
Abstract
Purpose
Glioblastoma (GBM) carries a dismal prognosis despite standard multimodal treatment with surgery, chemotherapy and radiation. Immune checkpoint inhibitors, such as PD1 blockade, for treatment of GBM failed to show clinical benefit. Rational combination strategies to overcome resistance of GBM to checkpoint monotherapy are needed to extend the promise of immunotherapy to GBM management. Emerging evidence suggests that protein phosphatase 2A (PP2A) plays a critical role in the signal transduction pathways of both adaptive and innate immune cells and that inhibition of PP2A could enhance cancer immunity. We investigated the use of a PP2A inhibitor, LB-100, to enhance antitumor efficacy of PD1 blockade in a syngeneic glioma model.
Methods
C57BL/6 mice were implanted with murine glioma cell line GL261-luc or GL261-WT and randomized into 4 treatment arms: (i) control, (ii) LB-100, (iii) PD1 blockade and (iv) combination. Survival was assessed and detailed profiling of tumor infiltrating leukocytes was performed.
Results
Dual PP2A and PD1 blockade significantly improved survival compared with monotherapy alone. Combination therapy resulted in complete regression of tumors in about 25% of mice. This effect was dependent on CD4 and CD8 T cells and cured mice established antigen-specific secondary protective immunity. Analysis of tumor lymphocytes demonstrated enhanced CD8 infiltration and effector function.
Conclusion
This is the first preclinical investigation of the effect of combining PP2A inhibition with PD1 blockade for GBM. This novel combination provided effective tumor immunotherapy and long-term survival in our animal GBM model.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Glioblastoma (GBM) is the most common primary malignancy of the central nervous system for patients older than 40 years of age [1]. Current standard of care, includes surgical resection combined with adjuvant chemotherapy and radiation, confers a dismal prognosis with a median survival of less than 2 years [2]. Novel strategies to improve GBM treatment are critically needed.
Immunotherapy, in particular checkpoint inhibitors targeting programmed dealth-1 (PD-1), can induce durable long-term immune responses in subsets of patients in many human cancers [3]. However, a large portion of patients fail to respond to monotherapy as varied mechanisms enable tumors to evade immune surveillance. Multiple clinical trials are currently on-going to investigate the use of PD-1/PD-L1 blockade for treatment of GBM. CheckMate 143 (NCT 02017717) is the first large randomized controlled trial to evaluate the safety and efficacy of nivolumab for treatment of recurrent GBM comparing with bevacizumab. However, results reported in April of 2017 at the WFNOS conference revealed a failure of nivolumab to extend overall survival and this arm was prematurely terminated [4]. GBM, like other cancer types, is able to escape immunosurveillance via multiple mechanisms. It has been shown to induce tumor infiltrating lymphocyte anergy, recruit immunosuppressive regulatory T cells (Treg) and activate multiple immune checkpoints [5]. In addition, the blood brain barrier poses unique challenges to treatment using monoclonal antibodies, which is substantially larger than what is expected to cross an intact BBB [4, 6]. Current evidence in GBM preclinical model suggest that antibody-mediated blockade of PD1/PD-L1 interaction occurs peripherally in circulating effector T-cells [4, 7]. The inability to access tumor infiltrating T-cells is posited as an underlying mechanism for the clinical ineffectiveness of nivolumab treatment for GBM. It is therefore critical to evaluate novel combination strategies with checkpoint inhibitors in preclinical studies using GBM models that could recapitulate the tumor immune microenvironment.
Protein phosphatase 2A (PP2A) is a ubiquitous serine/threonine phosphatase involved in diverse cellular processes. Emerging evidence suggests that PP2A plays a critical role in the signal transduction pathways of immune cells [8,9,10,11,12,13,14]. PP2A was first found to have a potential role as a negative regulator of cytotoxic T-cell effector function, as inhibition of PP2A resulted in enhanced antigen-specific cytotoxicity of lymphocytes [12]. It was later shown that PP2A mediated the inhibitory signaling of CTLA-4 by dephosphorylating Akt in activated T cells [15]. More recently, an in vivo shRNA screen for novel immunotherapy targets showed that inhibiting a regulatory subunit of PP2A, Ppp2r2d, produced the greatest enhancement of tumor infiltrating lymphocyte (TILs) proliferation [11]. In addition, PP2A activity was found to be essential for the function of Tregs. Transgenic deletion of PP2A resulted in Treg dysfunction and impaired immunosuppressive capability [13]. Our group demonstrated that pharmacologic inhibition of PP2A using a small molecule inhibitor, LB-100, could synergize the anti-tumor effect of PD1 blockade in a murine CT26 colon cancer model via preferential activation of the mTORC1 pathway [16]. Recently, we have also demonstrated that LB-100 could enhance the efficacy of CAR-T cells targeting carbonic anhydrase IX (CAIX) in preclinical models of GBM [17]. Taken together, inhibition of PP2A is a promising strategy to enhance anti-cancer immunity and its synergy with PD1 blockade has only been recently explored. LB-100 is a first-in-class small molecule inhibitor of PP2A. In a completed Phase 1 study, LB-100 was shown to be well tolerated in adult patients bearing progressive solid tumors [18]. We hypothesize that inhibiting PP2A, using LB-100, could enhance immune activation and synergize with PD1 blockade for treatment of GBM. While this strategy has previously been shown to be effective in subcutaneous preclinical models of colon cancer and melanoma [16], it is critical to demonstrate it applicability to GBM using an orthotropic, immunocompetent model given the unique challenges of GBM tumor microenvironment in the brain. In addition, we also hypothesize that, mechanistically, LB-100 sensitizes GBM treatment by enhancing tumor inflammation by increasing interferon-gamma (IFNγ) production by T cells. In turn, there is an adaptive increase in tumor PD-L1 expression, which render the tumor more amenable to PD1 blockade. To our knowledge, this is the first study demonstrating preclinical efficacy of combining PP2A inhibition and PD1 blockade for treatment of GBM.
Materials and methods
Drugs
LB-100 was obtained from Lixte Biotechnology.
Antibody clones used are listed in Supplementary Table S1.
Cells
GL261-WT cells, GL261-Luc cells and B16 melanoma were obtained from NCI, PerkinElmer and ATCC respectively. Tumor cells were cultured in complete RPMI medium. All cells lines used were tested and shown to be negative for mycoplasma contamination using PCR amplification.
Syngeneic tumor models
Mice were maintained and experiments were conducted with the approval of the NINDS and NCI Animal Use and Care Committees. C57BL/6 mice were purchased from Charles River Laboratory. To implant the tumor, the mouse was anesthetized and the head was fixated in a stereotactic apparatus. A longitudinal incision was made between the occiput and forehead. Using a high-speed drill, a small burr hole was made. 2 μl of single cell suspension was injected with a 28G microsyringe (Hamilton) into the right striatum (2 mm right of the midline, 2 mm posterior to the coronal suture, and 2 mm deep). After implantation, the animals were monitored daily by animal facility staff. The animals were euthanized when they reached survival endpoint, which was defined as (1) loss of greater than 20% of body weight, (2) hunched posture, or (3) head tilt.
GL261-WT tumor
6–8 weeks female mice were inoculated with 130,000 GL261 cells. Mice were randomized 8 days after implantation into four groups: PBS, LB-100 (0.16 mg/kg), anti-PD-1 (10 mg/kg) and combination. Treatments were given every 2 days for 40 days or until mice reached survival endpoint. GL261-Luc tumor: 6–8 weeks old female mice were implanted with 130,000 GL261-Luc cells as above. Mice underwent bioluminescent imaging (BLI) every 2 days and were randomized when BLI intensity was between 1.3 million and 2.4 million p/s/mm2 into four groups: PBS, LB-100 (0.16 mg/kg), anti-PD-1 (10 mg/kg) and combination. Tumor burden was assessed every 2 days until survival endpoint or complete regression of tumors.
In vivo BLI
Mice were anesthetized and given i.p. injection with 150 mg/kg D-luciferin. After 15 min, animals were imaged using an In-Vivo Xtreme II imaging station (Bruker). Regions of interest (ROI) were defined and the total photons/s/mm2 (photons per second per square mm) were recorded.
T cell depletion
Mice were implanted with GL261-WT cells as described. After 8 days, mice were randomized to control (PBS), anti-CD8 alone (10 mg/kg), anti-CD4 alone (10 mg/kg), combination, anti-CD4 + anti-CD8, combination + anti-CD8, combination + anti-CD4 and combination + anti-CD4 + anti-CD8. Anti-CD8 and anti-CD4 depleting antibodies were given on day 6 and 8 after inoculation then weekly until survival endpoints. Combination was started 8 days after inoculation and given every 2 days until survival end point.
Re-challenge study
Mice with complete regression (CR) and age-matched naïve mice were inoculated with either 1.3 × 106 GL261-WT or 1.3 × 106 B16 melanoma cells in the brain. Mice were monitored until endpoint was reached.
Flow cytometry
Cells were surface stained in PBS for surface markers. For both intracytoplasmic and intranuclear staining, cells were stained for surface molecules followed by fixation and permeabilization (eBioscience). For intracellular staining, cells were first stimulated with Cell Stimulation Cocktail (eBioscience) containing PMA/Ionomycin and protein transport inhibitor prior to undergoing staining. Cells were analyzed by flow cytometry (LSRII; BD Bioscience). Data analysis was performed using FlowJo software (TreeStar).
Isolation of TILs
C57BL/6 mice were injected with 130,000 GL261-WT cells as above. Mice were randomized 8 days after implantation into four groups: PBS, LB-100, anti-PD-1 and combination. After four treatments, given every 2 days, mice were sacrificed and tumors were harvested. Mice with grossly undetectable tumors from the combination group were excluded from analysis. Tumors were subjected to mechanical disruption using a GentleMACS Dissociator (Miltenyi Biotec) in presence of enzymatic digestion using Tumor Dissociation Kit (Miltenyi Biotec). Single cell suspensions were stained for flow cytometry analysis.
Splenic T cells IFNγ assay
CD8 + cells were isolated using CD8 + isolation kit (StemCell) from splenocytes of CR and naïve mice. CD8+ T cells (5 × 105) were co-cultured with either GL261 cells (5 × 105) or B16 melanoma cells (5 × 105) for 24 h in the presence of 0.5 ug of soluble anti-mouse CD28 (Biolegend). Protein transport inhibitor (ebioscience) was added for the last 4 h before cells were stained and analyzed with flow cytometry.
Transwell in vitro assay of PDL1 expression
CD8 + and CD4 + T cells were isolated using CD8 + and CD4 + isolation kits (StemCell) respectively from splenocytes of C57BL/6 mice. 5 × 105 T cells were activated in the presence of soluble anti-mouse CD28 (2 ug/ml) and immobilized anti-CD3 (10 ug/ml) for 3 h. T cells were then transferred into transwells (Falcon), which were physically separated from co-cultured GL261 cells (5 × 105). LB-100 was added at the time of activation. Controls included non-activated T cells, activated T cells exposed to 1 uM LB-100 in the presence of anti-IFNγ (2ug) and GL261 cells with LB-100 in the absence of T cells. LB-100 was replenished daily. Tumor and immune cells were collected and stained 72 h after activation.
Statistics
If not stated otherwise in the figure legend, samples were analyzed with GraphPad Prism software. Scatter dot plots and bar graphs are depicted as means with SEM. p ≤ 0.05 is considered as statistically significant.
Results
LB-100 and PD1 blockade combination treatment synergistically improves survival and induces regression of GL261 tumors
LB-100 is a selective inhibitor of PP2A. Its molecular structure has been reported [19]. The pharmacokinetics of LB-100 and its known metabolite endothall in mice [16] and in human [18] have been previously published. To test the hypothesis that LB-100 synergizes with PD1 blockade for treatment of GBM, we used GL261, a murine glioblastoma cell line. We have shown that at a dose of 0.16 mg/kg given every other day, LB-100 could effectively inhibit PP2A in T-cell by 40% [16].
To ensure that we can initiate treatment in mice with similar tumor burden, we first used GL261-Luc cell line. Cells were implanted into the right striatum. Ten days later, tumor burden was assessed every two days with BLI. When the tumors reached a BLI intensity between 1.3 to 2.4 million p/s/mm2, mice were randomized into one of the four treatment arms: control (PBS), PD1 blockade, LB-100, and combination. Mice were then treated and imaged every 2 days until survival endpoint or complete regression of tumors (Fig. 1a). There was no difference in tumor BLI between groups at the time of randomization, but after one cycle of combination treatment there was a statistically significant decrease in tumor burden compared to control (p < 0.01) (Fig. 1b and Supplementary Fig. S1). There was no significant difference in median survival between control and LB-100 (2 vs. 5 days, p = 0.07) or PD1 blockade monotherapy (2 vs. 4 days, p = 0.18) (Fig. 1c). Mice treated with combination therapy, however, demonstrated a significant increase in survival (13.5 days) compared to monotherapy (p < 0.005) or control (p < 0.001). More importantly, complete regression (CR) of tumor was achieved in 25% of combination treated mice while none was observed in other groups (Fig. 1c, d).

LB-100 and PD1 blockade elicit tumor rejection in murine glioblastoma. a C57BL/6 mice were inoculated with 1.3 × 105 GL261-Luc cells. When tumors reached 1.3 to 2.4 million p/s/mm2, mice were randomized and treated every 2 days until survival endpoint or tumor regression. b BLI on the day of randomization, day 0, and after one cycle of treatment, day 2. c Cumulative survival of mice over time. d Representative BLI images of mice treated with each therapy over time. e C57BL/6 mice were similarly inoculated with GL261-WT cells. Mice were randomized on post-operative day 8. f Cumulative survival of mice with GL261-WT tumor over time. Survival experiment with GL261-Luc were performed in duplicate with ≥ 4 mice per arm. The combined data was presented. Survival experiment with GL261-WT were performed in duplicate with 8 mice per arm. The combined data was presented. **p < 0.01, ***p < 0.001 (log-rank test)
To rule out the possibility that luciferase expression increased tumor immunogenicity, we repeated the experiment using GL261-WT cells. Eight days after implantation, mice were randomized into four treatment groups. Treatments were administered every 2 days for 40 days or until survival endpoint (Fig. 1e). While monotherapy had no effect on median survival compared to control (LB-100 vs. control: 18.5 days vs. 19 days, p = 0.88; PD1 blockade vs. control: 19 days vs. 19 days, p = 0.67), combination again significantly increased survival compared to all groups (26 days, p < 0.001 compared to control or single treatment groups) (Fig. 1f). Similar to the GL261-Luc tumors, 25% of the combination treated mice achieved CR.
Effect of LB-100 and PD1 blockade combination is dependent on CD8 + and CD4 + T cells
Given that PP2A is expressed in both tumor and immune cells, we would like to establish that the synergistic effect of LB-100 and PD1 blockade is T-cell dependent. GL261-WT bearing mice were subjected to temporary CD8 + and/or CD4 + T-cell ablation using depleting antibodies (Fig. 2a). When CD4 + and/or CD8+ T cells were depleted, combination treatment no longer conferred any survival benefit (Fig. 2b), suggesting that the potentiating effect of LB-100 on PD1 blockade was dependent on adaptive immunity.

LB-100 and PD-1 blockade synergistically elicit tumor rejection in a CD8 + and CD4 + T cell dependent manner. a C57BL/6 mice were inoculated with 1.3 × 106 GL261 cells and treated as above with combination therapy beginning on post-operative day 8. Mice were given CD8 + and/or CD4 + T cell depleting antibodies on post-operative day 6 and 8, then weekly until endpoint. b Cumulative survival of mice over time. All experiments were duplicated with 8 mice per group. The combined data was presented
Mice cured with combination therapy develop antigen-specific long-term memory
To confirm the ability to achieve tumor regression was an immune mediated process, we examined whether CR mice obtained protective antitumor immunity against GL261 tumors. CR mice were re-challenged with GL261 or B16 murine melanoma tumor by implanting cells into the left striatum at least 80 days after initial inoculation (Fig. 3a). Naïve mice were implanted with either GL261 or B16 cells in the same setting as controls. There was no difference in survival between CR and naïve mice implanted with B16 tumors (p = 0.06) and all mice from both groups died. However, CR mice re-challenged with GL261 cells had significantly prolonged survival compared to naïve mice injected with the same amount of GL261 cells (p < 0.01). More importantly, CR mice achieved 100% long term survival (Fig. 3b) compared to 0% of naïve mice. Histologic examination of brain specimens from GL261 re-challenged CR mice demonstrated no evidence of tumor formation (Supplementary Fig. S2). This suggests that CR mice developed a durable but antigen-specific secondary immune response to GL261 cells.

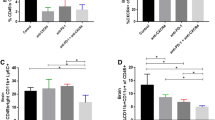
LB-100 and PD1 blockade result in a long-term antitumor antigen-specific memory of cured animals. a C57BL/6 CR and age-matched naïve control mice were re-challenged with 1.3 × 106 GL261 or B16 melanoma cells in the left striatum approximately 80 days after initial implantation. b Cumulative survival of mice over time. **p < 0.01 (log-rank test). c CD8+ T cells collected from CR or naïve mice were exposed to either GL261 or B16 melanoma cells in the presence of ⍺-CD28 for 24 h. Golgi block was added after 20 h. d Flow cytometry demonstrated enhanced IFNγ production by CD44 + CD8 + central memory T cells from cured mice when co-cultured with GL261 compared to B16. Memory T cells from cured mice had significantly enhanced IFNγ production when co-cultured with GL261 compared to memory T cells from naïve mice. e Representative FACS plots of IFNγ + CD8 + CD44 + T cells. Survival experiment was performed with 5 mice per group. CD8+ T cell data was obtained from the 5 surviving CR mice and 2 age-matched naïve controls. *p < 0.05, **p < 0.01 (one-way ANOVA with Tukey’s multiple comparison test)
To further investigate the immunologic memory of CR mice, we collected splenic CD8+ T cells from CR or naïve mice then co-cultured them with either GL261 or B16 cells (Fig. 3c). We examined the ability of CD44 + CD8 + central memory T cells to produce IFNγ when co-cultured with either GL261 or B16 tumors cells. Memory T cells from CR mice produced more IFNγ when exposed to GL261 than from naïve mice (p < 0.0001). There was no difference in IFNγ production in T cells from CR and naïve mice when exposed to B16 cells (p = 0.9755) (Fig. 3d, e). In addition, memory T cells from CR mice produced more IFNγ when exposed to GL261 compared to B16 cells, suggesting that the enhanced IFNγ production is specific to previously exposed cells. Taken together, these results suggest that CR mice achieve durable protective immunity by producing CD8+ T memory cells that recognize GL261 specific antigens.
Combination therapy enhanced proliferation and activation of TILs
To further address the cellular mechanism mediating tumor rejection by LB-100/PD1 blockade combination therapy, we performed a comprehensive analysis of the immune cells in the tumors. Mice were implanted with GL261-WT cells and treated as described above. After 4 cycles of treatment, tumors were harvested and analyzed by flow cytometry (Figs. 4, 5 and Supplementary Fig. S3). First, we examined the percentage of CD45 + cells of all live cells. There was a significant increase in overall CD45 + leukocytes in the combination group compared to control and LB-100 (50.9% vs. 35.8 and 36% respectively, p < 0.05) (Fig. 4a). Within the CD45 + population, there was as a significant increase in CD3 + T cells in the combination treated group compared to control, LB-100 and PD1 blockade alone (53.2 vs. 30.8, 32.6, and 39.4% respectively; p < 0.0005, p < 0.005, and p < 0.05) (Fig. 4b). More importantly, this increase in CD3 + T-cell population was attributed to a significant increase in CD8+ T cells relative to control, LB-100 and PD1 blockade alone (37.6 vs. 13.6, 12.4, and 19.6% respectively; p = 0.0001, p < 0.0001, and p < 0.005) (Fig. 4c, d), while the total CD4 + T-cell population remained unchanged (Fig. 4e). However, consistent with our previous finding that LB-100 alone enhanced Th1 differentiation of naïve CD4 cells, Tbet expression, as measured by MFI, was significantly increased in LB-100 and combination compared to control (1125, 1126 vs. 959 respectively; p < 0.05 for each comparison) (Fig. 4f). This result suggests that while combination is necessary to elicit enhanced infiltration of cytotoxic CD8 cells and tumor response, LB-100 alone had the effect of skewing CD4 towards Th1 differentiation, which in turn should increase tumor inflammation via enhanced IFNγ production. On the other hand, the ratio of CD8+ To CD4+ cells was markedly increased in the combination group compared to control, LB-100 and PD1 blockade alone (2.9 vs. 1.16, 1.1 and 1.4 respectively; p < 0.005, p < 0.005, and p = 0.01) (Fig. 4g). This indicated that combination resulted in enhanced CD8+ T cells infiltration to the tumor, which has consistently been shown to be one of the most important predictors of response to immunotherapy [20,21,22]. We further examined the subpopulation of CD8+ TILs by labeling the effector phenotype markers CD44 + and CD62L−. There was a significant increase in CD8 + CD44 + CD62L-T cells in combination relative to control (5.61 vs. 2.01; p < 0.05) (Fig. 4h).

LB-100 and PD1 blockade increase CD8 + infiltration and activation. Tumors the respective groups were harvested after 4 treatment cycles and analyzed by flow cytometry. a CD45 + cells as a percentage of live cells were significantly increased in combination compared to control and LB-100 alone. b CD3 + and c CD8 + cells as a percentage of CD45 + cells were significantly increased in combination compared to control or single treatments. d Representative FACS plots of CD8 + CD3 + T cells. e CD4+ cells as a percentage of CD45 + cells were not significantly changed. f Expression of Tbet by CD4+ cells were significantly increased in LB-100 alone and combination group compared to control. Tbet expression was measured by geometric Mean Fluorescent Intensity (MFI) in CD4+ cells. g Ratios of CD8+ To CD4 + T cells were significantly increased in combination compared to control or single treatment groups. h CD44 + CD62L− CD8+ T cells expressed were significantly increased in combination compared to control. i Summary of CD45 + immune cell subsets and CD45- cells. Subsets are depicted as percentage of all acquired live events (left) and CD45 + cells (right). Diagram on the right: CD4 + (blue; CD45 + CD3 + CD4 +), CD8 + (red, CD45 + CD3 + CD8 +), B cell (green; CD45 + CD19 +), NK cell (purple; CD45 + CD161 +), Monocyte (orange; CD45 + CD3− CD11clow CD11b + Ly6G- Ly6Chigh), Macrophage (grey; CD45 + CD3− CD11clow CD11b + Ly6G− Ly6Clow), DC (yellow; CD45 + CD11chigh). Experiments were duplicated with 4 mice per arm. The combined data was presented. *p < 0.05, **p < 0.01, ***p < 0.001, ****p < 0.0001 (one-way ANOVA with Tukey’s multiple comparison test)

LB-100 and PD1 blockade enhance CD8+ T cell effector function. a Representative FACS plots of IFNγ + CD8 + cells. b CD8 + IFNγ + cells were significantly increased in combination compared to control or single treatments. c IFNγ production as measured by geometric MFI in CD8 + cells was significantly increased in combination compared to PD1 blockade alone. d CD8+ TNFα + cells were significantly increased in combination compared to LB-100 alone and trended towards increase compared to control. e CD8 + IFNγ + TNFα + cells were significantly increased in combination compared to LB-100 and PD1 blockade alone and trended towards increase compared to control. f CD8 + Ki67 + T cells were significantly increased in combination compared to control or single treatments. g Ratios of CD8+ T cells to FoxP3 + CD25 + CD4 + T cells were significantly increased in combination compared to control. Experiments were duplicated with 4–5 mice per arm. The combined data was presented. *p < 0.05, **p < 0.01, ***p < 0.001 (one-way ANOVA with Tukey’s multiple comparison test)
We further characterized the immune components other than the lymphocytes population, including B-cells, dendritic cells, NK-cells, macrophages and monocytes [23] (Fig. 4i). The marked increase in CD8+ T cells contributed almost exclusively to the observed increase in CD45 + cells. Combination treatment did not synergistically alter the relative frequency of tumor infiltrating B-cells, NK-cells and dendritic cells. However, combination therapy did also decrease the CD45 + CD11clow CD11b + Ly6G- Ly6Chigh monocyte population (Supplementary Fig. S4), which has been found to be tumor promoting [24, 25] and therefore beneficial when diminished. This observation points to an unexplored effect of LB-100 on innate immune cells.
Next, we assessed the functional consequence of combination treatment in the CD8+ TILs. We analyzed intracellular expression of IFNγ in response to ex vivo stimulation with PMA/ionomycin. Combination treatment significantly enhanced IFNγ producing CD8+ TILs relative to control, LB-100 or PD1 blockade alone (24.5 vs. 5.7, 5.0 and 6.5% respectively; p < 0.005 for each comparison) (Fig. 5a, b). This increase was not simply driven by an increase in CD8 infiltration, as IFNγ production measured by MFI in CD8 + cells was significantly increased in combination compared to PD1 blockade alone (298 vs. 109; p < 0.05) (Fig. 5c) and a trend towards increase compared to control and LB-100 alone. In addition, the frequency of tumor necrosis factor-alpha TNFα-producing (Fig. 5d) CD8+ TILs were significantly increased with combination treatment compared to LB-100 alone (1.4 vs. 0.4%; p < 0.05 for each comparison) and a trend towards increase compared to control and PD1 blockade alone. CD8+ TILs doubly expressing both IFNγ and TNFα were also increased in the combination group compared to LB-100 or PD1 blockade alone (0.8 vs. 0.08 and 0.13% respectively; p < 0.05 for each comparison) and a trend towards increased compared to control (Fig. 5e). We also assessed the proliferative capacity of the CD8+ TILs by staining for the cell cycle associated protein Ki-67. The proportion of CD8 + Ki67 + TILs, which represented CD8 + cells that were proliferating in the tumor microenvironment were significantly increased in combination compared to control, LB-100 or PD1 blockade alone (25.4 vs. 5.9, 5.5 and 9.7% respectively; p = 0.0001, p = 0.0001, and p < 0.005) (Fig. 5f). We also examined the frequency of tumor infiltrating immunosuppressive Tregs. We found no difference in Treg frequency among CD3 + T cells (Supplementary Fig. S4g), but the relative CD8+ To Treg ratio remained significantly enhanced with combination treatment (Fig. 5g).
LB-100 upregulates PD-L1 expression in GL261 cells by enhancing T cell secretion of IFNγ in vitro
Next, we further investigated the mechanism underlying the synergy between PP2A inhibition and PD1 blockade. Previous studies have demonstrated that PP2A inhibition has a direct pro-inflammatory effect on T cells [11, 15, 16]. However, we failed to see any beneficial effect in vivo in GL261 or other syngeneic tumor models in our previous study [16] with LB-100 treatment alone. We hypothesized that while inhibiting PP2A increased T-cell activation, the enhanced production of IFNγ in the tumor microenvironment induced an increase in PD-L1 expression in the tumor that attenuated tumor immunity [26]. However, by enhancing PD-L1 expression, the tumor would in turn be sensitized to PD1 blockade, as PD-L1 expression has consistently been shown to be a predictive biomarker for PD1 blockade therapy [27].
We first examined in vitro if LB-100 alone has a direct effect on T cell proliferation. Mouse T cells were labeled with cytosolic dye CFSE. Cell proliferation was assessed 72 h after activation with titration concentration of LB-100. There was no significant increase in proliferation in CD8 + (Fig. 6a) or CD4+ cells (Supplementary Fig. S5a). However, IFNγ production was significantly enhanced in both CD8 + and CD4+ cells (Fig. 6b and Supplementary Fig. S5b) at concentration from 100 to 1000 nM. We then tested whether this increase in IFNγ could induce an increase in PD-L1 expression in GL261 cells in a paracrine fashion. Activated T cells were co-cultured with GL261 in a transwell plate in which T cells were physically separated from the tumor cells but secreted cytokines could pass freely in the shared medium. Cells were exposed to a titration concentration of LB-100 for 72 h and the PD-L1 expression of GL261 tumor cells was assessed by flow cytometry. PD-L1 positive GL261 cells were significantly increased when LB-100 was present from a concentration between 100 and 1000 nM (Fig. 6c). To confirm this effect was IFNγ mediated, we added IFNγ blocking antibody to the medium and the increase in PD-L1 expression was abolished (Fig. 6c). A similar pattern was seen in CD4+ co-culture, but was not statistically significant (Supplementary Fig. S5c). Given PP2A is expressed in tumor cells, to rule out the possibility that LB-100 has a direct effect on GL261 PDL-1 expression, GL261 cells were treated with similar concentration of LB-100 for 72 h. No change in PD-L1 expression was seen (Fig. 6d). In summary, we showed that LB-100 could enhance IFNγ secretion and that resulted in enhanced PD-L1 expression in GL261 tumors, which likely contributed to its sensitization to PD1 blockade.

LB-100 increases expression of PD-L1 on tumor cells by enhancing IFNγ secretion of CD8+ T cells. a LB-100 did not increase proliferation of CD8 T cells. Flow cytometry analyzing CFSE cytosolic dye as a marker of CD8+ T cell proliferation 72 h after activation. Representative FACS histogram. b LB-100 increased secretion of IFNγ by CD8 T cells. Flow cytometry analyzing expression of IFNγ in CD8+ T cells 72 h after activation in the presence of LB-100 dose titration. Representative FACS plots. c LB-100 increased expression of PD-L1 in tumor cells in transwell co-culture with CD8+ T cells. LB-100 mediated increase in tumor PD-L1 expression was abolished by IFNγ antibody in the culture medium. Representative FACS histogram. d LB-100 did not directly enhance PD-L1 expression in tumor in the absence of CD8+ T cells. Flow cytometry analyzing PD-L1 expression in tumor cells exposed to a titration concentration of LB-100 alone. CFSE experiments were repeated in duplicate and a representative repeat was presented. Trans-well experiments were repeated in triplicate and a representative repeat was presented. *p < 0.05, **p < 0.01, ***p < 0.001 (one-way ANOVA with Tukey’s multiple comparison test)
Discussion
Immunotherapy, in particular checkpoint inhibition, represents a milestone in the advancement of cancer therapy. Monoclonal antibodies against PD-1/PD-L1 are now approved by the FDA for many cancers. In a small subset of patients, checkpoint inhibitors confer dramatic responses even in advanced stages of disease. However, it is also clear that most patients have no or only partial response to mono-immunotherapy [28]. Rational combination to target non-redundant pathways of PD1/PD-L1 is critical to realize the full potential of checkpoint immunotherapy.
While kinases have been the prime pharmacologic targets for cancer therapy for many years, phosphatases have gained increasing attention as potential novel targets for treatment of cancer [29]. PP2A is a ubiquitous serine/threonine phosphatase that is implicated in a broad array of regulatory cellular functions [30]. We have initially shown that inhibiting PP2A can enhance the effect of chemo- or radiation- therapy in a number of preclinical models of human xenografts using immunocompromised mice [31,32,33]. However, there is mounting evidence to suggest PP2A plays a critical role in the signaling pathways of both the adaptive and innate immune system [8, 12,13,14,15, 34, 35]. PP2A was first identified as a negative regulator of T cell activation by de-phosphorylation of Akt [15]. PP2A, when inhibited, was identified in an in vivo shRNA screen to have the greatest effect of increasing T cell activation in TILs among all the genes examined [11]. Our group was first to demonstrate that pharmacologic inhibition of PP2A using a clinically relevant inhibitor, LB-100, could synergize anti-PD1 therapy in a preclinical model of colon cancer and melanoma [16]. The unique challenges of the tumor microenvironment of GBM justify a separate investigation into the feasibility of using LB-100/PD1 blockade combination as a novel strategy for GBM treatment. The recent reported failure of nivolumab to extend overall survival in recurrent GBM despite marked success in other cancers, is a stark reminder of the additional challenges GBM poses to effective immunotherapy. In particular, the BBB poses a significant barrier to the diffusion of large molecules such as monoclonal antibodies. While the bulk of the tumor has variably impaired BBB in GBM [36], the infiltrative edges of the tumor has intact BBB and are frequent source of recurrent disease [37]. The observation that combination therapy can regress established tumors in an orthotropic model of established GBM demonstrate that, at least in cured mice, the enhanced immunity is able to eradicate infiltrative margins of tumor with intact BBB. This is an important finding with translational significance for application to GBM treatment. Whether LB-100 derives its effect by activating circulating or tumor infiltrated lymphocytes or a combination of both remains to be elucidated. Pharmacokinetic studies of LB-100 in rats showed that the small molecule compound is able to penetrate the brain, but with significantly lower concentration than systemic circulation [16].
The enhancing effect of LB-100 on cancer immunity appeared to be more robust and durable than the sensitizing effect on chemotherapy and radiation, as a significant portion of tumors achieved complete regression with LB-100 and PD1 blockade combination. It is also of interest to note the relatively consistent portion of regressed GL261 tumor, about ~ 25%, which is lower than ~ 50% in CT26. The mechanism underlying selective durable response is unclear, but it is remarkably similar to the response characteristics seen in immune-mediated therapy reported in human clinical trials. For example, using anti-PD1 monotherapy, durable response is achieved in 10–19% and 16% of patients with melanoma and non-small cell lung cancer respectively [38]. Given the multitude of resistance pathways employed by tumors to escape immune surveillance, tumors could develop a number of adaptive changes to evade immune-checkpoint treatment, including shifting in antigen repertoire or mutations leading to defects in interferon-receptor signaling and antigen presentation. The addition of LB-100 to PD1 blockade significantly enhanced the durable response rate but further rational combination would be necessary to achieve even higher response rate.
We have chosen to use a significantly lower dose of LB-100 for immune activation than studies for chemo- or radio sensitization. Based on our previous in vitro finding, low dose of LB-100 enhanced activation of lymphocytes, while higher dose had the opposite effect [16]. It is known that PP2A inhibition, using other chemical inhibitor, has a dose-dependent dualistic response between inducing apoptosis and cell proliferation. In hepatocytes, low dose PP2A inhibition could promote cell proliferation by activating kinases such as MAPK, while high dose PP2A inhibition could induce cell death via blockage of cell cycle at G1/S [39]. The specific differential downstream pathways altered by different level of PP2A inhibition in lymphocytes has yet to be elucidated. The in vivo dose of LB-100 used to synergize with PD1 blockade was significantly lower than the human-equivalent maximum tolerated dose demonstrated in the phase 1 clinical trial [18], suggesting that the synergism of LB-100 with checkpoint inhibitors is achievable using a clinically relevant dose.
We have previously demonstrated that inhibiting PP2A in T cells could hyperactivate mTORC1 signaling and induce naive CD4 + T cells towards Th1 and away from Treg differentiation [16]. While we did see an increase in Th1 differentiation of CD4 + TILs of GL261 tumor, we did not observe a decrease in Tregs as in CT26. The significance of this is unclear. Tregs were initially reported to correlate with poor prognosis in ovarian cancer [40], but more recent studies suggest that the prognostic significance of Tregs is mixed and cancer type dependent [41]. In GBM, a study showed that Tregs, as measured in tumor and blood, did not correlate with patient survival and analysis of TCGA data also demonstrated no correlation between FoxP3 expression and survival [42]. It is also important to note that the relative frequency of Treg may not be reflective of its immunosuppressive capacity. Tregs with PP2A transgenically knocked down has diminished ability to suppress proliferation of effector T cells compared to wild-type in vitro [13]. It is possible that despite a relatively unchanged frequency of Tregs among TILS in GL261 tumors, Tregs in LB-100 treated group could have diminished functional capacity to suppress cytotoxic CD8 cells. Further functional characterization of Tregs in LB-100 treated tumors would be needed to address this important question.
Our previous study also did not elucidate the mechanism underlying the synergy between PP2A inhibition and PD1 blockade. It is unclear why the combination of LB-100 and PD1 blockade is required to achieve a dramatic immunologic response when neither monotherapy is able to. In this study, we demonstrated a plausible explanation. Similar to its human counterpart, GL261 glioblastoma responded poorly to PD1 blockade alone. While there are many steps within the cancer-immunity cycle [43, 44] that could confer resistance to checkpoint therapy, it is well accepted that expression of PD-L1 in tumors is one of the critical biomarkers for predicting response to PD1 blockade treatment [27]. On the other hand, expression of PD-L1 in tumors is known to be induced by IFNγ secreted by cytotoxic lymphocytes as an adaptive mechanism to suppress immune activation [45]. We hypothesized that while LB-100 alone could induce greater immune activation, this effect was blunted by an increase in tumor PD-L1 expression that suppressed T cell activation. This is supported by our in-vivo TILS analysis showing that LB-100 alone induced a more robust Th1 differentiation in tumor infiltrating CD4 cells, which is associated with enhanced IFNγ production and would thereby induce a higher PD-L1 expression in tumor. Therefore, LB-100 sensitized a previously PD1 blockade non-responding tumor into a responding one. We tested this hypothesis in a transwell co-culture experiment in which tumor cells were exposed to activated CD8+ T cells without direct contact. T cells treated with LB-100 induced enhanced production of IFNγ that was sufficient to induce greater expression of PD-L1 in tumors. Importantly, we showed that this effect was completely abolished by blocking anti-IFNγ antibody, confirming that IFNγ mediated the increase in PD-L1 expression. Taken together, we propose that one of the mechanisms mediating LB-100 synergy with PD1 blockade is by inducing increased expression tumor PD-L1 through enhanced paracrine secretion of IFNγ from cytotoxic T-cells.
A potential shortfall in our study is that we have not fully explored the effect of PP2A inhibition on glioma-associated macrophages and microglia (GAMs). It is estimated that GAMs constitute up to 30–50% of total cellular composition of GBM and play a critical role in shaping an immunosuppressive TME [46]. Since PP2A is also highly expressed in myeloid cells, it is possible that PP2A inhibition in GAMs contributed to the antitumor response. In fact, a recent study showed that PP2A is implicated in inhibiting Type I interferon (IFN) signaling in macrophages. Knock out of PP2A in macrophage resulted in enhanced IFN production in vitro and protection against lethal VSV infection in vivo [34], suggestive of an immunostimulatory effect in the setting of an antiviral response. If inhibiting PP2A can similarly enhance IFN production by GAMs, it would be highly complementary to its effect on T-cells. Further investigation is needed to dissect the relative impact of PP2A inhibition on different immune cell types and their contribution to enhancing immune mediated antitumor response.
Conclusion
Our study demonstrates that PP2A is a novel pharmacologic target to enhance checkpoint inhibition for treatment of glioblastoma. We believe our results carry great translational potential and argue for a clinical trial to evaluate LB-100 in combination with PD-1 blockade.
Data availability
The datasets used and/or analyzed during the current study are available from the corresponding author on reasonable request.
References
Li K, Lu D, Guo Y, Wang C, Liu X, Liu Y, Liu D (2018) Trends and patterns of incidence of diffuse glioma in adults in the United States, 1973–2014. Cancer Med 7:5281–5290
Stupp R, Hegi ME, Mason WP, van den Bent MJ, Taphoorn MJB, Janzer RC, Ludwin SK, Allgeier A, Fisher B, Belanger K et al (2009) Effects of radiotherapy with concomitant and adjuvant temozolomide versus radiotherapy alone on survival in glioblastoma in a randomised phase III study: 5-year analysis of the EORTC-NCIC trial. Lancet Oncol 10:459–466
Topalian SL, Taube JM, Anders RA, Pardoll DM (2016) Mechanism-driven biomarkers to guide immune checkpoint blockade in cancer therapy. Nat Rev Cancer 16:275–287
Fillet AC, Henriquez M, Dey M (2017) Recurrent glioma clinical trial, CheckMate-143: the game is not over yet. Oncotarget 8:91779–91794
Kim JE, Patel MA, Mangraviti A, Kim ES, Theodros D, Velarde E, Liu A, Sankey EW, Tam A, Xu H et al (2017) Combination therapy with anti-PD-1, anti-TIM-3, and focal radiation results in regression of murine gliomas. Clin Cancer Res 23:124–136
Banks WA (2009) Characteristics of compounds that cross the blood-brain barrier. BMC Neurol 9:S3
Reardon DA, Gokhale PC, Klein SR, Ligon KL, Rodig SJ, Ramkissoon SH, Jones KL, Conway AS, Liao X, Zhou J et al (2016) Glioblastoma eradication following immune checkpoint blockade in an orthotopic immunocompetent model. Cancer Immunol Res 4:124–135
Sun L, Pham TT, Cornell TT, McDonough KL, McHugh WM, Blatt NB, Dahmer MK, Shanley TP (2017) Myeloid-specific gene deletion of protein phosphatase 2A magnifies MyD88- and TRIF-dependent inflammation following endotoxin challenge. J Immunol 198:404–416
Trotta R, Ciarlariello D, Dal Col J, Mao H, Chen L, Briercheck E, Yu J, Zhang J, Perrotti D, Caligiuri MA (2011) The PP2A inhibitor SET regulates granzyme B expression in human natural killer cells. Blood 117:2378–2384
Trotta R, Ciarlariello D, Dal Col J, Allard J, Neviani P, Santhanam R, Mao H, Becknell B, Yu J, Ferketich AK et al (2007) The PP2A inhibitor SET regulates natural killer cell IFN-gamma production. J Exp Med 204:2397–2405
Zhou P, Shaffer DR, Alvarez Arias DA, Nakazaki Y, Pos W, Torres AJ, Cremasco V, Dougan SK, Cowley GS, Elpek K et al (2014) In vivo discovery of immunotherapy targets in the tumour microenvironment. Nature 506:52–57
Taffs RE, Redegeld FA, Sitkovsky M (1991) V Modulation of cytolytic T lymphocyte functions by an inhibitor of serine/threonine phosphatase, okadaic acid. Enhancement of cytolytic T lymphocyte-mediated cytotoxicity. J Immunol 147:722–728
Apostolidis SA, Rodríguez-Rodríguez N, Suárez-Fueyo A, Dioufa N, Ozcan E, Crispín JC, Tsokos MG, Tsokos GC (2016) Phosphatase PP2A is requisite for the function of regulatory T cells. Nat Immunol 17:556–564
Shanley TP, Vasi N, Denenberg A, Wong HR (2001) The serine/threonine phosphatase, PP2A: endogenous regulator of inflammatory cell signaling. J Immunol 166:966–972
Parry RV, Chemnitz JM, Frauwirth KA, Lanfranco AR, Braunstein I, Kobayashi SV, Linsley PS, Thompson CB, Riley JL (2005) CTLA-4 and PD-1 receptors inhibit T-cell activation by distinct mechanisms. Mol Cell Biol 25:9543–9553
Ho WS, Wang H, Maggio D, Kovach JS, Zhang Q, Song Q, Marincola FM, Heiss JD, Gilbert MR, Lu R et al (2018) Pharmacologic inhibition of protein phosphatase-2A achieves durable immune-mediated antitumor activity when combined with PD-1 blockade. Nat Commun 9:1–15
Cui J, Wang H, Medina R, Zhang Q, Xu C, Indig IH, Zhou J, Song Q, Dmitriev P, Sun MY et al (2020) Inhibition of PP2A with LB-100 enhances efficacy of CAR-T cell therapy against glioblastoma. Cancers (Basel) 12:1–15
Chung V, Mansfield AS, Braiteh F, Richards D, Durivage H, Ungerleider RS, Johnson F, Kovach JS (2017) Safety, tolerability, and preliminary activity of LB-100, an inhibitor of protein phosphatase 2A, in patients with relapsed solid tumors: an open-label, dose escalation, first-in-human, Phase I trial. Clin Cancer Res 23:3277–3284
Chang K-E, Wei B-R, Madigan JP, Hall MD, Simpson RM, Zhuang Z, Gottesman MM (2015) The protein phosphatase 2A inhibitor LB100 sensitizes ovarian carcinoma cells to cisplatin-mediated cytotoxicity. Mol Cancer Ther 14:90–100
Galon J, Costes A, Sanchez-Cabo F, Kirilovsky A, Mlecnik B, Lagorce-Pagès C, Tosolini M, Camus M, Berger A, Wind P et al (2006) Type, density, and location of immune cells within human colorectal tumors predict clinical outcome. Science 313:1960–1964
Sato E, Olson SH, Ahn J, Bundy B, Nishikawa H, Qian F, Jungbluth AA, Frosina D, Gnjatic S, Ambrosone C et al (2005) Intraepithelial CD8+ tumor-infiltrating lymphocytes and a high CD8+/regulatory T cell ratio are associated with favorable prognosis in ovarian cancer. Proc Natl Acad Sci USA 102:18538–18543
Zhang L, Conejo-Garcia JR, Katsaros D, Gimotty PA, Massobrio M, Regnani G, Makrigiannakis A, Gray H, Schlienger K, Liebman MN et al (2003) Intratumoral T cells, recurrence, and survival in epithelial ovarian cancer. N Engl J Med 348:203–213
Rose S, Misharin A, Perlman H (2012) A novel Ly6C/Ly6G-based strategy to analyze the mouse splenic myeloid compartment. Cytometry A 81A:343–350
Augier S, Ciucci T, Luci C, Carle GF, Blin-Wakkach C, Wakkach A (2010) Inflammatory blood monocytes contribute to tumor development and represent a privileged target to improve host immunosurveillance. J Immunol 185:7165–7173
Wilcox RA, Wada DA, Ziesmer SC, Elsawa SF, Comfere NI, Dietz AB, Novak AJ, Witzig TE, Feldman AL, Pittelkow MR et al (2009) Monocytes promote tumor cell survival in T-cell lymphoproliferative disorders and are impaired in their ability to differentiate into mature dendritic cells. Blood 114:2936–2944
Mandai M, Hamanishi J, Abiko K, Matsumura N, Baba T, Konishi I (2016) Dual faces of ifnγ in cancer progression: a role of pd-l1 induction in the determination of pro- and antitumor immunity. Clin Cancer Res 22:2329–2334
Patel SP, Kurzrock R (2015) PD-L1 Expression as a predictive biomarker in cancer immunotherapy. Mol Cancer Ther 14:847–856
Sanmamed MF, Chen L (2018) A paradigm shift in cancer immunotherapy: from enhancement to normalization. Cell 175:313–326
Mullard A (2018) Phosphatases start shedding their stigma of undruggability. Nat Rev Drug Discov 17:847–849
Baskaran R, Velmurugan BK (2018) Protein phosphatase 2A as therapeutic targets in various disease models. Life Sci 210:40–46
Ho WS, Feldman MJ, Maric D, Amable L, Hall MD, Feldman GM, Ray-Chaudhury A, Lizak MJ, Vera J-C, Robison RA et al (2016) PP2A inhibition with LB100 enhances cisplatin cytotoxicity and overcomes cisplatin resistance in medulloblastoma cells. Oncotarget 7:12447–12463
Ho WS, Sizdahkhani S, Hao S, Song H, Seldomridge A, Tandle A, Maric D, Kramp T, Lu R, Heiss JD et al (2018) LB-100, a novel Protein Phosphatase 2A (PP2A) inhibitor, sensitizes malignant meningioma cells to the therapeutic effects of radiation. Cancer Lett 415:217–226
Hong CS, Ho W, Zhang C, Yang C, Elder JB, Zhuang Z (2015) LB100, a small molecule inhibitor of PP2A with potent chemo- and radio-sensitizing potential. Cancer Biol Ther 16:821–833
Long L, Deng Y, Yao F, Guan D, Feng Y, Jiang H, Li X, Hu P, Lu X, Wang H et al (2014) Recruitment of phosphatase PP2A by RACK1 adaptor protein deactivates transcription factor IRF3 and limits type I interferon signaling. Immunity 40:515–529
Sheu J-R, Chen Z-C, Hsu M-J, Wang S-H, Jung K-W, Wu W-F, Pan S-H, Teng R-D, Yang C-H, Hsieh C-Y (2018) CME-1, a novel polysaccharide, suppresses iNOS expression in lipopolysaccharide-stimulated macrophages through ceramide-initiated protein phosphatase 2A activation. J Cell Mol Med 22:999–1013
Nduom EK, Yang C, Merrill MJ, Zhuang Z, Lonser RR (2013) Characterization of the blood-brain barrier of metastatic and primary malignant neoplasms. J Neurosurg 119:427–433
Sarkaria JN, Hu LS, Parney IF, Pafundi DH, Brinkmann DH, Laack NN, Giannini C, Burns TC, Kizilbash SH, Laramy JK et al (2018) Is the blood-brain barrier really disrupted in all glioblastomas? A critical assessment of existing clinical data. Neuro Oncol 20:184–191
Borcoman E, Kanjanapan Y, Champiat S, Kato S, Servois V, Kurzrock R, Goel S, Bedard P, Le Tourneau C (2019) Novel patterns of response under immunotherapy. Ann Oncol Off J Eur Soc Med Oncol 30:385–396
Gehringer MM (2004) Microcystin-LR and okadaic acid-induced cellular effects: a dualistic response. FEBS Lett 557:1–8
Curiel TJ, Coukos G, Zou L, Alvarez X, Cheng P, Mottram P, Evdemon-Hogan M, Conejo-Garcia JR, Zhang L, Burow M et al (2004) Specific recruitment of regulatory T cells in ovarian carcinoma fosters immune privilege and predicts reduced survival. Nat Med 10:942–949
deLeeuw RJ, Kost SE, Kakal JA, Nelson BH (2012) The prognostic value of FoxP3+ tumor-infiltrating lymphocytes in cancer: a critical review of the literature. Clin Cancer Res 18:3022–3029
Thomas AA, Fisher JL, Rahme GJ, Hampton TH, Baron U, Olek S, Schwachula T, Rhodes CH, Gui J, Tafe LJ et al (2015) Regulatory T cells are not a strong predictor of survival for patients with glioblastoma. Neuro Oncol 17:801–809
Sharma P, Hu-Lieskovan S, Wargo JA, Ribas A (2017) Primary, adaptive, and acquired resistance to cancer immunotherapy. Cell 168:707–723
Chen DS, Mellman I (2013) Oncology meets immunology: the cancer-immunity cycle. Immunity 39:1–10
Schumacher TN, Schreiber RD (2015) Neoantigens in cancer immunotherapy. Science 348:69–74
Wei J, Chen P, Gupta P, Ott M, Zamler D, Kassab C, Bhat KP, Curran MA, de Groot JF, Heimberger AB (2020) Immune biology of glioma-associated macrophages and microglia: functional and therapeutic implications. Neuro Oncol 22:180–194
Funding
The research was supported by the Intramural Research Program of the NINDS and NCI of the National Institutes of Health.
Author information
Authors and Affiliations
Contributions
Conception and design—WH, RL, ZZ. Development of methodology—WH, RL, ZZ. Acquisition of data (provided animals, acquired and managed patients, provided facilities, etc.)—RB, DM, WH, SW, HW, JC. Analysis and interpretation of data (e.g., statistical analysis, biostatistics, computational analysis)—RB, DM, WH, RL, ZZ, JH, MG. Writing, review, and/or revision of the manuscript—WH, RB, ZZ, JH, MG, JK. Administrative, technical, or material support (i.e., reporting or organizing data, constructing databases)—WH, DM, ZZ, JH.
Corresponding authors
Ethics declarations
Conflict of interest
WH is a board member and has stock options for Lixte Biotechnology Holdings Inc. JK is a board member and own stocks for Lixte Biotechnology Holdings Inc. WH, HW, RL, JK, ZZ have pending patents related to this work. No potential conflicts of interest were disclosed by the other authors.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
About this article

Cite this article
Maggio, D., Ho, W.S., Breese, R. et al. Inhibition of protein phosphatase-2A with LB-100 enhances antitumor immunity against glioblastoma. J Neurooncol 148, 231–244 (2020). https://doi.org/10.1007/s11060-020-03517-5
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s11060-020-03517-5