
Abstract
Purpose
Primary benign and malignant central nervous system (CNS) tumors are the most frequent solid tumors in the pediatric age and represent the leading cause of death by cancer in children in high income countries. However, information regarding specific causes of death in this population is still limited. The objective of this work was to investigate mortality in a large cohort of children diagnosed at our institution.
Methods
We identified patients consecutively diagnosed with CNS tumor and treated at a Tertiary Care Canadian Children’s Hospital between January 2000 and December 2017. Patient charts were reviewed and different variables such as tumor diagnosis, location, gender, age at diagnosis, age at death and cause of death collected.
Results
Of 1274 patients, 306 (24%) succumbed to their disease. Mortality rate varied significantly according to tumor subtype, ranging from 3.1% in low grade glioma (LGG) to 97.8% in diffuse intrinsic pontine glioma (DIPG). While high grade gliomas (HGG) and DIPG represented only 6.3 and 7.1% of total diagnoses respectively, together they accounted for 49.3% of total deaths (n = 151). Median time from diagnosis to death was 15 months (4 days to 15 years) and shortest for DIPG (11 months). Two hundred and ninety patients (94.8%) died as a result of the primary disease, 4 of treatment-related toxicity, two patients’ deaths were unrelated to the primary disease (idiopathic encephalopathy and domestic fire) whereas 10 patients succumbed to a secondary malignancy. Of note, four of these ten patients had a confirmed underlying cancer predisposition syndrome.
Conclusion
Disease progression is the main cause of death in children with brain tumor, while treatment related mortality is low in this series. Research should continue to focus on improving treatment strategies for patients whose prognosis remains dismal.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Primary central nervous system (CNS) tumors are the most common solid tumors in children, and they represent the second most common pediatric malignancies after leukemia, accounting for almost 25% of all childhood cancers in high income countries [1,2,3]. Yet, they are infrequent, with an estimated incidence which varies by country from 1.12 to 5.57 cases per 100,000 children each year [4].
Although major advances have been made over the last decades in the field of pediatric brain tumors, with new genetic discoveries, new therapies, and new technologies [5] both mortality and morbidity remain a serious concern in this population. Unfortunately, improvement in the outcome of the pediatric neuro-oncology population has been less favorable when compared to other pediatric malignancies [6, 7]. While mortality in children with leukemia and lymphoma has remarkably decreased over the recent decades, the percentage of cancer deaths due to CNS tumor has proportionally increased making brain tumors the leading cause of cancer related death among the 1–19 years old population [8, 9].
However, little is known about specific causes of death in pediatric neuro-oncology, and this aspect has remained under investigated [10]. The aim of this study was to describe the causes of deaths in a large cohort of pediatric patients treated at a single institution over a 17-year period.
Material and methods
Using our in-hospital neuro-oncology database, we conducted a retrospective chart review of clinical and pathological data of children diagnosed and treated for a CNS tumor at the Hospital for Sick Children, and who died during the course of their treatment or follow up period. Patients were eligible if they were 0–18 years old at the time of diagnosis, diagnosed from January 1st, 2000 to December 31st, 2017 and had sufficient data for analysis.
The study entry point was the day of the initial diagnosis by either the date of diagnostic imaging or the date of biopsy/tumor resection.
General demographics, information on tumor histology and location, metastatic status at diagnosis, time from diagnosis to death, specific causes of death and place of death were collected. Genetic tests were reviewed when available to identify patients with predisposing cancer condition. The cause of death was obtained either from the medical chart or from death certificates and was divided into four subcategories: disease-related, treatment related, secondary malignancy and death due to causes unrelated to the neoplastic diagnosis. Since patients are not followed beyond the age of 18 at our institution, we also reviewed all correspondences from adult long-term follow-up clinics in order to identify any death that occurred after transfer to adult care.
The study was approved by the institutional research ethic board. The need for informed consent and assent was waived given the retrospective nature of the study and the study cohort of deceased patients.
Statistical analysis
Kaplan Meier survival curves were generated to compare survival in the major tumor types and the overall significance for the difference between the groups used the log rank test. SAS 9.4 & R version 3.6.0 was used for this analysis.
For the comparisons of gender and metastatic/disseminated disease at diagnosis by major tumor type, a chi-square test was used to assess the difference in proportions of males and females in the tumor groups. For the comparison of the median age at diagnosis, a Kruskal–Wallis test was run to test the difference in the major tumor groups.
The cohort was split by diagnosis date (January 1st 2000–December 31st 2008, January 1st 2009–December 31st 2017) and comparisons made between the two eras. Differences in the proportion of patients deceased, gender, cause of death, and location of death were assessed with a chi-square test. Differences in the median age at diagnosis and the median time until death amongst deceased patients between the two eras was assessed using a Kruskal–Wallis test. Differences in the survival time after diagnosis between the two eras was assessed using a log rank test.
Results
Of the initial cohort of 1349 patients, 75 were excluded for lack of sufficient information. Hence, we included 1274 patients with central nervous system tumors diagnosed and treated at our institution during the study period. Of them, 306 (24%) died and were included in the study cohort. Demographic information of the study cohort is shown in Table 1.
The male/female ratio of the entire cohort is 1.19 (693 males, 581 females) which differed according to tumor subtype (p-value = 0.008), and ranged from 0.64 for diffuse intrinsic pontine glioma (DIPG) patients, 2.04 for medulloblastoma patients to 3.55 for patients with intracranial germ cell tumors. The male/female ratio of the deceased cohort (n = 306) was 1.04 (156 males, 150 females) and again differed according to tumor subtype ranging from 0.58 for DIPG to 2.38 for medulloblastoma patients.
The median age at diagnosis of the entire neuro-oncology population was 7.56 years (1 day to 18 years), and differed according to tumor subtype (p-value < 0.0001), ranging from 1.72 years in children with atypical teratoid rhabdoid tumors (AT/RT) to 10.7 years in patients with non-brainstem high grade gliomas (HGG).
For the cohort of patients who died, the median age at diagnosis was 6.8 years (range 1 day to 18.8 years). Median age at death was 8.9 years (13 days to 24.4 years). When dividing by age group, the mortality rate was 40.3% in patients less than 1 year at the time of diagnosis (25 deceased / 62 diagnosed), 24.4% in the 1–5 years cohort (108 deceased/442 diagnosed), 26.7% in the 6–10 years old (102 deceased/382 diagnosed), and 18.3% in patients eleven years old and older (71 deceased/388 diagnosed).
The median length of time elapsed from diagnosis to death was 15 months. It was significantly different according to tumor type (p-value < 0.0001), the shortest (11 months, range 12 days to 5 years) in children with DIPG, and the longest in those with ependymoma (4 years, range 4 days to 15.1 years) (Table 1).
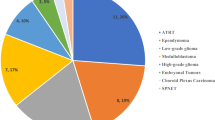
Table 2 depicts prevalence and mortality rates of all tumor types of the study cohort. While HGG and DIPG represented only 6.3% and 7.1% of total diagnoses, respectively, together they accounted for 49.3% of total deaths (n = 151). Medulloblastoma (MB) was the third most common cause of death (n = 44, 14.4%), followed by ependymoma (n = 30, 9.8%), other embryonal tumors (including “primitive neuroectodermal tumors”, n = 25, 8.2%) and AT/RT (n = 15, 4.9%). Mortality rate varied significantly according to tumor type, ranging from 3.1% in low grade glioma (LGG) (16 death of 508 patients), to 97.8% in DIPG (89 deaths of 91 patients). Other tumor types with high mortality rate were AT/RT (50%), and embryonal tumor other than AT/RT and medulloblastoma (58.1%). Of note, all patients with melanocytic tumors (n = 5) succumbed to the disease. Other tumors in our cohort, such as craniopharyngioma and germ cell tumors, were characterized by a lower mortality (Table 2). Importantly, for some brain tumors categories, no mortalities were observed (i.e.: SEGA, n = 24, meningioma, n = 15, spinal cord ependymomas, n = 14, hemangioblastoma, n = 6, pituitary adenoma, n = 6). Figure 1 depicts the Kaplan Meier curves for each of the most frequent brain tumors demonstrating a significant difference between survival curves for the different tumor types (p-value < 0.0001).

Survival according to tumor subtype
The main cause of death was disease related (n = 290; 94.8%). Most of these patients died of progressive disease (n = 282; 92.2%); 5 patients (1.6%) died as a result of intra-tumoral hemorrhage which was the presenting symptom of their tumor (3 HGG, 2 Medulloblastoma). All 3 deaths in the craniopharyngioma cohort were related to metabolic and endocrine disorders secondary to severe hypothalamic damage resulting from the tumor and/or its management. Of the 16 patients with LGG, 14 died as a result of disease progression and two passed due to other causes (domestic fire, hematological malignancy). Five patients had histologically confirmed malignant transformation, while the remaining 9 patients were either not biopsied more than once or showed low grade features in a second biopsy and/or post mortem examination. Importantly, none of the five patients with pathological evidence of malignant transformation had neither received prior radiation, nor was identified with a cancer predisposition syndrome
Treatment-related mortality in our cohort was 1.3% (n = 4), all of whom occurred in patients with embryonal tumor following sequential high dose chemotherapy and autologous stem cell rescue (HD/ASCR). Specifically, two patients died of pulmonary hypertension (1 AT/RT, 1 medulloblastoma) while 2 patients (1 pineoblastoma, 1 medulloblastoma) developed overwhelming sepsis and multiorgan failure. The mortality rate following HD/ASCR in our series was 3.1% (4/129 patients) for a total number of 412 transplants (median 3, range 1–5).
Ten patients (3.2%) of the deceased cohort succumbed to a secondary tumor (Glioblastoma multiforme n = 5, Lymphoma n = 2, Myelodysplastic syndrome = 1, DIPG = 1, Osteosarcoma n = 1). Mean time to second cancer was 5 years (range 1.8–10.9 years). In two cases, the cause of death was unrelated to the primary disease (idiopathic encephalopathy and domestic fire).
Twenty patients (6.5%) in the deceased patient cohort were known to harbor a pathogenic cancer predisposing germline mutation. TP53 germline mutation, confirming a diagnosis of Li-Fraumeni syndrome, was the most prevalent, and was identified in 6 patients. Rhabdoid predisposition syndrome type 1 (germline mutation of SMARCB1) was identified in 5 cases of AT/RT. Constitutional (bi-allelic) mismatch repair deficiency was confirmed in two cases of HGG, while in one patient with HGG, only one allele of mismatch repair gene was found to be mutated (heterozygous), in keeping with the diagnosis of Lynch syndrome. 3 patients had Neurofibromatosis type 1, and one patient each had DICER1 mutation, Ataxia telangiectasia and CHEK2 germline mutation. The rate of cancer genetic predisposition was higher in the high grade glioma group (n = 8/62, 13%). Cancer predisposition syndrome was confirmed in 40% (n = 4) of patients who were diagnosed with a secondary tumor; of them, one had a glioblastoma multiforme, whereas the 3 other patients developed extracranial secondary tumor (Table 3).
The location of death could be identified for 277 patients. Location was almost equally balanced between hospital (n = 135, 48.4%), and home (n = 133, 47.6%). Nine patients died at the local pediatric hospice in downtown Toronto (3.6%), since its opening in 2013 and one at a hospice in another province.
Amongst hospital deaths, most occurred on the oncology ward/palliative care room but 25 children died in the Intensive Care Unit (ICU).
When dividing the entire cohort of patients into two groups according to the period of treatment (before or after January 1st, 2009), differences with regards to death rate (29.6% versus 19.1%) and median survival since diagnosis (16 versus 14 months) were noted (Table 4).
Discussion
This study describes a cohort of pediatric patients who died following a diagnosis of brain tumors in a large tertiary hospital. The overall mortality rate of 24% is in keeping with previous reports and CTBRUS and SEER data with 5 and 10 years survival rates of 73.3 and 69.9%, respectively, in the pediatric age range (0–14 years) for the period 2000–2015 [11]. Although significant improvement in survival was observed in the years 1970–2000, there has been no obvious change in survival rates over the last 2 decades [12, 13].
Not surprisingly, while representing only 7.2% of total diagnoses, DIPG were characterized by the highest mortality (n = 89/91, 97.8%); all DIPG patients died of progressive disease, with a median survival time of 11 months. This is comparable to an overall median survival of 11.2 months reported in a retrospective study by Cooney et al., who analyzed survival endpoints in a large cohort of patients diagnosed with DIPG (n = 372) [14]. Similar to other series, we observed 2 long-term survivors in the DIPG cohort. Both patients had classical clinical-radiological features and were treated with focal radiotherapy only. Although rare, similar outcomes have been reported in the literature [15].
The influence of age is noticeable. As expected, our data show a higher mortality rate among children diagnosed during the first year of life (40%). There are large variations observed in the literature regarding infants and comparisons are difficult due to differences in eligibility criteria. However, our results compare with the recent report from Toescu et al. who analyzed a series of 98 children less than 1 year of age treated at a large tertiary UK institution over the period 1997–2014. In this series, 42 patients died during the study period and the 5- and 10-years survival rates were 44 and 28%, respectively [16]. This underscores the multiple challenges—in particular technical and ethical—associated with the management of central nervous system tumors in this very young age group.
Notably in less than 5% of cases, the cause of death was unrelated to disease progression. In particular, treatment-related mortality (TRM) as defined by death caused by treatment toxicity and not related to disease was low (n = 4, 1.3%). The difference of TRM to data recently reported relates to the difference in definition. The authors defined TRM as “any death occurring in the absence of progressive cancer” and stated a TRM in pediatric neuro-oncology as high as 13% [17, 18]. In a review of the causes of death in pediatric cancer patients in the Netherlands between 2003 and 2012, Loeffen et al. identified 8 out of 121 (6.6%) deaths as TRM in the pediatric brain tumor population [19]. Differences in the rate of toxic deaths between reports relate to a lack of consensus on the definition of TRM [18]. All toxic deaths in our cohort occurred in the context of high dose chemotherapy. Current prospective randomized trials comparing single and sequential high dose chemotherapy protocols may aid to better delineate risk of toxicity versus increased survival chances.
No intraoperative death occurred in our series. Five patients died due to massive intratumoral hemorrhage at initial presentation; while all underwent emergency surgery, none of the children was rescuable.
Interestingly, the mortality rate in the low-grade glioma cohort (3.1%) in this study compares favorably to numbers in the literature which range higher from 5–20%. Bandopadhayay et al.described the long term outcome of 4,040 patients diagnosed with low grade glioma in the pediatric age and found a 20-year overall survival (OS) of 87% and a 20 year cumulative incidence of death related to the glioma of 12% [20]. The major risk factor for death in her multivariate analysis was treatment with radiation (hazard ratio = 3.9) associated with increased risk of transformation and/or secondary malignancies, as observed by others [21, 22]. Indeed, in contrast to adults, malignant transformation of pediatric low-grade glioma is a rare event in pediatrics [23, 24] and linked to genetically distinct subtypes [25]. Of note, none of the 5 LGG patients showing transformation received radiotherapy upfront, as avoidance of radiation in pediatric LGG patients has been a guiding principle at our institution.
A germline cancer gene mutation was identified in 6.5% (n = 20) of our study population, confirming a cancer predisposition syndrome. However, this figure maybe an underestimation, as a large proportion of patients were not tested or families opted against genetic testing despite a previous history highly suggestive of a cancer predisposition syndrome. Screening for cancer predisposition has become more established over recent years but accurate numbers relating to patients undergoing screening over the whole study period couldn’t reliably determined. Our cohort’s profile is consistent with previous reports which identified Li-Fraumeni syndrome as the most common cancer predisposing condition [26,27,−28]. Similarly, the proportion of ATRT children with SMARCB1 germline mutation (5 out of 15 patients) is in keeping with the 35% predisposing rate reported by Bourdeaut et al. [29].
Importantly, for almost half of the patients who experienced a secondary malignancy (4/10), a genetic cancer susceptibility was identified. Regrettably, the other six patients did not undergo genetic testing. This strengthens the need for the implementation of screening for cancer genetic susceptibility in this population, as identifying an underlying genetic predisposition can ultimately improve the outcome of these patients and their families through surveillance for new and secondary tumors [30] [31] and may also affect treatment strategies [32, 33]. The observation of secondary high grade gliomas in 3 medulloblastoma patients at 5.3, 5.6 and 8.1 years confirms the importance of histological confirmation of late relapses in this tumor type [34].
Finally, almost half of the children passed away either at the hospital or at home (135 vs. 133 patients respectively), which was in most cases in accordance to families’ wishes. Of the 10 patients who died in hospice, nine were supported at the local facility which opened in 2013 and has since had an increased utilization by patients and families. These results are in keeping with a result of a recent Canadian study [17]. Of note, location of death was evenly distributed among the different brain tumors, with only a slight predominance of death at home in the DIPG group (44 patients died at home and 36 at the hospital, while location of death was unknown for 9 patients). Only 8% (25) of all deaths took place in the ICU, and half of these ICU admissions were related to acute complications such as intracranial hemorrhage or therapy refractory seizures. Albeit thorough “do not resuscitate” reviews with parents are a standard practice when curative chances have been exhausted, transfer to ICU has not always been preventable despite treating team’s palliative intentions [35].
Our study has several limitations. The major one is the retrospective nature of the study with its inherent difficulties related to incomplete and missing data. Furthermore, data from a large tertiary center may have intrinsic bias, such as the risk of an enrichment secondary to provincial referrals of harder to treat patients. One should also mention that data on the presence of metastatic disease at diagnosis could be an underestimation, particularly for LGG.
In conclusion, our data show that the cause of death in an overwhelming percentage of our patients was disease-related. Additionally, our study confirms the high proportion of deaths in patients with HGG (including DIPG) and to a lower extent in the AT/RT population. For patients affected by these diagnoses, new treatment approaches are urgently needed. In contrast, only a small proportion of patients with LGG succumbed to their disease, and in this patient population, clinicians should focus on maintaining quality of life and aim to minimize treatment burden and sequalae.
Data availability
Yes.
Code availability
Yes.
References
Linabery AM, Ross JA (2008) Trends in childhood cancer incidence in the U.S. (1992–2004). Cancer 112:416–432. https://doi.org/10.1002/cncr.23169
Kaatsch P (2010) Epidemiology of childhood cancer. Cancer Treat Rev 36:277–285. https://doi.org/10.1016/j.ctrv.2010.02.003
Fischer C, Petriccione M, Donzelli M, Pottenger E (2016) Improving care in pediatric neuro-oncology patients: an overview of the unique needs of children with brain tumors. J Child Neurol 31:488–505. https://doi.org/10.1177/0883073815597756
Johnson KJ, Cullen J, Barnholtz-Sloan JS, Ostrom QT, Langer CE, Turner MC, McKean-Cowdin R, Fisher JL, Lupo PJ, Partap S, Schwartzbaum JA, Scheurer ME (2014) Childhood brain tumor epidemiology: a brain tumor epidemiology consortium review. Cancer Epidemiol Biomark Prev 23:2716–2736. https://doi.org/10.1158/1055-9965.Epi-14-0207
Gajjar A, Bowers DC, Karajannis MA, Leary S, Witt H, Gottardo NG (2015) Pediatric brain tumors: innovative genomic information is transforming the diagnostic and clinical landscape. J Clin Oncol 33:2986–2998. https://doi.org/10.1200/jco.2014.59.9217
Chan MH, Boop F, Qaddoumi I (2015) Challenges and opportunities to advance pediatric neuro-oncology care in the developing world. Child's Nerv Syst 31:1227–1237. https://doi.org/10.1007/s00381-015-2771-x
Wohrer A, Waldhor T, Heinzl H, Hackl M, Feichtinger J, Gruber-Mosenbacher U, Kiefer A, Maier H, Motz R, Reiner-Concin A, Richling B, Idriceanu C, Scarpatetti M, Sedivy R, Bankl HC, Stiglbauer W, Preusser M, Rossler K, Hainfellner JA (2009) The Austrian Brain Tumour Registry: a cooperative way to establish a population-based brain tumour registry. J Neurooncol 95:401–411. https://doi.org/10.1007/s11060-009-9938-9
Baldwin RT, Preston-Martin S (2004) Epidemiology of brain tumors in childhood—a review. Toxicol Appl Pharmacol 199:118–131. https://doi.org/10.1016/j.taap.2003.12.029
Curtin SC, Minino AM, Anderson RN (2016) Declines in Cancer Death Rates Among Children and Adolescents in the United States, 1999–2014. NCHS data brief: 1–8
Upadhyaya SA, Ghazwani Y, Wu S, Broniscer A, Boop FA, Gajjar A, Qaddoumi I (2018) Mortality in children with low-grade glioma or glioneuronal tumors: a single-institution study. Pediatr Blood Cancer. https://doi.org/10.1002/pbc.26717
Siegel R, Naishadham D, Jemal A (2013) Cancer statistics, 2013. CA Cancer J Clin 63:11–30. https://doi.org/10.3322/caac.21166
Bleyer WA (1999) Epidemiologic impact of children with brain tumors. Child's nervous system. ChNS 15:758–763. https://doi.org/10.1007/s003810050467
Heath JA, Zacharoulis S, Kieran MW (2012) Pediatric neuro-oncology: current status and future directions. Asia-Pac J Clin Oncol 8:223–231. https://doi.org/10.1111/j.1743-7563.2012.01558.x
Cooney T, Lane A, Bartels U, Bouffet E, Goldman S, Leary SES, Foreman NK, Packer RJ, Broniscer A, Minturn JE, Shih CS, Chintagumpala M, Hassall T, Gottardo NG, Dholaria H, Hoffman L, Chaney B, Baugh J, Doughman R, Leach JL, Jones BV, Fouladi M, Warren KE, Monje M (2017) Contemporary survival endpoints: an International Diffuse Intrinsic Pontine Glioma Registry study. Neuro-oncology 19:1279–1280. https://doi.org/10.1093/neuonc/nox107
Hoffman LM, Veldhuijzen van Zanten SEM, Colditz N, Baugh J, Chaney B, Hoffmann M, Lane A, Fuller C, Miles L, Hawkins C, Bartels U, Bouffet E, Goldman S, Leary S, Foreman NK, Packer R, Warren KE, Broniscer A, Kieran MW, Minturn J, Comito M, Broxson E, Shih CS, Khatua S, Chintagumpala M, Carret AS, Escorza NY, Hassall T, Ziegler DS, Gottardo N, Dholaria H, Doughman R, Benesch M, Drissi R, Nazarian J, Jabado N, Boddaert N, Varlet P, Giraud G, Castel D, Puget S, Jones C, Hulleman E, Modena P, Giagnacovo M, Antonelli M, Pietsch T, Gielen GH, Jones DTW, Sturm D, Pfister SM, Gerber NU, Grotzer MA, Pfaff E, von Bueren AO, Hargrave D, Solanki GA, Jadrijevic Cvrlje F, Kaspers GJL, Vandertop WP, Grill J, Bailey S, Biassoni V, Massimino M, Calmon R, Sanchez E, Bison B, Warmuth-Metz M, Leach J, Jones B, van Vuurden DG, Kramm CM, Fouladi M (2018) Clinical, radiologic, pathologic, and molecular characteristics of long-term survivors of diffuse intrinsic pontine glioma (DIPG): A Collaborative Report From the International and European Society for Pediatric Oncology DIPG Registries. J Clin Oncol 36:1963–1972. https://doi.org/10.1200/jco.2017.75.9308
Toescu SM, James G, Phipps K, Jeelani O, Thompson D, Hayward R, Aquilina K (2019) Intracranial neoplasms in the first year of life: results of a third cohort of patients from a single institution. Neurosurgery 84:636–646. https://doi.org/10.1093/neuros/nyy081
Pole JD, Gibson P, Ethier MC, Lazor T, Johnston DL, Portwine C, Silva M, Alexander S, Sung L (2017) Evaluation of treatment-related mortality among paediatric cancer deaths: a population based analysis. Br J Cancer 116:540–545. https://doi.org/10.1038/bjc.2016.443
Alexander S, Pole JD, Gibson P, Lee M, Hesser T, Chi SN, Dvorak CC, Fisher B, Hasle H, Kanerva J, Moricke A, Phillips B, Raetz E, Rodriguez-Galindo C, Samarasinghe S, Schmiegelow K, Tissing W, Lehrnbecher T, Sung L (2015) Classification of treatment-related mortality in children with cancer: a systematic assessment. Lancet Oncol 16:e604–610. https://doi.org/10.1016/s1470-2045(15)00197-7
Loeffen EAH, Knops RRG, Boerhof J, Feijen E, Merks JHM, Reedijk AMJ, Lieverst JA, Pieters R, Boezen HM, Kremer LCM, Tissing WJE (2019) Treatment-related mortality in children with cancer: Prevalence and risk factors. Eur J Cancer (Oxford, Engl: 1990) 121:113–122. https://doi.org/10.1016/j.ejca.2019.08.008
Bandopadhayay P, Bergthold G, London WB, Goumnerova LC, Morales La Madrid A, Marcus KJ, Guo D, Ullrich NJ, Robison NJ, Chi SN, Beroukhim R, Kieran MW, Manley PE (2014) Long-term outcome of 4,040 children diagnosed with pediatric low-grade gliomas: an analysis of the Surveillance Epidemiology and End Results (SEER) database. Pediatr Blood Cancer 61:1173–1179. https://doi.org/10.1002/pbc.24958
Benesch M, Lackner H, Sovinz P, Suppan E, Schwinger W, Eder HG, Dornbusch HJ, Moser A, Triebl-Roth K, Urban C (2006) Late sequela after treatment of childhood low-grade gliomas: a retrospective analysis of 69 long-term survivors treated between 1983 and 2003. J Neurooncol 78:199–205. https://doi.org/10.1007/s11060-005-9091-z
Krishnatry R, Zhukova N, Guerreiro Stucklin AS, Pole JD, Mistry M, Fried I, Ramaswamy V, Bartels U, Huang A, Laperriere N, Dirks P, Nathan PC, Greenberg M, Malkin D, Hawkins C, Bandopadhayay P, Kieran MW, Manley PE, Bouffet E, Tabori U (2016) Clinical and treatment factors determining long-term outcomes for adult survivors of childhood low-grade glioma: a population-based study. Cancer 122:1261–1269. https://doi.org/10.1002/cncr.29907
Murphy ES, Leyrer CM, Parsons M, Suh JH, Chao ST, Yu JS, Kotecha R, Jia X, Peereboom DM, Prayson RA, Stevens GHJ, Barnett GH, Vogelbaum MA, Ahluwalia MS (2018) Risk factors for malignant transformation of low-grade glioma. Int J Radiat Oncol Biol Phys 100:965–971. https://doi.org/10.1016/j.ijrobp.2017.12.258
Broniscer A, Baker SJ, West AN, Fraser MM, Proko E, Kocak M, Dalton J, Zambetti GP, Ellison DW, Kun LE, Gajjar A, Gilbertson RJ, Fuller CE (2007) Clinical and molecular characteristics of malignant transformation of low-grade glioma in children. J Clin Oncol 25:682–689. https://doi.org/10.1200/jco.2006.06.8213
Mistry M, Zhukova N, Merico D, Rakopoulos P, Krishnatry R, Shago M, Stavropoulos J, Alon N, Pole JD, Ray PN, Navickiene V, Mangerel J, Remke M, Buczkowicz P, Ramaswamy V, Guerreiro Stucklin A, Li M, Young EJ, Zhang C, Castelo-Branco P, Bakry D, Laughlin S, Shlien A, Chan J, Ligon KL, Rutka JT, Dirks PB, Taylor MD, Greenberg M, Malkin D, Huang A, Bouffet E, Hawkins CE, Tabori U (2015) BRAF mutation and CDKN2A deletion define a clinically distinct subgroup of childhood secondary high-grade glioma. J Clin Oncol 33:1015–1022. https://doi.org/10.1200/jco.2014.58.3922
Zhang J, Walsh MF, Wu G, Edmonson MN, Gruber TA, Easton J, Hedges D, Ma X, Zhou X, Yergeau DA, Wilkinson MR, Vadodaria B, Chen X, McGee RB, Hines-Dowell S, Nuccio R, Quinn E, Shurtleff SA, Rusch M, Patel A, Becksfort JB, Wang S, Weaver MS, Ding L, Mardis ER, Wilson RK, Gajjar A, Ellison DW, Pappo AS, Pui CH, Nichols KE, Downing JR (2015) Germline mutations in predisposition genes in pediatric cancer. N Engl J Med 373:2336–2346. https://doi.org/10.1056/NEJMoa1508054
Parsons DW, Roy A, Yang Y, Wang T, Scollon S, Bergstrom K, Kerstein RA, Gutierrez S, Petersen AK, Bavle A, Lin FY, López-Terrada DH, Monzon FA, Hicks MJ, Eldin KW, Quintanilla NM, Adesina AM, Mohila CA, Whitehead W, Jea A, Vasudevan SA, Nuchtern JG, Ramamurthy U, McGuire AL, Hilsenbeck SG, Reid JG, Muzny DM, Wheeler DA, Berg SL, Chintagumpala MM, Eng CM, Gibbs RA, Plon SE (2016) Diagnostic yield of clinical tumor and germline whole-exome sequencing for children with solid tumors. JAMA Oncol 2:616–624. https://doi.org/10.1001/jamaoncol.2015.5699
Mody RJ, Wu YM, Lonigro RJ, Cao X, Roychowdhury S, Vats P, Frank KM, Prensner JR, Asangani I, Palanisamy N, Dillman JR, Rabah RM, Kunju LP, Everett J, Raymond VM, Ning Y, Su F, Wang R, Stoffel EM, Innis JW, Roberts JS, Robertson PL, Yanik G, Chamdin A, Connelly JA, Choi S, Harris AC, Kitko C, Rao RJ, Levine JE, Castle VP, Hutchinson RJ, Talpaz M, Robinson DR, Chinnaiyan AM (2015) Integrative clinical sequencing in the management of refractory or relapsed cancer in youth. JAMA 314:913–925. https://doi.org/10.1001/jama.2015.10080
Bourdeaut F, Dufour C, Delattre O (2010) Rhadboid tumours: hSNF/INI1 deficient cancers of early childhood with aggressive behaviour. Bull Cancer 97:37–45. https://doi.org/10.1684/bdc.2009.1024
Brodeur GM, Nichols KE, Plon SE, Schiffman JD, Malkin D (2017) Pediatric cancer predisposition and surveillance: an overview, and a tribute to Alfred G. Knudson Jr. Clin Cancer Res 23:e1–e5. https://doi.org/10.1158/1078-0432.Ccr-17-0702
Villani A, Shore A, Wasserman JD, Stephens D, Kim RH, Druker H, Gallinger B, Naumer A, Kohlmann W, Novokmet A, Tabori U, Tijerin M, Greer ML, Finlay JL, Schiffman JD, Malkin D (2016) Biochemical and imaging surveillance in germline TP53 mutation carriers with Li-Fraumeni syndrome: 11 year follow-up of a prospective observational study. Lancet Oncol 17:1295–1305. https://doi.org/10.1016/s1470-2045(16)30249-2
Banerjee A, Jakacki RI, Onar-Thomas A, Wu S, Nicolaides T, Young Poussaint T, Fangusaro J, Phillips J, Perry A, Turner D, Prados M, Packer RJ, Qaddoumi I, Gururangan S, Pollack IF, Goldman S, Doyle LA, Stewart CF, Boyett JM, Kun LE, Fouladi M (2017) A phase I trial of the MEK inhibitor selumetinib (AZD6244) in pediatric patients with recurrent or refractory low-grade glioma: a Pediatric Brain Tumor Consortium (PBTC) study. Neuro-oncology 19:1135–1144. https://doi.org/10.1093/neuonc/now282
Bouffet E, Larouche V, Campbell BB, Merico D, de Borja R, Aronson M, Durno C, Krueger J, Cabric V, Ramaswamy V, Zhukova N, Mason G, Farah R, Afzal S, Yalon M, Rechavi G, Magimairajan V, Walsh MF, Constantini S, Dvir R, Elhasid R, Reddy A, Osborn M, Sullivan M, Hansford J, Dodgshun A, Klauber-Demore N, Peterson L, Patel S, Lindhorst S, Atkinson J, Cohen Z, Laframboise R, Dirks P, Taylor M, Malkin D, Albrecht S, Dudley RW, Jabado N, Hawkins CE, Shlien A, Tabori U (2016) Immune checkpoint inhibition for hypermutant glioblastoma multiforme resulting from germline biallelic mismatch repair deficiency. J Clin Oncol 34:2206–2211. https://doi.org/10.1200/jco.2016.66.6552
Packer RJ, Zhou T, Holmes E, Vezina G, Gajjar A (2013) Survival and secondary tumors in children with medulloblastoma receiving radiotherapy and adjuvant chemotherapy: results of Children's Oncology Group trial A9961. Neuro-oncology 15:97–103. https://doi.org/10.1093/neuonc/nos267
Arland LC, Hendricks-Ferguson VL, Pearson J, Foreman NK, Madden JR (2013) Development of an in-home standardized end-of-life treatment program for pediatric patients dying of brain tumors. J Spec Pediatr Nurs 18:144–157. https://doi.org/10.1111/jspn.12024
Funding
None.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
The authors declare that they have no conflict of interest.
Ethical approval
This study was approved by the Research Ethics Board of the Hospital for Sick Children, Toronto.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
About this article

Cite this article
Renzi, S., Michaeli, O., Ramaswamy, V. et al. Causes of death in pediatric neuro-oncology: the sickkids experience from 2000 to 2017. J Neurooncol 149, 181–189 (2020). https://doi.org/10.1007/s11060-020-03590-w
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s11060-020-03590-w