [61, 62]. It is also transiently impaired following cerebral ischemia, with concomitant deposition of polyubiquitinated proteins [63, 64], and UPS-mediated structural remodeling of dendrites suppresses excitability arising after transient ischemia [14]. These data suggest that UCHL1 inhibition, through the depletion of ubiquitin turnover, leads to UPS impairment and alterations in the postsynaptic density. In this context, hypersynchronous activity arising from SE is exacerbated.
Finally, we suggest that SE-induced downregulation of UCHL1 is a post-transcriptional event. Uchl1 transcription was not altered at 8 or 24 h following SE in any hippocampal subfield. Post-transcriptional regulation of UCHL1 expression might be attained through translational induction by AsUchl1, a pro-translational lncRNA [21]. We confirm for the first time that AsUchl1 positively regulates the expression of UCHL1 in vivo and is downregulated following SE. Furthermore, as AsUchl1 export into the cytoplasm increases following mTOR inhibition [21], we confirmed that rapamycin, a potent mTOR inhibitor and anti-epileptogenic agent [20, 65], increased UCHL1 expression. MTOR is a master regulator of several cellular processes, with implications for cell death, excitability and epileptogenesis [65]. Hyperactivation of mTOR and deregulation of upstream signalling components can precipitate neurological disorders involving encephalopathy and epilepsy [66–68] and is epileptogenic in animal models [69, 70]. Rapamycin can attenuate processes associated with epileptogenesis, including hippocampal cell death, axonal sprouting, neurogenesis and spontaneous seizure onset [71]. Hyperactivation of mTOR following SE appears to favour complexation with raptor in the mTOR complex 1 (mTORC1), the primary effector of downstream perturbations to dendritic dysregulation and cognitive deficits [20]. Intriguingly, UCHL1 has been implicated as a regulator of the balance between mTORC1 and rictor-containing mTORC2, where overactivation of mTORC1 is particularly deleterious to cell survival [72]. The regulation of AsUchl1 localisation by mTOR coupled with the regulation of mTORC1:mTORC2 balance by UCHL1 suggests that UCHL1 is involved in an important regulatory loop of mTOR signalling. We propose that dysregulation of this loop, as supported here and elsewhere [20, 72], would favour overactivation of mTORC1 and suppression of UCHL1 following SE.
While our experimental approach lacks direct therapeutic relevance, we are confident that the data demonstrate that the levels of UCHL1 can influence injury following SE, and there are noteworthy clinical implications in considering UCHL1 function. Two independent groups have identified elevated CSF levels of UCHL1 following epilepsy-induced neuronal damage [73] and within 48 h of epileptic seizure [74]. UCHL1 is a highly abundant neuronal protein, and while its elevation in CSF is likely a result of widespread cell damage, our data suggest that the loss of UCHL1 expression may also precipitate injury. Our data also implicate rapamycin in the regulation of UCHL1. Rapamycin is remarkably effective in reducing seizures in patients suffering familial focal epilepsies, reducing seizure frequency in Pretzel Syndrome patients [75] and effectively controlling convulsions in two patient cohorts suffering Tuberous Sclerosis Complex [76, 77]. The therapeutic potential of UCHL1 overexpression has been demonstrated in other neurological conditions also, including Alzheimer’s disease [78] and other tauopathies [79].
In sum, these data demonstrate that proteomic analysis of SE is a powerful discovery tool for uncovering novel regulators of epilepsy pathophysiology. The main target pursued here, UCHL1, appears to protect against prolonged high-amplitude, low-frequency discharges and seizure-induced cell death. Further, we describe for the first time in vivo that UCHL1 translation is positively regulated by the lncRNA, AsUchl1. AsUchl1 is downregulated following SE (where Uchl1 transcription continues) and is activated by mTOR inhibition [18]. The dysregulation of mTOR after SE and during epilepsy is well documented; however, the regulatory loop with UCHL1 represents an underappreciated regulatory modality of mTOR signalling cascades and the possible role of UCHL1 upregulation in the efficacy of rapamycin may be one worth exploring.
References
Brodie MJ (2010) Antiepileptic drug therapy the story so far. Seizure 19:650–655
Pitkänen A, Lukasiuk K (2011) Mechanisms of epileptogenesis and potential treatment targets. Lancet Neurol 10:173–186
Simonato M, Löscher W, Cole AJ et al (2012) Finding a better drug for epilepsy: preclinical screening strategies and experimental trial design. Epilepsia 53:1860–1867
Dirnagl U, Simon RP, Hallenbeck JM (2003) Ischemic tolerance and endogenous neuroprotection. Trends Neurosci 26:248–254
Jimenez-Mateos EM, Henshall DC (2009) Seizure preconditioning and epileptic tolerance: models and mechanisms. Int J Physiol Pathophysiol Pharmacol 1:180–191
Hatazaki S, Bellver-Estelles C, Jimenez-Mateos EM et al (2007) Microarray profile of seizure damage-refractory hippocampal CA3 in a mouse model of epileptic preconditioning. Neuroscience 150:467–477
Tanaka K, Jimenez-Mateos EM, Matsushima S et al (2010) Hippocampal damage after intra-amygdala kainic acid-induced status epilepticus and seizure preconditioning-mediated neuroprotection in SJL mice. Epilepsy Res 88:151–161
Jimenez-Mateos EM, Hatazaki S, Johnson MB et al (2008) Hippocampal transcriptome after status epilepticus in mice rendered seizure damage-tolerant by epileptic preconditioning features suppressed calcium and neuronal excitability pathways. Neurobiol Dis 32:442–453
Jimenez-Mateos EM, Bray I, Sanz-Rodriguez A et al (2011) miRNA Expression profile after status epilepticus and hippocampal neuroprotection by targeting miR-132. Am J Pathol 179:2519–2532
Reynolds JP, Miller-Delaney SFC, Jimenez-Mateos EM et al (2015) Transcriptional response of polycomb group genes to status epilepticus in mice is modified by prior exposure to epileptic preconditioning. Front Neurol 6:46
Miller-Delaney SFC, Das S, Sano T et al (2012) Differential DNA methylation patterns define status epilepticus and epileptic tolerance. J Neurosci 32:1577–1588
McKiernan RC, Jimenez-Mateos EM, Sano T et al (2012) Expression profiling the microRNA response to epileptic preconditioning identifies miR-184 as a modulator of seizure-induced neuronal death. Exp Neurol 237:346–354
Stapels M, Piper C, Yang T et al (2010) Polycomb group proteins as epigenetic mediators of neuroprotection in ischemic tolerance. Sci Signal 3:ra15
Meller R, Thompson SJ, Lusardi TA et al (2008) Ubiquitin proteasome-mediated synaptic reorganization: a novel mechanism underlying rapid ischemic tolerance. J Neurosci 28:50–59
Liu J, Ye J, Zou X et al (2014) CRL4A(CRBN) E3 ubiquitin ligase restricts BK channel activity and prevents epileptogenesis. Nat Commun 5:3924
Paemka L, Mahajan VB, Ehaideb SN et al (2015) Seizures are regulated by ubiquitin-specific peptidase 9 X-linked (USP9X), a de-ubiquitinase. PLoS Genet 11:e1005022
Engel T, Martinez-Villarreal J, Henke C, et al (2017) Spatiotemporal progression of ubiquitin-proteasome system inhibition after status epilepticus suggests protective adaptation against hippocampal injury. Mol Neurodegener 12:21
Engel T, Hatazaki S, Tanaka K et al (2010) Deletion of Puma protects hippocampal neurons in a model of severe status epilepticus. Neuroscience 168:443–450
Lindsten K, Menéndez-Benito V, Masucci MG, Dantuma NP (2003) A transgenic mouse model of the ubiquitin/proteasome system. Nat Biotechnol 21:897–902
Brewster AL, Lugo JN, Patil VV et al (2013) Rapamycin reverses status epilepticus-induced memory deficits and dendritic damage. PLoS One 8:e57808
Carrieri C, Cimatti L, Biagioli M et al (2012) Long non-coding antisense RNA controls Uchl1 translation through an embedded SINEB2 repeat. Nature 491:454–457
Fic E, Kedracka-Krok S, Jankowska U et al (2010) Comparison of protein precipitation methods for various rat brain structures prior to proteomic analysis. Electrophoresis 31:3573–3579
Silva JC, Gorenstein MV, Li G-Z et al (2006) Absolute quantification of proteins by LCMSE: a virtue of parallel MS acquisition. Mol Cell Proteomics 5:144–156
Li G-Z, Vissers JPC, Silva JC et al (2009) Database searching and accounting of multiplexed precursor and product ion spectra from the data independent analysis of simple and complex peptide mixtures. Proteomics 9:1696–1719
Paxinos G, Franklin KBJ (2004) The mouse brain in stereotaxic coordinates. Elsevier Academic Press, San Diego
Beissbarth T, Speed TP (2004) GOstat: find statistically overrepresented Gene Ontologies within a group of genes. Bioinformatics 20:1464–1465
Huang DW, Sherman BT, Lempicki RA (2009) Systematic and integrative analysis of large gene lists using DAVID bioinformatics resources. Nat Protoc 4:44–57
Cline MS, Smoot M, Cerami E et al (2007) Integration of biological networks and gene expression data using Cytoscape. Nat Protoc 2:2366–2382
Merico D, Isserlin R, Stueker O et al (2010) Enrichment map: a network-based method for gene-set enrichment visualization and interpretation. PLoS One 5:e13984
Gong B, Cao Z, Zheng P et al (2006) Ubiquitin hydrolase Uch-L1 rescues beta-amyloid-induced decreases in synaptic function and contextual memory. Cell 126:775–788
Mani M, Lee SY, Lucast L et al (2007) The dual phosphatase activity of synaptojanin1 is required for both efficient synaptic vesicle endocytosis and reavailability at nerve terminals. Neuron 56:1004–1018
Xie Y, Vessey JP, Konecna A et al (2007) The GTP-binding protein Septin 7 is critical for dendrite branching and dendritic-spine morphology. Curr Biol 17:1746–1751
Kim IH, Racz B, Wang H et al (2013) Disruption of Arp2/3 results in asymmetric structural plasticity of dendritic spines and progressive synaptic and behavioral abnormalities. J Neurosci 33:6081–6092
Silva-Santos S, van Woerden GM, Bruinsma CF et al (2015) Ube3a reinstatement identifies distinct developmental windows in a murine Angelman syndrome model. J Clin Invest 125:2069–2076
Liu Y, Lashuel HA, Choi S et al (2003) Discovery of inhibitors that elucidate the role of UCH-L1 activity in the H1299 lung cancer cell line. Chem Biol 10:837–846
Cartier AE, Ubhi K, Spencer B et al (2012) Differential effects of UCHL1 modulation on alpha-synuclein in PD-like models of alpha-synucleinopathy. PLoS One 7:e34713
Hamaue N, Minami M, Terado M et al (2004) Comparative study of the effects of isatin, an endogenous MAO-inhibitor, and selegiline on bradykinesia and dopamine levels in a rat model of Parkinson’s disease induced by the Japanese encephalitis virus. Neurotoxicology 25:205–213
Tan Y-Y, Zhou H-Y, Wang Z-Q, Chen S-D (2008) Endoplasmic reticulum stress contributes to the cell death induced by UCH-L1 inhibitor. Mol Cell Biochem 318:109–115
Stenzel-Poore MP, Stevens SL, Xiong Z et al (2003) Effect of ischaemic preconditioning on genomic response to cerebral ischaemia: similarity to neuroprotective strategies in hibernation and hypoxia-tolerant states. Lancet 362:1028–1037
Greene NDE, Bamidele A, Choy M et al (2007) Proteome changes associated with hippocampal MRI abnormalities in the lithium pilocarpine-induced model of convulsive status epilepticus. Proteomics 7:1336–1344
Rohe M, Nebrich G, Klein O et al (2011) Kainate promotes alterations in neuronal RNA splicing machinery. J Proteome Res 10:1459–1467
Liu X-Y, Yang J-L, Chen L-J et al (2008) Comparative proteomics and correlated signaling network of rat hippocampus in the pilocarpine model of temporal lobe epilepsy. Proteomics 8:582–603
Krapfenbauer K, Berger M, Lubec G, Fountoulakis M (2001) Changes in the brain protein levels following administration of kainic acid. Electrophoresis 22:2086–2091
Satoh K, Takeuchi M, Oda Y et al (2002) Identification of activity-regulated proteins in the postsynaptic density fraction. Genes Cells 7:187–197
Setsuie R, Wada K (2007) The functions of UCH-L1 and its relation to neurodegenerative diseases. Neurochem Int 51:105–111
Yasuda T, Nihira T, Ren Y-R et al (2009) Effects of UCH-L1 on alpha-synuclein over-expression mouse model of Parkinson’s disease. J Neurochem 108:932–944
Bilguvar K, Tyagi NK, Ozkara C et al (2013) Recessive loss of function of the neuronal ubiquitin hydrolase UCHL1 leads to early-onset progressive neurodegeneration. Proc Natl Acad Sci USA 110:3489–3494
Coulombe J, Gamage P, Gray MT, et al (2014) Loss of UCHL1 promotes age-related degenerative changes in the enteric nervous system. Front Aging Neurosci 6:129
Osaka H, Wang Y-L, Takada K et al (2003) Ubiquitin carboxy-terminal hydrolase L1 binds to and stabilizes monoubiquitin in neuron. Hum Mol Genet 12:1945–1958
Walters BJ, Campbell SL, Chen PC et al (2008) Differential effects of Usp14 and Uch-L1 on the ubiquitin proteasome system and synaptic activity. Mol Cell Neurosci 39:539–548
Cartier AE, Djakovic SN, Salehi A et al (2009) Regulation of synaptic structure by ubiquitin C-terminal hydrolase L1. J Neurosci 29:7857–7868
Stowell C, Wang L, Arbogast B et al (2010) Retinal proteomic changes under different ischemic conditions—implication of an epigenetic regulatory mechanism. Int J Physiol Pathophysiol Pharmacol 2:148–160
Saigoh K, Wang YL, Suh JG et al (1999) Intragenic deletion in the gene encoding ubiquitin carboxy-terminal hydrolase in gad mice. Nat Genet 23:47–51
Wobst H, Förster S, Laurini C et al (2012) UCHL1 regulates ubiquitination and recycling of the neural cell adhesion molecule NCAM. FEBS J 279:4398–4409
Martínez-Villarreal J, García Tardón N, Ibáñez I et al (2012) Cell surface turnover of the glutamate transporter GLT-1 is mediated by ubiquitination/deubiquitination. Glia 60:1356–1365
de Juan-Sanz J, Núñez E, López-Corcuera B, Aragón C (2013) Constitutive endocytosis and turnover of the neuronal glycine transporter GlyT2 is dependent on ubiquitination of a C-terminal lysine cluster. PLoS One 8:e58863
Migaud M, Charlesworth P, Dempster M et al (1998) Enhanced long-term potentiation and impaired learning in mice with mutant postsynaptic density-95 protein. Nature 396:433–439
Frasca A, Aalbers M, Frigerio F et al (2011) Misplaced NMDA receptors in epileptogenesis contribute to excitotoxicity. Neurobiol Dis 43:507–515
Tai H-C, Schuman EM (2008) Ubiquitin, the proteasome and protein degradation in neuronal function and dysfunction. Nat Rev Neurosci 9:826–838
Jiang X, Litkowski PE, Taylor AA et al (2010) A role for the ubiquitin-proteasome system in activity-dependent presynaptic silencing. J Neurosci 30:1798–1809
Rubinsztein DC (2006) The roles of intracellular protein-degradation pathways in neurodegeneration. Nature 443:780–786
Ortega Z, Díaz-Hernández M, Maynard CJ et al (2010) Acute polyglutamine expression in inducible mouse model unravels ubiquitin/proteasome system impairment and permanent recovery attributable to aggregate formation. J Neurosci 30:3675–3688
Magnusson K, Wieloch T (1989) Impairment of protein ubiquitination may cause delayed neuronal death. Neurosci Lett 96:264–270
Asai A, Tanahashi N, Qiu J-H et al (2002) Selective proteasomal dysfunction in the hippocampal CA1 region after transient forebrain ischemia. J Cereb Blood Flow Metab 22:705–710
Wong M (2010) Mammalian target of rapamycin (mTOR) inhibition as a potential antiepileptogenic therapy: From tuberous sclerosis to common acquired epilepsies. Epilepsia 51:27–36
Kwon C-H, Zhu X, Zhang J, Baker SJ (2003) mTor is required for hypertrophy of Pten-deficient neuronal soma in vivo. Proc Natl Acad Sci USA 100:12923–12928
Meikle L, Pollizzi K, Egnor A et al (2008) Response of a neuronal model of tuberous sclerosis to mammalian target of rapamycin (mTOR) inhibitors: effects on mTORC1 and Akt signaling lead to improved survival and function. J Neurosci 28:5422–5432
Zhou J, Blundell J, Ogawa S et al (2009) Pharmacological inhibition of mTORC1 suppresses anatomical, cellular, and behavioral abnormalities in neural-specific Pten knock-out mice. J Neurosci 29:1773–1783
Buckmaster PS, Ingram EA, Wen X (2009) Inhibition of the mammalian target of rapamycin signaling pathway suppresses dentate granule cell axon sprouting in a rodent model of temporal lobe epilepsy. J Neurosci 29:8259–8269
Zeng L-H, Rensing NR, Wong M (2009) The mammalian target of rapamycin signaling pathway mediates epileptogenesis in a model of temporal lobe epilepsy. J Neurosci 29:6964–6972
Zeng L-H, McDaniel S, Rensing NR, Wong M (2010) Regulation of cell death and epileptogenesis by the mammalian target of rapamycin (mTOR): a double-edged sword? Cell Cycle 9:2281–2285
Hussain S, Feldman AL, Das C et al (2013) Ubiquitin hydrolase UCH-L1 destabilizes mTOR complex 1 by antagonizing DDB1-CUL4-mediated ubiquitination of raptor. Mol Cell Biol 33:1188–1197
Li Y, Wang Z, Zhang B et al (2013) Cerebrospinal fluid ubiquitin C-terminal hydrolase as a novel marker of neuronal damage after epileptic seizure. Epilepsy Res 103:205–210
Mondello S, Palmio J, Streeter J et al (2012) Ubiquitin carboxy-terminal hydrolase L1 (UCH-L1) is increased in cerebrospinal fluid and plasma of patients after epileptic seizure. BMC Neurol 12:85
Parker WE, Orlova KA, Parker WH et al (2013) Rapamycin prevents seizures after depletion of STRADA in a rare neurodevelopmental disorder. Sci Transl Med 5:182ra53
Krueger DA, Wilfong AA, Holland-Bouley K et al (2013) Everolimus treatment of refractory epilepsy in tuberous sclerosis complex. Ann Neurol 74:679–687
Canpolat M, Per H, Gumus H et al (2014) Rapamycin has a beneficial effect on controlling epilepsy in children with tuberous sclerosis complex: results of 7 children from a cohort of 86. Childs Nerv Syst 30:227–240
Zhang M, Cai F, Zhang S, et al (2014) Overexpression of ubiquitin carboxyl-terminal hydrolase L1 (UCHL1) delays Alzheimer’s progression in vivo. Sci Rep 4:7298
Xie M, Han Y, Yu Q et al (2016) UCH-L1 inhibition decreases the microtubule-binding function of tau protein. J Alzheimers Dis 49:353–363
Acknowledgements
Breeding pairs of mice constitutively expressing green fluorescent protein with an N-terminal ubiquitin (ubiquitinG76V−GFP) were generously gifted by J. J. Lucas, Universidad Autónoma de Madrid, Spain. This colony of mice was maintained by Tobias Engel and Amaya Sanz-Rodriguez.
Funding sources
This study was supported by funding from the Health Research Board Ireland (PHD/2007/11, HRA_POR/2011/41), Science Foundation Ireland (08/IN.1./B1875), the NIH National Institute for Neurological Disorders and Stroke (U54 NS060659) and an endowment from the Morehouse School of Medicine (to AZ).
Author Contributions
JR, EJM, AZ and DH designed research; JR, EJM, MA and LC performed research; JR, EJM, LC, FB and SMD analysed data; JR, AZ and DH wrote the paper.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
The authors declare no competing financial interests.
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
About this article

Cite this article
Reynolds, J.P., Jimenez-Mateos, E.M., Cao, L. et al. Proteomic Analysis After Status Epilepticus Identifies UCHL1 as Protective Against Hippocampal Injury. Neurochem Res 42, 2033–2054 (2017). https://doi.org/10.1007/s11064-017-2260-6
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s11064-017-2260-6