
Abstract
IGFBP6, a member of the insulin-like growth factor-binding proteins family that contains six high affinity IGFBPs, modulates insulin-like growth factor (IGF) activity and also showed an independent effect of IGF, such as growth inhibition and apoptosis. However, the role of IGFBP6 in spinal cord injury (SCI) remains largely elusive. In this study, we have performed an acute SCI model in adult rats and investigated the dynamic changes of IGFBP6 expression in the spinal cord. Our results showed that IGFBP6 was upregulated significantly after SCI, which was paralleled with the levels of apoptotic proteins p53 and active caspase-3. Immunofluorescent labeling showed that IGFBP6 was co-localizated with active caspase-3 and p53 in neurons. To further investigate the function of IGFBP6, an apoptosis model was established in primary neuronal cells. When IGFBP6 was knocked down by specific short interfering RNA (siRNA), the protein levels of active caspase-3 and Bax as well as the number of apoptotic primary neurons were significantly decreased in our study. Taken together, our findings suggest that the change of IGFBP6 protein expression plays a key role in neuronal apoptosis after SCI.
Similar content being viewed by others
Avoid common mistakes on your manuscript.
Introduction
Traumatic spinal cord injury (SCI) is one of the foremost reason for neurological disability in the world, with an annual prevalence of 15–52.5 cases per million [1–3]. SCI is caused by direct or indirect trauma and includes two main pathological stages: primary injury and secondary injury [4–6]. The primary injury initiated a cascade of biochemical and cellular processes [7], including electrolyte imbalance, edema, excitotoxicity, and endoplasmic reticulum (ER) stress, leading to the secondary injury and resulting in neuronal apoptosis [8–10]. The apoptosis of neurons may mediate a series of subsequent neurological dysfunction following spinal cord injury [11, 12]. However, the signaling regulatory mechanisms that lead to neuronal apoptosis after SCI are still unknown and need further exploration.
IGFBP6, a member of the insulin-like growth factor-binding proteins family that contains six high affinity IGFBPs, modulates insulin-like growth factor (IGF) activity and also showed an independent effect of IGF [13]. IGFBP6 is characteristic of a 50-fold binding preference for IGF-II over IGF-I [14]. It can inhibit IGF-II availability in vitro and in vivo by preventing IGF from binding to cell surface receptors [15]. Studies suggest that IGFBP6 is a tumor suppressor that inhibits the growth of a number of IGF-II-dependent tumors. IGFBP6 increased apoptosis of non-small-cell lung cancer cells [16]. Constitutive up-regulation of IGFBP6 inhibited neuroblastoma xenograft growth in vivo, attributing to increased apoptosis [17]. Although IGFBP6’s major action appears to be inhibiting IGF-II activity, a number of studies suggest that it may also have IGF-independent actions, such as growth inhibition and apoptosis [15]. As a tumour suppressor in nasopharyngeal cancer, nuclear localization of IGFBP6 is dependent on a C-terminal domain NLS via importin-α and may play a role in rhabdomyosarcoma (RMS) cells cell survival [18]. Previous studies have demonstrated the important role of IGFBP6 in cell apoptosis, However, whether IGFBP6 could affect the apoptosis of neurons after SCI or not remains ambiguous.
In this study, we investigated temporal-spatial patterns of IGFBP6 at protein level and its colocalization with active caspase-3 and p53 using an acute SCI model on adult rats. In vitro, we studied the role of IGFBP6 in neuronal apoptosis by using the IGFBP6-specific siRNA. Our experiments were conducted to gain an insight into the functions of IGFBP6 and its potential role in neuronal apoptosis after SCI.
Materials and Methods
Animals, Surgery and Locomotor Function
Male adult Sprague-Dawley (SD) rats weighing from 250 to 300 g were used in this study. Dorsal laminectomies were carried out under anesthetsia with Ketamine (90 mg/kg)/xylazine (10 mg/kg) at the level of the ninth thoracic vertebra (T9) and surgery was performed under aseptic conditions. Ketoprofen (5 mg/kg) was administered to minimize postsurgical discomfort and pain. Trauma injuries (n = 54) were performed by the NYU impactor [19]. The exposed spinal cord of rat was contused by dropping a rod 2.0 mm in diameter and 10 g in weight from a height of 75 mm. The overlying muscles and skin were, respectively, closed in layers after injury with 4–0 silk sutures and staples, and the rats were placed on a 30 °C heating pad to recover. To prevent urinary tract infection, post-operative treatments included saline (2.0 cc, s.c.) for rehydration and Baytril (0.3 cc, 22.7 mg/ml, s.c., twice daily). Bladders were manually expressed twice daily until reflex bladder emptying returned. With free access to food and water, rats were housed under a 12 h (h) light/dark cycle in apathogen-free area. The spontaneous recovery of locomotor function after SCI was examined using the Basso, Beattie and Bresnahan (BBB) rating scale [20]. Animals were sacrificed at 6, 12 h, 1, 3, 5, 7 and 14 days after injury. Sham-operated animals (n = 12) served as sham controls. They were deeply anesthetized with chloral hydrate described above and surgery was performed under aseptic conditions. Dorsal laminectomies were carried out at T9, while contusion injuries were not performed. Surgical interventions and post-operative rats care were performed in accordance with the Guide for the Care and Use of Laboratory Animals (National Research Council, 1996, USA) and were approved by the Chinese National Committee to the Use of Experimental Animals for Medical Purposes, Jiangsu Branch. All efforts were made to minimize suffering and the number of rats.
Western Blot Analysis
The sham or injured spinal cords were excised to obtain samples for Western blot analysis. The portion of the spinal cord extending 5 mm rostral and 5 mm caudal to the injury epicenter was dissected out and immediately frozen at −80 °C until use. Frozen spinal cord samples were minced with eye scissors on ice to prepare lysates. The samples were homogenized in a lysis buffer (1% sodium dodecyl sulfate (SDS), 1% Triton X-100, 50 mmol/L Tris, 1% NP-40, pH 7.5, 5 mmol/L EDTA, 1% sodium deoxycholate, 1 μg/ml leupeptin, 10 μg/ml aprotinin, and 1 mmol/LPMSF), centrifuged at 12,000×g and 4 °C for 20 min to collect the supernatant and stored at −80 °C until use. Cell cultures for Western blot were lysed with sodium lauryl sulfate loading buffer and stored at −80 °C until use. After determining the protein concentration with the Bradford assay (Bio-Rad), protein samples were subjected to SDS–polyacrylamide gel electrophoresis (SDS-PAGE) and transferred to a polyvinylidine diflouride filter (PVDF) membrane (Millipore, Bedford, MA, USA). The membranes were blocked with 5% skim milk for 2 h at room temperature and then incubated overnight at 4 °C with antibodies for active caspase-3 and Bax (1:200; Cell Signaling, MA, USA), IGFBP6 (1:500), p53 (1:800; Santa Cruz, CA, USA), LaminB, Tubulin (1:200;Cell Signaling). Immunoreactive bands were detected by incubation with horseradish peroxidase (HRP)-conjugated anti-rabbit IgG (1:2000; Southern-Biotech, Alabama, USA) for 2 h and then visualized using an enhanced chemiluminescence system (ECL, Pierce Company, Woburn, MA, USA). For each experiment, the gels were run in triplicate. The western blot bands intensity were measured by Image J.
Reverse Transcription-Polymerase Chain Reaction (RT-PCR)
Total RNA was isolated from the frozen specimens at different survival times using Trizol Reagent (Life Technologies, Rockville, MD, USA). Complementary DNA (cDNA) was synthesized from total RNA with Revert- Aid™ First Strand cDNA Synthesis Kit (Fermentas Life Sciences) according to the manufacturer’s protocol. A specific cDNA fragment was amplified by Tag DNA polymerase (Life Technologies) using specific primers for IGFBP6 (sense 5′-ATCCGCCCAAGGACGACGAG-3′, antisense 5′-TGCCTGGATTCCTCTGTTGGTC-3′) and GAPDH (sense 5′-ACCACAGTCCATGCCATCAC-3′, antisense 5′-TCCACCACCCTGTTGCTGTA-3′). Each reaction (25 μl) contained 2 μl of cDNA, 2.5 μl of 10 × PCR buffer, 2 mM MgCl2, 0.2 mM dNTPs, 1 μl of each primer(10 μM), and 2.5 U of Taq DNA polymerase. Amplification protocol was: initial denaturation 3 min (94 °C); repeated cycles of denaturation 30 s (94 °C), annealing 45 s (58 °C for IGFBP6 and 57 °C for GAPDH), extension 45 s (72 °C); and final extension 7 min (72 °C). Number of cycles was selected from the exponential range for each primer pair: 25 (GAPDH) and 28 (IGFBP6). PCR products were electrophoresed through 2% agarose gel containing ethidium bromide (0.5 μg/ml). Gels were visualized under UV light, photographed and optical densities of the bands were analyzed with molecular dynamics densitometer (Scion).
Immunohistochemistry
At the designated time points, the normal and injured rats were terminally anesthetized and perfused through the ascending aorta with physiological saline followed by 4% paraformaldehyde. After perfusion, for immunohistochemistry staining, the sham and injured spinal cords were removed and post-fixed in the same fixative for 12 h and then replaced with 20% sucrose for 1 day, followed with 30% sucrose for 2–3 days. After treatment with sucrose solutions, the tissues were embedded in O. C. T. compound. Then, 7-μm frozen cross-sections at two spinal cord levels (2 mm rostral and caudal to the epicenter of injury) were prepared and examined. All of the sections were blocked with using 10% donkey serum with 0.3% Triton-X100 and 1% bovine serum albumin (BSA) for 2 h at room temperature (RT). After blocking, the membranes were incubated overnight at 4 °C with an anti-IGFBP6 antibody (1:200; Santa Cruz, CA, USA), Phosphate Buffered Saline (PBS, 0.1 M) instead of primary antibody for negative control, followed by incubation by incubation in biotinylated secondary antibody (Vector Laboratories, Burlingame, CA), followed by incubating with a biotinylated secondary antibody. Staining was visualized using DAB. The stained sections were examined using a Leica light microscope (Germany). The results of immunohistochemistry were measured by Image J.
Double Immunofluorescent Staining
After air-dried for 1 h at RT, sections were blocked with 10% normal serum blocking solution-species the same as the secondary antibody, containing 3% (w/v) BSA and 0.1% Triton-X100 and 0.05% Tween-20 2 h at RT to avoid unspecific staining. Then, the sections were incubated with the primary antibodies for IGFBP6 (1:100; Santa Cruz, CA, USA), p53, active caspase-3 (1:100; Cell Signaling, MA, USA) or different phenotype-specific markers as follows: NeuN (anti-rabbit or anti-mouse, neuron marker, 1:600; Chemicon), CD11b (anti-rabbit or anti-mouse, microglia marker, 1:200; Sigma) and GFAP (anti-rabbit or anti-mouse, astrocytes marker, 1:200; Sigma).PBS (0.1 M) was used as negative control instead of primary antibody. Sections were incubated with the primary antibodies or PBS (0.1 M) overnight at 4 °C, followed by a mixture of the FITC- and TRITC-conjugated secondary antibodies for 2 h at 4 °C. The stained sections were examined with Leica fluorescence microscope (Leica, DM 5000B, Leica CTR 5000, Germany). Cells were fixed at 4 °C for 20 min with ice-cold PBS (0.1 M) that contained 4% formaldehyde, permeabilized with 0.1% Triton X-100 for 10 min, and then blocked by 1% BSA for 2 h. After washing in PBS, the cells were incubated for 1 h with rabbit primary antibodies anti-IGFBP6 (1:100; Santa Cruz, CA, USA), anti-p53 (1:100; Cell Signaling, MA, USA) and goat polyclonal antibody anti-active caspase-3 (1:100; Cell Signaling, MA, USA) antibody. Then the cells were washed with PBS five times and incubated with FITC- and TRITC-conjugated secondary antibodies for 30 min at 4 °C. Following covered with DAPI (0.1 mg/ml in PBS; blue, Sigma, MO, USA) for 30 min at 30 °C, the cells were washed with PBS (0.1 M) and reversed on glass slides with glycerol and PBS (1:1), then examined under a Leica fluorescence microscope (Leica, DM 5000B, Leica CTR 5000, Germany).
Rat Primary Neurons
Primary neuronal cultures were prepared from the spinal cord of 1-day-old Sprague-Dawley rat pups, and the tissues were gently minced by forceps and then dissociated by incubating at 37 °C for 30 min, with occasional mixing, in 10 ml of phosphate-buffered saline. After centrifugation at 1200 rpm for 5 min, the tissues were removed in 5 ml DMEM containing 10% FBS and gently aspirated several times to inactivate the trypsin. The procedures were carried out similarly to those previously described. After the medium had been decanted, the cells were resuspended in a Neurobasal medium (Gibco) supplemented with B27 (Gibco) containing 0.5 mM glutamine. Cells were plated at a density of 4 × 104 cells/ml on poly-L-lysine-coated 6-well culture plates. In order to study apoptosis, cells were incubated in a low concentration of serum (1% horse serum) for 24 h prior to treatment with 300 nmol/L H2O2 for different times.
Nuclear and Cytoplasmic Extraction
Cell pellets from a culture were incubated in a hypotonic buffer (10 mM HEPES pH 7.2, 10 mM KCl, 1.5 mM MgCl2, 0.1 mM EGTA, 20 mM NaF, 100 μM Na3VO4, and protease inhibitor mixture) for 30 min at 4 °C on a rocking platform. Cells were homogenized (Dounce, 30 strokes), and their nuclei were pelleted by centrifugation (10 min × 14,000×g, 4 °C). The supernatant was saved as the cytosolic fraction, and nuclear pellets were incubated in nuclear lysis buffer (10 mM Tris–HCl pH 7.5, 150 mM NaCl, 5 mM EDTA, and 1% Triton X-100) for 1 h at 4 °C on a rocking platform. The nuclear fraction was collected by centrifugation (10 min × 14,000×g, 4 °C). Prior to immunoblotting, cells were washed twice with ice-cold PBS (0.1 M), resuspensed in 2 × lysis buffer (50 mM Tris–HCl, 120 mM NaCl, 0.5% Nonidet P-40, 100 mM NaF, 200 μM Na3VO4, and protease inhibitor mixture), and incubated for 20 min at 4 °C while rocking. Lysates were cleared by centrifugation (10 min × 12,000×g, 4 °C) and 50 μg total proteins was resolved by SDS-PAGE and transferred on to a poly (vinylidene difluoride) membrane filter (Immbilon; Millipore). The membranes were firstly blocked and then incubated with the primary antibody described above for 2 h at room temperature. After washing three times, filters were incubated with horseradish peroxidaseconjugated human anti-mouse or anti-rabbit antibodies (Pierce) for 1 h at room temperature. Immuno-complexes were detected with an enhanced chemiluminescence system (NEN Life Science Products, Boston, MA, USA).
Annexin-V/PI Analysis of Primary Neurons Apoptosis
After the indicated treatment, primary neurons were washed by PBS for twice and centrifuged at 1000 rpm for 5 min to collect cells. Then 100 μl 1 × Annexin V Binding Buffer was used to resuspend the cells and add Annexin V-fluorescein isothiocyanate (AV) and PI in the dark. Finally, the solution was incubated for 20 min and added another 400 μl 1 × Annexin V Binding Buffer before test. The apoptosis assay was performed by Annexin-V-FLUOS staining kit (Roche Diagnostics, Basel, Switzerland) obeying the manufacturer’s instructions.
siRNA and Transfection
Small interfering RNA for IGFBP6 was obtained from Santa Cruz Biotechnology. The sequences for the three siRNAs are as follows: IGFBP6-siRNA#1: 5′-CCGCUGUUGAUGCUGUUAATT-3′,5′-UUAACAGCAUCAACAGCGGTT-3′.IGFBP6-siRNA#2:5′-GGAAGAGACUACCAAGGAGTT-3′,5′-CUCCUUGGUAGUCUCUUCCTT-3′.IGFBP6-siRNA#3:5′-GCUCUAUGUGCCAAACUGUTT-3′,5′-ACAGUUUGGCACAUAGAGCTT-3′.Transfections with siRNA were performed using lipofectamine 2000 (Invitrogen) in accordance with the manufacturer’s protocol. For transient transfection, the IGFBP6 siRNA vector and the non-specific vector were carried out using lipofectamine 2000 and plus reagent in OptiMEM (Invitrogen). Transfected cells were used for the subsequent experiments 36 h after transfection. The siRNA transfection efficiency was 76%.
Quantitative and Statistical Analysis
The numbers of IGFBP6-positive cells in the spinal cord 2 mm rostral to the epicenter were counted in a 500 × 500 μm measuring frame. For each animal, a measure was taken in a section through the dorsal horn, the lateral funiculus and the ventral horn. To avoid counting the same cell in more than one section, we counted every fifth section (50 μm apart). The cell counts were then used to determine the total number of IGFBP6-positive cells per square millimeter. Cells double labeled for IGFBP6 and the other phenotypic-specific markers used in the experiment were also quantified in each section. All data were analyzed with Stata7.0 statistical software. All values were expressed as the mean ± SEM. One-way ANOVA followed by the Tukey’s post hoc multiple comparison tests and un-paired t-test for double comparison were used for statistical analysis. P-values less than 0.05 were considered statistically significant.
Results
Behavioral Changes After Traumatic Spinal Cord Injury
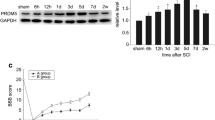
To observe behavioral changes following spinal cord injury, spontaneous recovery of locomotor function for rats were tested by using the BBB rating scale [20]. After SCI, all animals lacked hind-limb locomotion. Spontaneous improvement in function occurred from 12 h after SCI (Fig. 1).

Time course and degree of functional recovery in rats after SCI. Rats (n = 3) were killed at each time points (6, 12 h, 1, 3, 5, 7, and 14 days post-injury). The data are reported as mean ± values of open-field locomotion BBB scores
The Expression Profiles of IGFBP6 Following Spinal Cord Injury
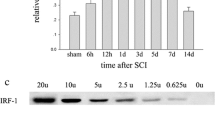
RT-PCR analysis was performed to show the expression of IGFBP6 at mRNA level after SCI. IGFBP6 mRNA had a progressive increase which peaked after 5 day of injury and then gradually decreased (Fig. 2a).Western blot was performed to investigate IGFBP6 expression after spinal cord contusion injury. IGFBP6 protein level was low in sham group, significantly increased beginning 12 h after injury, and reaching a peak at 5 days (P < 0.05). Then, IGFBP6 expression gradually decreased in the next 7 days (Fig. 2b). These results suggested that IGFBP6 protein might be up-regulated by injury. Moreover, IGFBP6 mRNA level was consistent with changes in its protein expression, indicating that IGFBP6 may be regulated at a transcriptional level.

The expression changes of IGFBP6 in adult rat spinal cord following contusion injury. a RT-PCR was performed to show the mRNA level of IGFBP6 in the spinal cord tissues at different survival times after SCI. IGFBP6 mRNA level was low in the sham-operated spinal cord, peaked at 5 days, and then gradually reduced thereafter. b Western blotting was performed to assess the protein level of IGFBP6 in the spinal cord tissues at different survival times after SCI. The IGFBP6 protein had a progressive increase which peaked after 5 day of injury and declined back to baseline level thereafter. The bar chart below demonstrates the ratio of IGFBP6 relative to GAPDH expression for each time point. The data are means ± SEM (n = 3, *P < 0.05, significantly different from the sham group)
IGFBP6 Expression and Distribution in Rat Spinal Cord
To identify the distribution of IGFBP6 after SCI, Immunohistochemistry with anti-IGFBP6 polyclonal antibody was performed on transverse cryosections of the spinal cord. IGFBP6 was widely expressed both in the gray and white matter (Fig. 3a, b), regardless of whether the spinal cord were injured or sham. Notably, IGFBP6 was expressed in the cytoplasm and nuclear, and immunostaining of IGFBP6 was mainly increased in neurons after SCI (Fig. 3c–f). In addition, we can found that there are an significant difference of the number of IGFBP6-positive cells between sham-operated spinal cords and day 5 in dorsal top rather than in ventral bottom after injury(P < 0.05), which was consistent with our western blot results (Fig. 3g).

The distribution changes of IGFBP6 in adult rat spinal cord. The distribution of IGFBP6 in adult rat spinal cord, 2 mm rostral to injury epicenter, was examined by immunohistochemistry. Low-power views of cross-sections immunostained with antibody specific for IGFBP6 in sham spinal cord (a) and day 5 after injury (b). Higher-power views in the gray matter (c and d) and white matter (e and f) of the sham and injured spinal cord. Scale bars 200 μm (a and b) and 20 μm (c–f). g Quantitative analysis of IGFBP6-positive cells/mm2 between sham and day 5 after SCI. Asterisk indicates significant difference at P < 0.05 compared with sham spinal cord. Error bars represent SEM
The Colocalization of IGFBP6 with Different Phenotype-Specific Markers by Double Immunofluorescent Staining
To further address the expression of IGFBP6 in the spinal cord, we performed double immunofluorescent microscopy studies intransverse cryosections of spinal cord tissues within 2 mm distance from the lesion site by colabeling with NeuN(neuronal marker), GFAP(astrocytic marker) and CD11b(microglial marker). As shown in Fig. 4, IGFBP6 expression was increased more significantly in neurons at 5 day (Fig. 4a–f, P < 0.05) after SCI compared with the sham spinal cord rather than astrocytes (Fig. 4g–i) and microglia (Fig. 4m–r).

Double immunofluorescence staining for IGFBP6 and different phenotype-specific markers in the spinal cord. In the adult rat spinal cord 2 mm to epicenter at day 5 after SCI, horizontal sections were labeled with IGFBP6 (red) and different phenotype-specific markers (green), such as NeuN, GFAP and CD11b. The colocalization of IGFBP6 with different phenotype-specific markers (yellow) is shown in (c, f, i, l, o and r). Scale bars 20 μm (a–r). Quantitative analysis of different phenotype-specific markers positive cells expressing IGFBP6 (%) in sham spinal cord and at 5 day after SCI. Changes of IGFBP6 expression after SCI were increased in neurons (n = 3 in sham group, n = 3 in injured group; asterisk indicates significant difference at P < 0.05 compared with sham spinal cord) (s). Error bars represent SEM. (Color figure online)
Detection of Neuronal Apoptosis and Relative IGFBP6 Changes in the Adult Rat Spinal Cord After SCI
To verify the relationship between IGFBP6 and apoptotic cells, western blot analyses were performed to examine the expression of several apoptosis-related proteins, such as active caspase-3, p53 in the process of SCI. Active caspase-3 expression was significantly increased at 12 h, reaching a peak at 5 day (P < 0.05) after SCI, then gradually reducing to basal levels at 2 weeks. The similar results were found in the expression of p53 and active caspase-3, which were positively correlated with the protein level of IGFBP6 (Fig. 5a, b). All these data above indicated that the changes of IGFBP6 expression might be associated with neuronal apoptosis. To confirm the distribution and colocalization of IGFBP6, p53 and active caspase-3 in the injured spinal cords at 5 day post-injury, we next performed double-labeling immunofluorescent staining of IGFBP6 with p53 and active caspase-3 in injured and sham spinal cords, respectively (Fig. 5c–n). We found that the colocalization of IGFBP6 with p53 and active caspase-3 was observed at 5 day after SCI (Fig. 5h, n), while there was hardly any expression of active caspase-3 and p53 detected by immunofluorescent staining in sham groups (Fig. 5d, j).

Association of IGFBP6 with apoptosis after SCI. Western blot detection of p53 and active caspase-3 in the injured rat spinal cords at indicated time points (a). Semiquantitative analysis (relative density) of the intensity of the proteins to GAPDH at the indicated time points (b). The data are means ± SEM (n = 3; *, #P < 0.05, significantly different from the sham groups). Double immunofluorescence staining is for IGFBP6 with p53 and active caspase-3 in sham and injured spinal cord (c–n). Sections labeled with IGFBP6 (c, f, i, l), p53 (d and g) and active caspase-3 (j and m); the colocalization of IGFBP6 with p53 and active caspase-3 is shown in the Merged photographs (e, h, k, n). Scale bars 20 μm (c-n)
IGFBP6 Expression in H2O2-Induced Primary Neurons Apoptosis
To further determine whether IGFBP6 was associated with neuronal apoptosis, H2O2 was applied to induce primary neuronal cells apoptosis in vitro, and protein expressions were evaluated by western blot. After H2O2 treatment, a significant increase was found in the expression of IGFBP6 as well as Bax and active caspase-3, which all reached a peak at 9 h (Fig. 6a, b, P < 0.05). All of these suggested that IGFBP6 play a role in neuronal apoptosis. In addition to the increased expression of IGFBP6, there is also an important regulation at the level of IGFBP6 subcellular localization. Western blot showed that IGFBP6 significantly decreased in the cytoplasm fraction and increased in the nuclear fraction after H2O2 treatment (Fig. 6c). These findings suggest that nuclear localization of IGFBP6 may play a key role in the process of H2O2-induced neuronal apoptosis.

Detection of IGFBP6 expression in H2O2-treated primary neuronal cells. Western blot analysis of IGFBP6, Bax and active caspase-3 expression in H2O2-treated primary neuronal cells at the indicated time points (a). Semiquantitative analysis (relative density) of the intensity of the proteins to GAPDH at the indicated time points (b). Nuclear and cytosolicproteins were immunoblotted for IGFBP6. The levels of LaminB and tubulin in the nuclear and cytosolic fractions, respectively, were also immunoblotted to confirm the purity of the subcellular fractions (c). The data are means ± SEM (n = 3; *, #, ˆ P < 0.05, significantly different from the control groups). Each band shown was representative of three experiments with similar results
Correlation of IGFBP6 and Neuronal Apoptosis in Vitro
To further determine the function of IGFBP6 in the process of H2O2-induced neuronal apoptosis, in our study, we used siRNA to knock IGFBP6 down in primary neurons. IGFBP6-siRNA and non-specific siRNA were tested 36 h post-transfection. The efficiency of the IGFBP6-siRNA was tested by Western blot analysis. Western blot analysis showed IGFBP6-siRNA#2 markedly reduced the level of IGFBP6 expression compared with IGFBP6-siRNA#1, but the difference between IGFBP6-siRNA#2 and IGFBP6-siRNA#3 were not significant (Fig. 7a, b).

IGFBP6 regulates the expression of active caspase-3 and neuronal apoptosis. Western blot analysis showing that IGFBP6-siRNA#2 treatment markedly decreases IGFBP6 levels at 36 h after siRNA transfection primary neurons (a). Bar chart showing the ratio of IGFBP6 to GAPDH (b). Western blot analysis showing that IGFBP6-siRNA#2 treatment markedly decreases Bax and active caspase-3 levels at 36 h after siRNA transfection in primary neurons (c). Bar chart showing the ratio of Bax and active caspase-3 to GAPDH (d). Primary neurons were respectively transfected by nothing (normal control), control-siRNA, and IGFBP6-siRNA#2 for 36 h, then treated with H2O2 for 9 h, next subjected to flow cytometrical analysis, cells were transfected by IGFBP6-siRNA#2 shown great role in reducing neuronal apoptosis (e). Bar chart showing the ratio of the apoptosis of primary neurons (n = 9; *significant difference at P < 0.05 compared with control group; error bars SEM) (f)
To judge the relationship between IGFBP6 neuronal apoptosis, we examine the protein expression of Bax and active caspase-3 with H2O2 stimulus after IGFBP6 silencing. After treating IGFBP6-knockdown primary neurons with H2O2 for 9 h, Bax and active caspase-3 was reduced remarkably (Fig. 7c, d, P < 0.05). In order to further understand the relationship between IGFBP6 and apoptosis, we use Annexin-V/PI to analyze apoptosis in H2O2-treated primary neurons after IGFBP6 silencing. We found that apoptosis cells were obviously declined in IGFBP6-siRNA#2 cells compared with control group (Fig. 7e, f, P < 0.05). Taken together, we speculated that IGFBP6 might promote neuronal apoptosis after SCI.
Discussion
In the present study, we revealed the expression profiles of IGFBP6 in adult rat after spinal cord injury. Western blot analysis showed that IGFBP6 was up-regulated after injury, and its expression was increased significantly in neurons rather than in astrocytes and microglia. Moreover, the expression of p53 and active caspase-3 were parallel with that of IGFBP6, which intimated the potential role of IGFBP6 in regulating neuronal apoptosis. Finally, siRNA experiment showed that IGFBP6 played an important role in apoptosis in vitro. These data suggested that IGFBP6 might be associated with neuronal apoptosis after SCI.
Traumatic SCI is a devastating neurologic disorder which could not be improved prognosis by present therapy. After SCI the secondary pathogenesis caused by neuronal cell apoptosis is the most critical period of the disease [21–23]. Following Traumatic SCI Many stress-response signaling pathways are rapidly activated in neurons, such as p53, NF-kB and mTOR signaling pathways [24, 25], the activation of these signaling pathways was a double-edged sword in regulateing neuronal survival and apoptosis [26–28]. The multi-domain Bcl-2 family member Bax, one of the p53 specific target gene, has been recognized as a major mediator of apoptosis [29–31]. When Bax translocates into the mitochondria, cytochrome C will be released and then form an apoptotic complex with Apaf-1, ATP, and pro-caspase-9 [32, 33]. Subsequently, a series of molecular cascade of events associated with apoptosis is activated, and finally activate caspase-3 and trigger the cellular apoptosis [22].
IGFBP6 is a member of the insulin-like growth factor-binding proteins family which is characteristic of a 50-fold binding preference for IGF-II over IGF-I [14]. IGFBP6 can prevent IGF from binding to cell surface receptors, leading to inhibit IGF-II availability in vitro and in vivo. In addition to its ability to bind and sequester IGF-II, recent in vitro studies suggest that IGFBP6 also has IGF-independent actions [34], such as growth inhibition and apoptosis [15]. In endothelial cells the expression of IGFBP-6 was upregulated by hypoxia via HIF-1α signal pathway. When overexpressed, IGFBP-6 can inhibit angiogenesis in rhabdomyosarcoma xenografts and zebrafish embryos, suggesting that IGFBP-6 contributes to limit hypoxia-induced angiogenesis [35].IGFBP6 increased apoptosis of non-small-cell lung cancer cells [16]. In lung cancer cells, the expression levels of IGFBP-6 were increased by the tumour suppressor p53 and SEMA3B, which may mediate its pro-apoptotic effect [36, 37]. Nuclear localization of IGFBP6 may play a role in RMS cell survival and as a tumour suppressor in nasopharyngeal cancer [18]. IGFBP6 inhibited vitamin D-mediated osteoblast differentiation by interacting with the vitamin D receptor in the nucleus and inhibiting its transcriptional activity [15]. In our study, western blot and immunohistochemical analysis of injured tissue sections showed more staining for IGFBP6 than the control, however, the specific role of IGFBP6 in SCI is still unknown. Immunofluorescent showed that IGFBP6 was mainly co-localized with p53 and caspase-3 in neurons, suggesting that the function of IGFBP6 might be related to apoptosis. To further investigate the function of IGFBP6, an apoptosis model was established in primary neuronal cells. Western blot analysis showed that knockdown of IGFBP6 apparently decreased the level of Bax and active caspase-3, demonstrating its probable proapoptotic role in neuronal apoptosis. Moreover, it has been reported that nuclear localization of IGFBP6 can regulate transcriptional activity of some specific target genes. Sequence analysis revealed that IGFBP6 also possesses candidate phosphorylation sites for CK2 and a DNA repair protein DNA-PK involved in apoptosis [38]. When IGFBP6 was knocked down by specific siRNA, the protein levels of Bax and active caspase-3 as well as the number of apoptotic primary neuronal cells were significantly decreased in our study, suggesting that IGFBP6 may be relevant to neuronal apoptosis via the caspase-3-dependent, Bax-related apoptotic pathway. Taken together, it is significant to speculate whether IGFBP6 plays a key role in neuronal apoptosis following trauma spinal cord injury.
In conclusion, we reported the expression changes of IGFBP6 after acute spinal cord injury in adult rat. The present study suggested IGFBP6 might participate in regulating the biological and physiological function of neurons after SCI. We propose that IGFBP6 might play an important part in neuronal apoptosis after SCI based on these findings. However, underlying mechanism remains to be elucidated about how IGFBP6 regulates neuronal apoptosis after trauma SCI.
References
Zhang J, Feng G, Bao G, Xu G, Sun Y, Li W, Wang L, Chen J, Jin H, Cui Z (2015) Nuclear translocation of PKM2 modulates astrocyte proliferation via p27 and -catenin pathway after spinal cord injury. Cell cycle 14(16):2609–2618. doi:10.1080/15384101.2015.1064203
Khalatbary AR, Zarrinjoei GR (2012) Anti-inflammatory effect of oleuropein in experimental rat spinal cord trauma. Iranian Red Crescent Med J 14 (4):229–234
Saadoun S, Bell BA, Verkman AS, Papadopoulos MC (2008) Greatly improved neurological outcome after spinal cord compression injury in AQP4-deficient mice. Brain 131 (Pt 4):1087–1098. doi:10.1093/brain/awn014
DePaul MA, Palmer M, Lang BT, Cutrone R, Tran AP, Madalena KM, Bogaerts A, Hamilton JA, Deans RJ, Mays RW, Busch SA, Silver J (2015) Intravenous multipotent adult progenitor cell treatment decreases inflammation leading to functional recovery following spinal cord injury. Scientific reports 5:16795. doi:10.1038/srep16795
Wells JE, Rice TK, Nuttall RK, Edwards DR, Zekki H, Rivest S, Yong VW (2003) An adverse role for matrix metalloproteinase 12 after spinal cord injury in mice. J Neurosci 23(31):10107–10115
Alkabie S, Boileau AJ (2016) The role of therapeutic hypothermia after traumatic spinal cord injury-a systematic review. World Neurosurg 86:432–449. doi:10.1016/j.wneu.2015.09.079
Bareiss SK, Dugan E, Brewer KL (2015) PI3K mediated activation of GSK-3beta reduces at-level primary afferent growth responses associated with excitotoxic spinal cord injury dysesthesias. Mol Pain 11:35. doi:10.1186/s12990-015-0041-2
Wu MF, Zhang SQ, Liu JB, Li Y, Zhu QS, Gu R (2015) Neuroprotective effects of electroacupuncture on early- and late-stage spinal cord injury. Neural Regen Res 10(10):1628–1634. doi:10.4103/1673-5374.167762
Casha S, Zygun D, McGowan MD, Bains I, Yong VW, Hurlbert RJ (2012) Results of a phase II placebo-controlled randomized trial of minocycline in acute spinal cord injury. Brain 135 (Pt 4):1224–1236. doi:10.1093/brain/aws072
Byrnes KR, Stoica BA, Fricke S, Di Giovanni S, Faden AI (2007) Cell cycle activation contributes to post-mitotic cell death and secondary damage after spinal cord injury. Brain 130 (Pt 11):2977–2992. doi:10.1093/brain/awm179
Niu C, Yip HK (2011) Neuroprotective signaling mechanisms of telomerase are regulated by brain-derived neurotrophic factor in rat spinal cord motor neurons. J Neuropathol Exp Neurol 70(7):634–652. doi:10.1097/NEN.0b013e318222b97b
Choo AM, Liu J, Dvorak M, Tetzlaff W, Oxland TR (2008) Secondary pathology following contusion, dislocation, and distraction spinal cord injuries. Exp Neurol 212(2):490–506. doi:10.1016/j.expneurol.2008.04.038
Xu Y, Zhang L, Sun SK, Zhang X (2014) CC chemokine ligand 18 and IGF-binding protein 6 as potential serum biomarkers for prostate cancer. Tohoku J Exp Med 233(1):25–31
Hwa V, Oh Y, Rosenfeld RG (1999) The insulin-like growth factor-binding protein (IGFBP) superfamily. Endocr Rev 20(6):761–787. doi:10.1210/edrv.20.6.0382
Cui J, Ma C, Qiu J, Ma X, Wang X, Chen H, Huang B (2011) A novel interaction between insulin-like growth factor binding protein-6 and the vitamin D receptor inhibits the role of vitamin D3 in osteoblast differentiation. Mol Cell Endocrinol 338(1–2):84–92. doi:10.1016/j.mce.2011.03.011
Sueoka N, Lee HY, Wiehle S, Cristiano RJ, Fang B, Ji L, Roth JA, Hong WK, Cohen P, Kurie JM (2000) Insulin-like growth factor binding protein-6 activates programmed cell death in non-small cell lung cancer cells. Oncogene 19(38):4432–4436. doi:10.1038/sj.onc.1203813
Grellier P, Berrebi D, Peuchmaur M, Babajko S (2002) The IGF system in neuroblastoma xenografts: focus on IGF-binding protein-6. J Endocrinol 172(3):467–476
Iosef C, Gkourasas T, Jia CY, Li SS, Han VK (2008) A functional nuclear localization signal in insulin-like growth factor binding protein-6 mediates its nuclear import. Endocrinology 149(3):1214–1226. doi:10.1210/en.2007-0959
Gruner JA (1992) A monitored contusion model of spinal cord injury in the rat. J Neurotrauma 9 (2):123–126; discussion 126–128. doi:10.1089/neu.1992.9.123
Basso DM, Beattie MS, Bresnahan JC (1995) A sensitive and reliable locomotor rating scale for open field testing in rats. J Neurotrauma 12(1):1–21
Bethea JR, Dietrich WD (2002) Targeting the host inflammatory response in traumatic spinal cord injury. Curr Opin Neurol 15(3):355–360
Keane RW, Davis AR, Dietrich WD (2006) Inflammatory and apoptotic signaling after spinal cord injury. J Neurotrauma 23(3–4):335–344. doi:10.1089/neu.2006.23.335
Oyinbo CA (2011) Secondary injury mechanisms in traumatic spinal cord injury: a nugget of this multiply cascade. Acta Neurobiol Exp 71(2):281–299
Zhang J, Cui Z, Feng G, Bao G, Xu G, Sun Y, Wang L, Chen J, Jin H, Liu J, Yang L, Li W (2015) RBM5 and p53 expression after rat spinal cord injury: implications for neuronal apoptosis. Int J Biochem Cell Biol 60:43–52. doi:10.1016/j.biocel.2014.12.020
Morrison RS, Kinoshita Y, Johnson MD, Guo W, Garden GA (2003) p53-dependent cell death signaling in neurons. Neurochem Res 28(1):15–27
Miscusi M, Ebner F, Ceccariglia S, Menegazzi M, Mariotto S, Berra L, Del Fa A, Gangitano C, Lauretti L, Missori P, Delfini R, Suzuki H (2006) Early nuclear factor-kappaB activation and inducible nitric oxide synthase expression in injured spinal cord neurons correlating with a diffuse reduction of constitutive nitric oxide synthase activity. J Neurosurg Spine 4(6):485–493. doi:10.3171/spi.2006.4.6.485
Kotipatruni RR, Dasari VR, Veeravalli KK, Dinh DH, Fassett D, Rao JS (2011) p53- and Bax-mediated apoptosis in injured rat spinal cord. Neurochem Res 36(11):2063–2074. doi:10.1007/s11064-011-0530-2
Yuan B, Liu D, Liu X (2014) Spinal cord stimulation exerts analgesia effects in chronic constriction injury rats via suppression of the TLR4/NF-kappaB pathway. Neurosci Lett 581:63–68. doi:10.1016/j.neulet.2014.08.023
Miller FD, Pozniak CD, Walsh GS (2000) Neuronal life and death: an essential role for the p53 family. Cell Death Differ 7(10):880–888. doi:10.1038/sj.cdd.4400736
Alshatwi AA, Subash-Babu P, Antonisamy P (2016) Violacein induces apoptosis in human breast cancer cells through up regulation of BAX, p53 and down regulation of MDM2. Exp Toxicol Pathol 68(1):89–97. doi:10.1016/j.etp.2015.10.002
Follis AV, Llambi F, Merritt P, Chipuk JE, Green DR, Kriwacki RW (2015) Pin1-induced proline isomerization in cytosolic p53 mediates BAX activation and apoptosis. Mol Cell 59(4):677–684. doi:10.1016/j.molcel.2015.06.029
Martinou JC, Youle RJ (2011) Mitochondria in apoptosis: Bcl-2 family members and mitochondrial dynamics. Dev Cell 21(1):92–101. doi:10.1016/j.devcel.2011.06.017
Bleicken S, Hofhaus G, Ugarte-Uribe B, Schroder R, Garcia-Saez AJ (2016) cBid, Bax and Bcl-xL exhibit opposite membrane remodeling activities. Cell Death Dis 7:e2121. doi:10.1038/cddis.2016.34
Zhang C, Lu L, Li Y, Wang X, Zhou J, Liu Y, Fu P, Gallicchio MA, Bach LA, Duan C (2012) IGF binding protein-6 expression in vascular endothelial cells is induced by hypoxia and plays a negative role in tumor angiogenesis. Int J Cancer 130(9):2003–2012. doi:10.1002/ijc.26201
Messmer-Blust A, An X, Li J (2009) Hypoxia-regulated angiogenic inhibitors. Trends Cardiovasc Med 19(8):252–256. doi:10.1016/j.tcm.2010.02.006
Koyama N, Zhang J, Huqun, Miyazawa H, Tanaka T, Su X, Hagiwara K (2008) Identification of IGFBP-6 as an effector of the tumor suppressor activity of SEMA3B. Oncogene 27(51):6581–6589
Kannan K, Amariglio N, Rechavi G, Jakob-Hirsch J, Kela I, Kaminski N, Getz G, Domany E, Givol D (2001) DNA microarrays identification of primary and secondary target genes regulated by p53. Oncogene 20(18):2225–2234. doi:10.1038/sj.onc.1204319
Han JJ, Huang BR, Wang X, Ma XL, Chen H (2009) Nuclear localization of insulin-like growth factor binding protein-6. Zhongguo yi xue ke xue yuan xue bao Acta Academiae Medicinae Sinicae 31 (6):735–739. doi:10.3881/j.issn.1000-503X.2009.06.017
Acknowledgements
This work was supported by the following funding, including the Health Research of the Jiangsu Province (Z201316); the Science and Technology of construction and people’s livelihood Foundation of Jiangsu Province (BL2014061); the ‘Top Six Types of Talents’ Financial Assistance of Jiangsu Province Grant (WSW-007).
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
We declare that we have no financial and personal relationships with other people or organizations that can inappropriately influence our work, there is no professional or other personal interest of any nature or kind in any product, service and company that could be construed as influencing the position presented in, or the review of, the manuscript entitled.
Additional information
Song Wang and Zhiming Cui are contributed equally to this work.
Rights and permissions
About this article

Cite this article
Wang, S., Liu, Y., Wu, C. et al. The Expression of IGFBP6 after Spinal Cord Injury: Implications for Neuronal Apoptosis. Neurochem Res 42, 455–467 (2017). https://doi.org/10.1007/s11064-016-2092-9
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s11064-016-2092-9