
Abstract
Introduction
Non-invasively distinguishing aggressive from non-aggressive brain tumors is an important clinical challenge. Intracellular pH (pHi) regulation is essential for normal cell function and is normally maintained within a narrow range. Cancer cells are characterized by a reversed intracellular to extracellular pH gradient, compared to healthy cells, that is maintained by several distinct mechanisms. Previous studies have demonstrated acute pH modulation in glioblastoma detectable by chemical exchange saturation transfer (CEST) magnetic resonance imaging (MRI) after blocking individual pH regulatory mechanisms. The purpose of the current study was to simultaneously block five pH regulatory mechanisms while also providing glucose as an energy substrate. We hypothesized that this approach would increase the acute pH modulation effect allowing the identification of aggressive cancer.
Methods
Using a 9.4 T MRI scanner, CEST spectra were acquired sensitive to pHi using amine/amide concentration independent detection (AACID). Twelve mice were scanned approximately 11 ± 1 days after implanting 105 U87 human glioblastoma multiforme cells in the brain, before and after intraperitoneal injection of a combination of five drugs (quercetin, cariporide, dichloroacetate, acetazolamide, and pantoprazole) with and without glucose.
Results
Two hours after combination drug injection there was a significant 0.1 ± 0.03 increase in tumor AACID value corresponding to a 0.4 decrease in pHi. After injecting the drug combination with glucose the AACID value increased by 0.18 ± 0.03 corresponding to a 0.72 decrease in pHi. AACID values were also slightly increased in contralateral tissue.
Conclusions
The combined drug treatment with glucose produced a large acute CEST MRI contrast indicating tumor acidification, which could be used to help localize brain cancer and monitor tumor response to chemotherapy.
Similar content being viewed by others
Avoid common mistakes on your manuscript.
Introduction
Glioblastoma multiforme (GBM) is a highly aggressive cancer and the most common primary brain tumor of the central nervous system (CNS). The current standard of treatment combines surgical resection of the primary tumor followed by concurrent radiation and chemotherapy [1,2,3,4,5]. However, due to the high migratory nature of GBM cancer cells, many escape treatment leading to recurrence in 90% of GBM patients [4]. As a result, GBM patients often survive only 12–18 months following diagnosis [2,3,4]. In the United States, glioblastomas represent the second leading cause of death among neurological diseases and one of the greatest challenges in the cure of cancer worldwide [1,2,3,4].
Cancer cells are characterized by a distinctive intracellular/extracellular pH gradient, that is reversed compared to normal cells. Specifically, in cancer cells, the extracellular space is acidic and the intracellular compartment is neutral or slightly basic [6,7,8]. This reversal of the pH gradient in tumors occurs because [9, 10] aerobic glycolysis even in the presence of oxygen, known as the Warburg effect [9, 10] produces a large quantity of acidic metabolites that are exported to the extracellular space. The acidification of the extracellular tumor microenvironment contributes to cancer cell evasion of apoptosis [11, 12], drug resistance [13], proliferation [12], and increased metastatic potential [6, 9, 11, 14]. Aggressive tumors often increase in size too quickly to develop adequate vasculature. Therefore, cancer cells that favor glycolysis tend to have an advantage [9, 11]. High glucose uptake, which is the basis of cancer detection using the glucose analog 18F-labeled fluorodeoxyglucose (18F FDG) positron emission tomography (PET) [9, 11, 15], and up-regulation of pro-glycolytic enzymes such as hexokinase [9] can lead to further extracellular acidification.
Conversely, purposefully decreasing tumor intracellular pH can have several potential beneficial applications for cancer patients. For example, the manipulation of tumor intracellular pH may predict tumor response to chemotherapy and radiation therapy [3, 16]. Additionally, decreasing intracellular pH in cancer cells may increase the efficacy of some anti-tumor treatments [17], and may directly induce cancer cell apoptosis [17]. Tumor acidification can be achieved by blocking the regulators that extrude H+ and lactate, which maintain a relatively alkaline tumor pHi. These regulators include carbonic anhydrase inhibitors, anion exchangers, the Cl−/HCO3− exchangers, Na+/HCO3– co-transporters, Na+/H+ exchangers, monocarboxylate transporters (MCT), and the vacuolar ATPase and ATP synthase [18]. We have previously shown that blocking specific regulators of pH can acidify the intracellular tumor environment within two hours of dosing in a U87 GBM mouse model. Specifically, we found the MCT inhibitor lonidamine decreased pHi by 0.25 at a dose of 50 mg/kg and decreased pHi by 0.45 at a dose of 100 mg/kg [19], the carbonic anhydrase inhibitor topiramate (120 mg/kg) decreased pHi by 0.17 [20], the MCT and V-ATPases inhibitor dichloroacetate (200 mg/kg) decreased pHi by 0.16 [21], the Na+/H+ exchange inhibitor cariporide (6 mg/kg) decreased pHi by 0.48 [22], and the MCT inhibitor quercetin (200 mg/kg) decreased pHi by 0.27 [23]. However, it is currently unknown whether combining two or more drugs to simultaneously block multiple pH regulatory mechanisms can produce greater tumor acidification.
The purpose of the current study was to determine whether acute glioblastoma intracellular acidification could be enhanced in vivo in the U87 GBM mouse model using a combined therapy that simultaneously blocked several major pH regulators, and to determine whether cancer cell apoptosis increased following short term acidification treatment. We hypothesize that combining five drugs targeting different pH regulatory pathways would induce greater acidification within tumors than that previously found with only a single drug. We further hypothesized that pre-treatment with glucose to provide additional substrate for aerobic glycolysis in combination with the five drug acidification treatment would further increase intracellular acidification. Finally, we hypothesized that short term acidiciation treatment would increase tumor apoptosis. In all studies, intracellular pH was monitored in vivo using amine/amide concentration independent detection (AACID) chemical exchange saturation transfer (CEST) magnetic resonance imaging (MRI).
Experimental
Subjects
Nineteen different female Crl:-Nu-Foxn1Nu (NU/NU) (Charles River Laboratories, Canada) adult mice were included in the current study. Mice were group housed in ventilated racks, on a 12 h/12 h light/dark cycle. All animal procedures were performed according to a protocol that was consistent with the guidelines established by the Canadian Council on Animal Care and was approved by the University of Western Ontario Animal Use Subcommittee.
For the assessment of the acute intracellular tumor acidification produced by the drug combination, six NU/NU mice with U87MG brain tumors were used (Group A1). An additional six NU/NU mice with U87MG brain tumors were used to evaluate the acute intracellular tumor acidification produced by the drug combination preceded by glucose administration (Group A2). To assess apoptosis following brief continuous treatment (8 days), three groups of mice were studied. The first group consisted of three mice from Group A1 that were treated with the five-drug combination (Group T1). The second group was a control group of three mice with brain tumors that received only vehicle (Group T2). The third group was also a control group of four mice without tumors that were treated with the five-drug combination to assess the impact on normal brain (Group T3).
Selection of drugs to block pH regulation
Intracellular acidification can be achieved by inhibiting different regulators of intracellular pH including vacuolar H+-ATPases [24], Na+/H+ exchangers [25], monocarboxylate transporters [9, 11], and carbonic anhydrases [26]. In most instances, drugs considered safe in humans have been shown to inhibit these mechanisms. We designed a combination therapy to simultaneously target these five pH regulatory mechanisms using mostly drugs approved for human use including quercetin, pantoprazole (PPZ), acetazolamide, dichloroacetate (DCA), and cariporide. Quercetin (Sigma-Aldrich, Canada) is an inhibitor of monocarboxylate transporters (MCTs) [27,28,29,30] with linear formula C15H10O7. It specifically inhibits MCT1 and MCT2 [28]. The quercetin dose used was 100 mg/kg every 8 h [31]. Pantoprazole (United States Pharmacopeia, Maryland, USA) is a proton pump inhibitor (PPI) with linear formula C16H14F2N3NaO4S·1.5H2O that targets the vacuolar H+-ATPase and has been shown to decrease intracellular pH and increase apoptotic cell death in cancer cells [32, 33]. The pantoprazole dose used was 2 mg/kg every 8 h. Acetazolamide (Sigma-Aldrich, Canada) with linear formula C4H6N4O3S2 inhibits carbonic anhydrase activity and decreases expression of the aquaporin-1 (AQP1) water channel [34,35,36,37,38]. It may also inhibit angiogenesis and endothelial cell proliferation in various cancers [39, 40]. The acetazolamide dose used was 50 mg/kg every 8 h. Dichloroacetate (Sigma–Aldrich, Canada) with linear formula Cl2CHCO2Na inhibits pyruvate dehydrogenase kinases (PDKs) and redirects pyruvate back into the mitochondria [41], which reverses the Warburg effect by activating pyruvate dehydrogenase (PDH). However, DCA also decreases the expression of monocarboxylate transporters (MCTs) and V-ATPase [42] in tumor cells, leading to reduced pHi in tumors. DCA has been shown to induce cell death in several different types of cancer [43, 44]. The DCA dose used was 100 mg/kg every 8 h. Finally, cariporide (Cayman Chemical Company, Michigan, USA) with linear formula C12H17N3O3S selectively inhibits the Na+/H+ exchange isoform 1 (NHE1) [45] with little effect on other ion transport systems [46] and has been shown to suppress the invasion and migration of cancer cells [47]. The cariporide dose used was 2 mg/kg every 8 h. Previous studies have shown that quercetin [48, 49], pantoprazole [50, 51], dichloroacetate [52, 53], and acetazolamide [54, 55] all cross the blood brain barrier, and therefore should easily be taken up by the tumor where the blood brain barrier is compromised and the vasculature is leaky. Cariporide is not expected to cross an intact blood brain barrier [56, 57], but it has been shown to enter affected tissue under ischemic conditions [56], and in cancer [58]. Therefore all agents used in the current study are expected to reach the tumor.
Drug delivery
All drugs were obtained in powder form and dissolved in dimethyl sulfoxide (DMSO). To assess the acute effects on pH of the drug combination using CEST MRI (Groups A1 and A2), the drugs were injected together in DMSO only without dilution. Specifically, the single injection contained drug doses equal to that used for eight hours of treatment as described above. To assess whether glucose increased tumor intracellular acidification using CEST MRI, glucose (Sigma-Aldrich, Canada) with linear formula C6H12O6 was dissolved in PBS and administered by i.p. injection (Group A2) at a dose of 5 g/kg in 0.2 ml. Glucose was provided 20 min before injection of the drug combination. For mice receiving treatment (Groups T1 and T3), the drugs were diluted with phosphate buffered saline (PBS) at a ratio of 1:19 and administered by intraperitoneal (i.p.) injection in a total volume of 1 ml every 8 h for 8 days. Control mice with brain tumors (Group T2) received DMSO + PBS at a ratio of 1:19 in a total volume of 1 ml every 8 h for 8 days.
Animal model of glioblastoma
The GBM animal model has been published previously but is provided for completeness [19, 21,22,23]. GBM brain tumors were induced in 22–27 g, NU/NU mice (N = 15) using U87MG glioma cells established from a human GBM (ATCC; Rockville, MD, USA) as described previously [59]. Briefly, U87MG cells were grown in Dulbecco’s modified Eagles' medium supplemented with 10% fetal bovine serum (Wisent Inc., St-Jean-Baptiste, QC, Canada) at 37 °C in a humidified incubator with 5% CO2 and passaged twice a week. On the day of injection, U87MG cells were washed and dissociated with versene solution (PBS plus 0.5 mM EDTA), then washed twice with PBS, counted and re-suspended to a final concentration of 1 × 105 cells in 2 mL PBS. Before injection, mice were anesthetized by inhalation of 4% isoflurane and maintained using 1.5% isoflurane. The mouse was placed in a stereotactic head frame (Stoelting instruments, Wood Dale, IL, USA). The scalp was swabbed with betadine, and an incision was made in the scalp to expose the bregma. A 1 mm diameter hole was drilled at coordinates measured from the bregma (1 mm anterior and 2 mm lateral). U87MG cells (2 µl) were injected at a rate of 0.5 μL/min, at a position 3 mm deep from the bregma into the right frontal lobe using a Hamilton (Reno, NV, USA) syringe with a 27-gauge needle attached.
Mouse preparation for in vivo imaging
Approximately 11 ± 1 days after cancer cell injection, the mice in Groups A1 and A2 were scanned on a 9.4 T small animal MRI system equipped with a 30 mm millipede volume coil (Agilent, Santa Clara, CA, USA). Anesthesia was induced using 4% isoflurane in oxygen and maintained with 1.5–2.5% isoflurane in oxygen. Each mouse was secured on a custom-built MRI-compatible stage, and the head was secured using a bite bar [20] and surgical tape to limit motion due to respiration. Animal temperature was monitored with a rectal temperature probe, and respiration was monitored with a respiratory sensor pad connected to a pressure transducer that was placed on the thoracic region. Body temperature was maintained at 36.9–37.1 °C throughout imaging by blowing warm air over the animal using a model 1025 small-animal monitoring and gating system (SA Instruments Inc., Stony Brook, NY, USA). Following baseline imaging, the mouse was injected with the drug combination inside the MRI through fine plastic tube ended with a needle to deliver into the peritoneum. Following imaging, three animals from Group A1 were treated with the five drug combination for 8 days (Group T1), while all others animals were sacrificed immediately after MR imaging.
In vivo magnetic resonance imaging and pH-weighted imaging
The imaging protocol used in this study has been published previously [19, 21, 22]. Briefly, T2-weighted images were used for tumor detection acquired using a 2-dimensional fast spin echo pulse sequence (FSE) with parameters: TR/TE = 3000/10 ms, ETL = 4, effective TE = 40 ms, FOV = 25.6 × 25.6 mm2, matrix size = 128 × 128, slice thickness = 1 mm. Two slices from the series of T2-weighted images with maximum tumor coverage were selected for CEST imaging. CEST images were acquired using a fast spin-echo (FSE) pulse sequence (TR/TE = 7000/7 ms, ETL = 32, effective TE = 7 ms, FOV = 25.6 × 25.6 mm2, matrix size = 64 × 64, slice thickness = 2 mm) preceded by a continuous wave radiofrequency (RF) pulse with amplitude 1.5 µT and 4 s duration. The CEST images were acquired at different saturation frequencies (from 1.2 to 4.5 (∆ = 0.1) ppm, from 5.4 to 6.6 (∆ = 0.1) ppm, and − 1000 and 1000 ppm images were acquired as a reference, total 49 images). A complete series of CEST images were acquired three times before and three times after drug injection to improve the signal-to-noise ratio. For B0 correction, the water saturation shift referencing (WASSR) technique was used [60]. A linearly spaced 37-point WASSR CEST spectrum with saturation frequencies ranging from − 0.6 to 0.6 ppm was acquired using the same pulse sequence except preceded by a short RF saturation pulse (100 ms) with low amplitude (0.2 µT).
CEST data processing
All acquired CEST MR data were processed on a pixel-by-pixel basis using custom MATLAB (Mathworks, Natick, MA, USA) code for analysis as previously described [19, 21,22,23]. Each WASSR spectrum and CEST spectrum was interpolated to achieve 1-Hz resolution. All CEST spectra were smoothed using the ‘smooth’ algorithm from the MATLAB curve fitting toolbox. Each CEST spectrum was then frequency shifted, using the corresponding WASSR spectrum, to account for B0 variation. B0 variations were corrected on a pixel-by-pixel basis. The three pre- and three post-injection CEST spectra were summed following B0 corrections to increase signal to noise ratio. As previously shown [19], the B1 variation in the CEST slice was less than 5%, so no B1 correction was applied [19].
Mapping tissue intracellular pH
Tissue pHi was monitored using amine and amide concentration-independent detection (AACID), which uses the ratio of CEST effects from amide (Δω = 3.5 ppm) and amine (Δω = 2.75 ppm) protons to generate pHi dependent contrast independent of tissue macromolecule concentration and temperature [61]. The CEST contrast originates from exchangeable amine and amide protons that are found in tissue proteins and peptides [62,63,64]. However the AACID CEST measurement of tissue pH is highly weighted to the intracellular compartment [63] because almost 90% of total protein content exists in the intracellular space [64]. AACID values were measured on a pixel-by-pixel basis using the associated B0-corrected and smoothed CEST spectra. The AACID value represents the ratio of the CEST effects of amine protons resonating at 2.75 ppm and amide protons at 3.50 ppm, normalized by MT effects measured after saturation at 6.0 ppm and is calculated using Eq. (1) [61].
Following drug administration, the change in pH was estimated by Eq. (2) obtained using the calibration provided by Eq. (8) in McVicar et al. [61].
Immunohistochemistry on mouse brains
On day 18–19 after injection of cancer cells, mice were euthanized and perfused with 4% paraformaldehyde in PBS as described previously [65]. After 24-h incubation in 4% paraformaldehyde at 4 °C, brains were cut using a vibratome into 80-µm coronal sections. Randomly chosen sections containing tumor were then immunostained as described previously [66], using an anti-cleaved caspase-3 (CC3) antibody (Cell Signaling, cat#9664, 1:500) and Alexa Fluor 488 secondary anti-rabbit antibody (Thermo Fisher, cat#A-11008), followed by nuclear staining with Hoechst 33342. Stained brain sections were mounted on glass slides using Immu-mount (Thermo Scientific, cat#9,990,402) and imaged using a FV1000 confocal microscope (Olympus) equipped with a 10 ×/0.4 or a 20 ×/0.75 objective. Images were stitched using FluoView software (Olympus) and analyzed using ImageJ Measure and Cell Counter plugins (NIH, Bethesda, MD). At least three sections per animal and at least three animals per condition were analyzed. Apoptosis was quantified using the number of CC3-positive cells per mm3.
Statistical analysis
Regions of interest (ROIs) containing tumor tissue and contralateral tissue were drawn manually in each mouse brain using the MATLAB (‘roipoly’ function) using the contrast observed in the T2-weighted images as a guide. Average AACID values were calculated before and after injection of the drug combination within each ROI. A paired t test was used to calculate differences in mean AACID values measured in the tumor and contralateral ROIs before and after injection of the drug combination and the drug combination plus glucose. The number of CC3 positive cells per mm3 were compared between treatment groups using a Student’s t test in Prism (GraphPad). In all comparisons, p < 0.05 was considered statistically significant.
Results
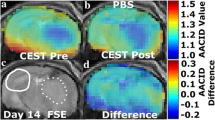
AACID CEST maps were acquired in all animals 11 ± 1 days after U87MG cancer cell implantation. Anatomical FSE MR images were successfully used to identify regions of interest in the tumor and on the contralateral side (Figs. 1c and 2c). AACID CEST maps showed the expected lower AACID value in the tumor region compared to the surrounding brain tissue (Figs. 1a and 2a) indicating a relatively basic intracellular tumor pH. Also, as expected, a large increase in tumor AACID value was observed within the tumor following the injection of the drug combination (Fig. 1b) indicating rapid intracellular acidification within 2 h of injection. As expected, the difference between the post and pre images in a single animal showed a greater effect in the tumor ROI compared to the contralateral ROI (Fig. 1d). Specifically, two hours after injection of the drug combination, there was an average increase in the AACID value of 0.10 ± 0.03 (N = 6, p < 0.05) in the tumor region (Fig. 3a), but no change in AACID value within the contralateral tissue (Fig. 3a). When providing glucose prior to the combined drug injection an even larger increase in AACID value was visible (Fig. 2b) suggesting even greater acidification was achieved. When pre-treating with glucose, there was a significant increase in the AACID value of 0.18 ± 0.03 (N = 6, p < 0.0001) two hours after the combination drug injection in the tumor region (Fig. 3b). However, there was also a small change in the AACID value within the contralateral tissue (Fig. 3b). The difference between the post and pre images in a single animal (Fig. 2d) showed a greater effect in the tumor ROI compared to the contralateral ROI as expected. The measured changes in the average AACID value within the tumor after the combination drug treatment corresponded to a 0.4 pH drop, while pretreatment with glucose produced a 0.72 pH drop, estimated using Eq. 2.

Mouse brain with GBM tumor 11 ± 1 days after implantation: a baseline AACID map prior to drug injection, b the AACID map two hours post drug injection, c coronal fast spin-echo anatomical image showing the ROIs in the tumor (dashed white line) and on the contralateral side (solid white line), and d AACID difference maps for the same brain post–pre drug injection

Mouse brain with GBM tumor 11 ± 1 days after implantation: a baseline AACID map prior to drug and glucose injections, b the AACID map two hours post drug and glucose injections, c coronal fast spin-echo anatomical image showing the ROIs in the tumor (dashed white line) and on the contralateral side (solid white line), and d AACID difference maps for the same brain post–pre drug and glucose injections

Average AACID value in tumor and contralateral ROIs: a pre and post intraperitoneal five drug combination injection (N = 6). b pre and post five drug combination + glucose intraperitoneal injection (N = 6). Error bars represent the standard error of the mean. The asterisks indicated p < 0.05 in repeated measures t test
All animals in Groups T1 and T3 completed 8 days of drug treatment. Representative immunostained brain sections showing the results of cleaved (active) caspase-3 (CC3) immunostaining as a marker of apoptotic cell death are provided in Fig. 4. Control mice without tumors showed almost no evidence of CC3 staining (Fig. 4a). However, within tumors, CC3 staining was increased in the control group (Fig. 4b, d) and after treatment (Fig. 4c, d) in the treatment group. In the mice treated with the five drug combination there was a significant increase in CC3 levels (p = 0.0023) (Figs. 4c, d).

Cleaved Caspase-3 (CC3) Increases in Tumor after Treatment. a–c Representative images of coronal brain sections of a non-tumor control mice b mice with U87 tumors, non-treated receiving vehicle only, and c mice with tumors, treated for 8 days with the five drugs combination. Blue corresponds to nuclear stain (Hoechst), red corresponds to CC3 (apoptotic marker). Scale bar: 0.4 mm. d Quantification of apoptotic cells in (a–c). **indicate p < 0.01
Discussion
This study demonstrates intracellular acidification in brain tumors two hours after injection of a single dose of five drugs designed to block different pH regulatory mechanisms. Tumor acidification was further enhanced by pretreatment with glucose. This in vivo study using an aggressive model of brain cancer is the first to demonstrate the efficacy of a specific combination of drugs and glucose, designed to acidify intracellular pH. The magnitude of these changes is larger than that reported previously for single drug treatment. In contrast, there was no change in brain tissue pH detected on the side contralateral to the tumor after combined drug injection, but there was a small decrease in pH on the contralateral side when glucose was given prior to the combined drug injection. In addition, we have shown that the continued use of these drugs in this animal model of GBM induces apoptosis. Specifically, 8 days of treatment with the five drug combination significantly increase the number of cells positive for cleaved caspase-3 within the tumor.
The drug combination used in the current study was designed to block multiple pH regulatory mechanisms to enhance the acidification produced by any single compound. Quercetin is a natural compound, and a plant product that has been used as a chemotherapeutic agent to treat many cancer types [29]. Quercetin is an MCT inhibitor [27,28,29] that specifically inhibits MCT1 and MCT2 [28]. Pantoprazole (PPZ) has also been shown to be an anti-cancer agent [67] due to its ability to induce cancer cell death by inhibiting the V-ATPase causing reversal of H+ homeostasis. Acetazolamide induces intracellular acidification by inhibiting carbonic anhydrase activity and decreasing aquaporin-1 (AQP1) water channel protein expression [35, 37, 38]. Dichloroacetate (DCA) is a small molecule and has low toxicity compared to other anti-cancer agents. DCA has been shown to induce cell death in several different types of cancer [43]. DCA inhibits pyruvate dehydrogenase kinases (PDKs) and redirects pyruvate back into the mitochondria [41], which reverses the Warburg effect by activating pyruvate dehydrogenase (PDH). However, DCA also decreases the expression of monocarboxylate transporters and V-ATPase [42] in cancer cells reducing pHi in tumors. Cariporide selectively inhibits the sodium proton (Na+/H+) exchange isoform 1 (NHE1) [45] with little effect on other ion transport systems [46]. In malignant tumors NHE1 plays a significant role in maintaining acidic pHe, alkaline pHi, and regulating cell volume [68]. It is activated by growth factors and cellular proliferation processes [69]. Cariporide effectively inhibits NHE1 and has been shown to suppress the invasion and migration of cancer cells [47]. Cariporide also decreased intracellular pH and down-regulated vascular endothelial growth factor (VEGF) secretion in K562 leukemia cells [70]. Therefore, using these specific drugs blocks several known pH regulatory mechanisms: Na+/H+ exchange, monocarboxylate transporters (MCTs), carbonic anhydrase and aquaporin-1(AQP1), pyruvate dehydrogenase kinases (PDKs) and V-ATPase.
In previous studies using the same methodology to evaluate the magnitude of tumor acidification, we found lonidamine decreased pHi by 0.25 at a dose of 50 mg/kg and decreased pHi by 0.45 at a dose of 100 mg/kg [19] while topiramate decreased pHi by 0.17 [20]. More recently we also showed that 200 mg/kg of dichloroacetate decreased pHi by 0.16 [21], and that 6 mg/kg of cariporide decreased pHi by 0.48 [22]. At the dose studied, the combination of drugs used in the current study decreased pHi by 0.4 alone and by 0.72 in combination with glucose. Although the five drug combination produced a pHi change similar to that shown previously with 100 mg/kg of lonidamine and 6 mg/kg of cariporide, the combination used in the current study used mostly drugs considered safe in humans and used a much lower dose of cariporide (one third of that used previously) and DCA (half of that used previously). The dose was lowered in the current study to reduce potential side effects and interactions. Studies in different cells types have shown that there is a critical threshold pHi that may be required to induce apoptosis [17, 71, 72]. Rather than defining this threshold, which in vivo may be variable and depend on factors such as cell type, perfusion status, temperature, and the condition of the microenvironment within the tumor, this study focused on maximizing intracellular acidification to achieve the greatest effect.
In an effort to further enhance intracellular acidification, glucose was injected 20 min prior to drug injection. Cancer cells readily take up and metabolize glucose more so than normal cells, which forms the basis of cancer detection using fluorodeoxyglucose (18F-FDG) positron emission tomography (PET). The end products of glucose metabolism are protons and lactate, which are normally removed from the cell by the pH regulatory mechanism targeted in this study. Blocking these mechanisms 20 min after glucose injection produced a much larger acute intracellular acidification effect. The application of several drugs together, in combination with glucose achieved greater tumor intracellular acidification than any previous single drug studied. Since pH is measured on a logarithmic scale, the decline of 0.72 pH units in the current study represents a twofold increase in [H+] within the cell compared to that previously achieved with cariporide alone [22] and a ninefold increase compared to that that achieved with topiramate [20] and dichloroacetate [21].
To study the long-term effect of intracellular acidification on cancer cell viability in vivo, the five-drug combination was given to mice three times per day for 8 days. Within tumor, there was a significant increase in the number of cells staining positive for cleaved caspase-3, suggesting an increase in the number of cells undergoing apoptosis. There were very few cells undergoing apoptosis in normal brain, after being given the same drug treatment. These in vivo results are consistent with previous studies of cancer cell lines, which have shown that cancer cell acidification can induce apoptosis in a variety of cancer models [73]. The results from the current study provide further evidence that tumor intracellular acidification may provide a benefit for cancer control and treatment.
The current study has several limitations that should be considered. First, the number of animals used was small. However, as in previous studies by our group, the effect sizes are large, and the ROI based pH measurements have low variability. Also, the test–retest design reduces inter-subject variation. Therefore, the animal numbers used were sufficient to determine whether the drug combination could produce a measurable pH effect. Second, we did not optimize the combination dose in the current study, only one dose of drugs was examined. Future studies should determine whether higher doses of these drugs could produce a greater effect, or if a similar effect would be produced with lower doses, decreasing the risk of potential side effects. It should also be established whether the effect is repeatable after multiple exposures and whether the treatment would be more effective is started earlier on in the development of the tumor. The timing and dose of glucose supplementation to increase cellular metabolism prior to drug injection should be studied to optimize intracellular acidification, and consequently cancer cell death. Future studies should also examine whether chronic intracellular acidification could enhance the efficacy of existing chemotherapies to reduce tumor volume, and whether the drug combination used in the current study effectively decreases pHi in other tumor models including patient derived xenograft models.
The use of CEST MRI contrast to detect changes in intracellular pH has many potential clinical applications in cancer detection and treatment evaluation [3, 63]. The results of the current study further demonstrate that acute CEST MRI contrast changes after administration of several drugs in combination with glucose could help localize brain cancer by rapidly and selectively inducing a shift in intracellular pH. The current study demonstrated that the magnitude of intracellular acidification (AACID) of the tumor after combined drug injection was larger when providing glucose as a substrate.
Abbreviations
- GBM:
-
Glioblastoma multiforme
- pHi :
-
Intracellular pH
- pHe :
-
Extracellular pH
- PBS:
-
Phosphate buffered saline
- CEST:
-
Chemical exchange saturation transfer
- MCTs:
-
Monocarboxylate transporters
- RF:
-
Radiofrequency
- MTRasym :
-
Asymmetric magnetization transfer ratio
- MT:
-
Magnetization transfer
- AACID:
-
Amine and amide concentration-independent detection
- FSE:
-
Fast spin-echo
- WASSR:
-
Water saturation shift referencing
- ROI:
-
Region of interest
References
Kanu OO, Mehta A, Di C, Lin N, Bortoff K, Bigner DD, Yan H, Adamson DC (2009) Glioblastoma multiforme: a review of therapeutic targets. Expert Opin Ther Targets 13:701–718. https://doi.org/10.1517/14728220902942348
Wen PY, Kesari S (2008) Malignant gliomas in adults. N Engl J Med 359:492–507. https://doi.org/10.1056/NEJMra0708126
Sagiyama K, Mashimo T, Togao O, Vemireddy V, Hatanpaa KJ, Maher EA, Mickey BE, Pan E, Sherry AD, Bachoo RM, Takahashi M (2014) In vivo chemical exchange saturation transfer imaging allows early detection of a therapeutic response in glioblastoma. Proc Natl Acad Sci USA 111:4542–4547. https://doi.org/10.1073/pnas.1323855111
Easaw JC, Mason WP, Perry J, Laperriere N, Eisenstat DD, Del Maestro R, Belanger K, Fulton D, Macdonald D, Canadian Glioblastoma Recommendations C (2011) Canadian recommendations for the treatment of recurrent or progressive glioblastoma multiforme. Curr Oncol 18:e126–e136
Hart MG, Grant R, Garside R, Rogers G, Somerville M, Stein K (2008) Temozolomide for high grade glioma. Cochrane Database Syst Rev. https://doi.org/10.1002/14651858.CD007415
Gerweck LE, Seetharaman K (1996) Cellular pH gradient in tumor versus normal tissue: potential exploitation for the treatment of cancer. Cancer Res 56:1194–1198
Maintz D, Heindel W, Kugel H, Jaeger R, Lackner KJ (2002) Phosphorus-31 MR spectroscopy of normal adult human brain and brain tumours. NMR Biomed 15:18–27
Lim H, Albatany M, Martínez-Santiesteban F, Bartha R, Scholl TJ (2018) Longitudinal measurements of intra- and extracellular pH gradient in a rat model of glioma. Tomography 4:46–54. https://doi.org/10.18383/j.tom.2018.00001
Gatenby RA, Gillies RJ (2004) Why do cancers have high aerobic glycolysis? Nat Rev Cancer 4:891–899. https://doi.org/10.1038/nrc1478
Liberti MV, Locasale JW (2016) The warburg effect: how does it benefit cancer cells? Trends Biochem Sci 41:211–218. https://doi.org/10.1016/j.tibs.2015.12.001
Webb BA, Chimenti M, Jacobson MP, Barber DL (2011) Dysregulated pH: a perfect storm for cancer progression. Nat Rev Cancer 11:671–677. https://doi.org/10.1038/nrc3110
Shrode LD, Tapper H, Grinstein S (1997) Role of intracellular pH in proliferation, transformation, and apoptosis. J Bioenerg Biomembr 29:393–399
Barar J, Omidi Y (2013) Dysregulated pH in Tumor Microenvironment Checkmates Cancer Therapy. Bioimpacts 3:149–162. https://doi.org/10.5681/bi.2013.036
Archetti M (2015) Heterogeneity and proliferation of invasive cancer subclones in game theory models of the Warburg effect. Cell Prolif 48:259–269. https://doi.org/10.1111/cpr.12169
Dang CV, Kim JW (2018) Convergence of Cancer Metabolism and Immunity: an Overview. Biomol Ther 26:4–9. https://doi.org/10.4062/biomolther.2017.194
Wood PJ, Sansom JM, Newell K, Tannock IF, Stratford IJ (1995) Reduction of tumour intracellular pH and enhancement of melphalan cytotoxicity by the ionophore Nigericin. Int J Cancer 60:264–268
Park HJ, Lyons JC, Ohtsubo T, Song CW (1999) Acidic environment causes apoptosis by increasing caspase activity. Br J Cancer 80:1892–1897. https://doi.org/10.1038/sj.bjc.6690617
Neri D, Supuran CT (2011) Interfering with pH regulation in tumours as a therapeutic strategy. Nat Rev Drug Discov 10:767–777. https://doi.org/10.1038/nrd3554
McVicar N, Li AX, Meakin SO, Bartha R (2015) Imaging chemical exchange saturation transfer (CEST) effects following tumor-selective acidification using lonidamine. NMR Biomed 28:566–575. https://doi.org/10.1002/nbm.3287
Marathe K, McVicar N, Li A, Bellyou M, Meakin S, Bartha R (2016) Topiramate induces acute intracellular acidification in glioblastoma. J Neurooncol 130:465–472. https://doi.org/10.1007/s11060-016-2258-y
Albatany M, Li A, Meakin S, Bartha R (2017) Dichloroacetate induced intracellular acidification in glioblastoma: in vivo detection using AACID-CEST MRI at 94 Tesla. J Neuro-Oncol 136(2):255–262. https://doi.org/10.1007/s11060-017-2664-9
Albatany M, Li A, Meakin S, Bartha R (2018) In vivo detection of acute intracellular acidification in glioblastoma multiforme following a single dose of cariporide. Int J Clin Oncol 23(5):812–819. https://doi.org/10.1007/s10147-018-1289-0
Albatany M, Meakin S, Bartha R (2018) The monocarboxylate transporter inhibitor quercetin induces intracellular acidification in a mouse model of glioblastoma multiforme: in-vivo detection using magnetic resonance imaging. Invest New Drugs. https://doi.org/10.1007/s10637-018-0644-3
Spugnini EP, Citro G, Fais S (2010) Proton pump inhibitors as anti vacuolar-ATPases drugs: a novel anticancer strategy. J Exp Clin Cancer Res 29:44. https://doi.org/10.1186/1756-9966-29-44
Slepkov ER, Rainey JK, Sykes BD, Fliegel L (2007) Structural and functional analysis of the Na+/H+ exchanger. Biochem J 401:623–633. https://doi.org/10.1042/BJ20061062
Boron WF (2004) Regulation of intracellular pH. Adv Physiol Educ 28:160–179. https://doi.org/10.1152/advan.00045.2004
Kim JH, Kim SH, Alfieri AA, Young CW (1984) Quercetin, an inhibitor of lactate transport and a hyperthermic sensitizer of HeLa cells. Cancer Res 44:102–106
Perez-Escuredo J, Van Hee VF, Sboarina M, Falces J, Payen VL, Pellerin L, Sonveaux P (2016) Monocarboxylate transporters in the brain and in cancer. Biochim Biophys Acta 1863:2481–2497. https://doi.org/10.1016/j.bbamcr.2016.03.013
Izumi H, Takahashi M, Uramoto H, Nakayama Y, Oyama T, Wang KY, Sasaguri Y, Nishizawa S, Kohno K (2011) Monocarboxylate transporters 1 and 4 are involved in the invasion activity of human lung cancer cells. Cancer Sci 102:1007–1013. https://doi.org/10.1111/j.1349-7006.2011.01908.x
Volk C, Kempski B, Kempski OS (1997) Inhibition of lactate export by quercetin acidifies rat glial cells in vitro. Neurosci Lett 223:121–124
Reddy NS, Nirmala P, Chidambaram N, Kumar P, Nagar A (2012) Quercetin in dimethyl benzanthracene induced breast cancer in rats. Am J Pharmacol Toxicol 7:68–72
Yeo M, Kim DK, Kim YB, Oh TY, Lee JE, Cho SW, Kim HC, Hahm KB (2004) Selective induction of apoptosis with proton pump inhibitor in gastric cancer cells. Clin Cancer Res 10:8687–8696. https://doi.org/10.1158/1078-0432.CCR-04-1065
Brana I, Ocana A, Chen EX, Razak AR, Haines C, Lee C, Douglas S, Wang L, Siu LL, Tannock IF, Bedard PL (2014) A phase I trial of pantoprazole in combination with doxorubicin in patients with advanced solid tumors: evaluation of pharmacokinetics of both drugs and tissue penetration of doxorubicin. Invest New Drugs 32:1269–1277. https://doi.org/10.1007/s10637-014-0159-5
Ameli PA, Madan M, Chigurupati S, Yu A, Chan SL, Pattisapu JV (2012) Effect of acetazolamide on aquaporin-1 and fluid flow in cultured choroid plexus. Acta Neurochir Suppl 113:59–64. https://doi.org/10.1007/978-3-7091-0923-6_13
Gao J, Wang X, Chang Y, Zhang J, Song Q, Yu H, Li X (2006) Acetazolamide inhibits osmotic water permeability by interaction with aquaporin-1. Anal Biochem 350:165–170. https://doi.org/10.1016/j.ab.2006.01.003
Altıntop MD, Sever B, Özdemir A, Kucukoglu K, Onem H, Nadaroglu H, Kaplancıklı ZA (2017) Potential inhibitors of human carbonic anhydrase isozymes I and II: design, synthesis and docking studies of new 1,3,4-thiadiazole derivatives. Bioorg Med Chem 25:3547–3554. https://doi.org/10.1016/j.bmc.2017.05.005
Said HM, Hagemann C, Carta F, Katzer A, Polat B, Staab A, Scozzafava A, Anacker J, Vince GH, Flentje M, Supuran CT (2013) Hypoxia induced CA9 inhibitory targeting by two different sulfonamide derivatives including acetazolamide in human glioblastoma. Bioorg Med Chem 21:3949–3957. https://doi.org/10.1016/j.bmc.2013.03.068
Cazzamalli S, Corso AD, Neri D (2017) Linker stability influences the anti-tumor activity of acetazolamide-drug conjugates for the therapy of renal cell carcinoma. J Control Release 246:39–45. https://doi.org/10.1016/j.jconrel.2016.11.023
Xiang Y, Ma B, Li T, Gao JW, Yu HM, Li XJ (2004) Acetazolamide inhibits aquaporin-1 protein expression and angiogenesis. Acta Pharmacol Sin 25:812–816
Mohammadpour R, Safarian S, Ejeian F, Sheikholya-Lavasani Z, Abdolmohammadi MH, Sheinabi N (2014) Acetazolamide triggers death inducing autophagy in T-47D breast cancer cells. Cell Biol Int 38:228–238. https://doi.org/10.1002/cbin.10197
Cairns RA, Papandreou I, Sutphin PD, Denko NC (2007) Metabolic targeting of hypoxia and HIF1 in solid tumors can enhance cytotoxic chemotherapy. Proc Natl Acad Sci USA 104:9445–9450. https://doi.org/10.1073/pnas.0611662104
Kumar A, Kant S, Singh SM (2013) Antitumor and chemosensitizing action of dichloroacetate implicates modulation of tumor microenvironment: a role of reorganized glucose metabolism, cell survival regulation and macrophage differentiation. Toxicol Appl Pharmacol 273:196–208. https://doi.org/10.1016/j.taap.2013.09.005
Ishiguro T, Ishiguro M, Ishiguro R, Iwai S (2012) Cotreatment with dichloroacetate and omeprazole exhibits a synergistic antiproliferative effect on malignant tumors. Oncol Lett 3:726–728. https://doi.org/10.3892/ol.2012.552
Kankotia S, Stacpoole PW (2014) Dichloroacetate and cancer: new home for an orphan drug? Biochim Biophys Acta 1846:617–629. https://doi.org/10.1016/j.bbcan.2014.08.005
Harguindey S, Arranz JL, Orozco JDP, Rauch C, Fais S, Cardone RA, Reshkin SJ (2013) Cariporide and other new and powerful NHE1 inhibitors as potentially selective anticancer drugs—an integral molecular/biochemical/metabolic/clinical approach after one hundred years of cancer research. Journal of Translational Medicine 11:282. https://doi.org/10.1186/1479-5876-11-282
Scholz W, Albus U, Counillon L, Gogelein H, Lang HJ, Linz W, Weichert A, Scholkens BA (1995) Protective effects of Hoe642, a selective sodium-hydrogen exchange subtype-1 inhibitor, on cardiac ischemia and reperfusion. Cardiovasc Res 29:260–268. https://doi.org/10.1016/S0008-6363(96)88579-8
Lv C, Yang X, Yu B, Ma Q, Liu B, Liu Y (2012) Blocking the Na+/H+ exchanger 1 with cariporide (HOE642) reduces the hypoxia-induced invasion of human tongue squamous cell carcinoma. Int J Oral Max Surg 41:1206–1210. https://doi.org/10.1016/j.ijom.2012.03.001
Ren SC, Suo QF, Du WT, Pan H, Yang MM, Wang RH, Liu J (2010) Quercetin permeability across blood-brain barrier and its effect on the viability of U251 cells. Sichuan Da Xue Xue Bao Yi Xue Ban 41(751–754):759
Moon YJ, Wang L, DiCenzo R, Morris ME (2008) Quercetin pharmacokinetics in humans. Biopharm Drug Dispos 29:205–217. https://doi.org/10.1002/bdd.605
Ortiz-Guerrero G, Amador-Muñoz D, Calderón-Ospina CA, López-Fuentes D, Nava Mesa MO (2018) Proton pump inhibitors and dementia: physiopathological mechanisms and clinical consequences. Neural Plast 2018:5257285. https://doi.org/10.1155/2018/5257285
Shin JM, Kim N (2013) Pharmacokinetics and pharmacodynamics of the proton pump inhibitors. J Neurogastroenterol Motil 19:25–35. https://doi.org/10.5056/jnm.2013.19.1.25
Bonnet S, Archer SL, Allalunis-Turner J, Haromy A, Beaulieu C, Thompson R, Lee CT, Lopaschuk GD, Puttagunta L, Bonnet S, Harry G, Hashimoto K, Porter CJ, Andrade MA, Thebaud B, Michelakis ED (2007) A mitochondria-K+ channel axis is suppressed in cancer and its normalization promotes apoptosis and inhibits cancer growth. Cancer Cell 11:37–51. https://doi.org/10.1016/j.ccr.2006.10.020
James MO, Stacpoole PW (2016) Pharmacogenetic considerations with dichloroacetate dosing. Pharmacogenomics 17:743–753. https://doi.org/10.2217/pgs-2015-0012
Hanson MA, Nye PC, Torrance RW (1981) The location of carbonic anhydrase in relation to the blood-brain barrier at the medullary chemoreceptors of the cat. J Physiol 320:113–125. https://doi.org/10.1113/jphysiol.1981.sp013938
Hampson AJ, Babalonis S, Lofwall MR, Nuzzo PA, Krieter P, Walsh SL (2016) A pharmacokinetic study examining acetazolamide as a novel adherence marker for clinical trials. J Clin Psychopharmacol 36:324–332. https://doi.org/10.1097/JCP.0000000000000529
O'Donnell ME, Chen YJ, Lam TI, Taylor KC, Walton JH, Anderson SE (2013) Intravenous HOE-642 reduces brain edema and Na uptake in the rat permanent middle cerebral artery occlusion model of stroke: evidence for participation of the blood-brain barrier Na/H exchanger. J Cereb Blood Flow Metab 33:225–234. https://doi.org/10.1038/jcbfm.2012.160
Mokgokong R, Wang S, Taylor CJ, Barrand MA, Hladky SB (2014) Ion transporters in brain endothelial cells that contribute to formation of brain interstitial fluid. Pflugers Arch 466:887–901. https://doi.org/10.1007/s00424-013-1342-9
Ballabh P, Braun A, Nedergaard M (2004) The blood-brain barrier: an overview: structure, regulation, and clinical implications. Neurobiol Dis 16:1–13
Li AX, Suchy M, Li C, Gati JS, Meakin S, Hudson RH, Menon RS, Bartha R (2011) In vivo detection of MRI-PARACEST agents in mouse brain tumors at 9.4 T. Magn Reson Med 66:67–72. https://doi.org/10.1002/mrm.22772
Kim M, Gillen J, Landman BA, Zhou J, van Zijl PC (2009) Water saturation shift referencing (WASSR) for chemical exchange saturation transfer (CEST) experiments. Magn Reson Med 61:1441–1450. https://doi.org/10.1002/mrm.21873
McVicar N, Li AX, Goncalves DF, Bellyou M, Meakin SO, Prado MA, Bartha R (2014) Quantitative tissue pH measurement during cerebral ischemia using amine and amide concentration-independent detection (AACID) with MRI. J Cereb Blood Flow Metab 34:690–698. https://doi.org/10.1038/jcbfm.2014.12
Zong X, Wang P, Kim SG, Jin T (2014) Sensitivity and source of amine-proton exchange and amide-proton transfer magnetic resonance imaging in cerebral ischemia. Magn Reson Med 71:118–132. https://doi.org/10.1002/mrm.24639
Zhou JY, Payen JF, Wilson DA, Traystman RJ, van Zijl PCM (2003) Using the amide proton signals of intracellular proteins and peptides to detect pH effects in MRI. Nat Med 9:1085–1090. https://doi.org/10.1038/nm907
Murray RKGD (2003) Membranes: structure & function. McGraw-Hill Companies Inc, New York, pp 415–433
Ostapchenko VG, Beraldo FH, Guimarães AL, Mishra S, Guzman M, Fan J, Martins VR, Prado VF, Prado MA (2013) Increased prion protein processing and expression of metabotropic glutamate receptor 1 in a mouse model of Alzheimer's disease. J Neurochem 127:415–425. https://doi.org/10.1111/jnc.12296
Ostapchenko VG, Chen M, Guzman MS, Xie YF, Lavine N, Fan J, Beraldo FH, Martyn AC, Belrose JC, Mori Y, MacDonald JF, Prado VF, Prado MA, Jackson MF (2015) The transient receptor potential melastatin 2 (TRPM2) channel contributes to β-amyloid oligomer-related neurotoxicity and memory impairment. J Neurosci 35:15157–15169. https://doi.org/10.1523/JNEUROSCI.4081-14.2015
Vishvakarma NK, Singh SM (2010) Immunopotentiating effect of proton pump inhibitor pantoprazole in a lymphoma-bearing murine host: implication in antitumor activation of tumor-associated macrophages. Immunol Lett 134:83–92. https://doi.org/10.1016/j.imlet.2010.09.002
McLean LA, Roscoe J, Jorgensen NK, Gorin FA, Cala PM (2000) Malignant gliomas display altered pH regulation by NHE1 compared with nontransformed astrocytes. Am J Physiol Cell Physiol 278:C676–688
Lagana A, Vadnais J, Le PU, Nguyen TN, Laprade R, Nabi IR, Noel J (2000) Regulation of the formation of tumor cell pseudopodia by the Na+/H+ exchanger NHE1. J Cell Sci 113:3649–3662
Gao W, Chang GQ, Wang J, Jin WN, Wang LH, Lin YN, Li HW, Ma L, Li QH, Pang TX (2011) Inhibition of K562 leukemia angiogenesis and growth by selective Na+/H+ exchanger inhibitor cariporide through down-regulation of pro-angiogenesis factor VEGF. Leukemia Res 35:1506–1511. https://doi.org/10.1016/j.leukres.2011.07.001
Chen Q, Benson RS, Whetton AD, Brant SR, Donowitz M, Montrose MH, Dive C, Watson AJ (1997) Role of acid/base homeostasis in the suppression of apoptosis in haemopoietic cells by v-Abl protein tyrosine kinase. J Cell Sci 110(Pt 3):379–387
Rich IN, Worthington-White D, Garden OA, Musk P (2000) Apoptosis of leukemic cells accompanies reduction in intracellular pH after targeted inhibition of the Na(+)/H(+) exchanger. Blood 95:1427–1434
De Milito A, Iessi E, Logozzi M, Lozupone F, Spada M, Marino ML, Federici C, Perdicchio M, Matarrese P, Lugini L, Nilsson A, Fais S (2007) Proton pump inhibitors induce apoptosis of human B-cell tumors through a caspase-independent mechanism involving reactive oxygen species. Cancer Res 67:5408–5417. https://doi.org/10.1158/0008-5472.CAN-06-4095
Acknowledgements
Funding for this study was provided by the Ontario Institute of Cancer Research (OICR) Smarter Imaging Program, the Canadian Institutes of Health Research (CIHR), the Canada First Research Excellence Fund, and Brain Canada. Thanks to Misan University-Ministry of Higher Education and Scientific Research, Iraq.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
The authors declare no potential conflicts of interest.
Ethical approval
All applicable national and institutional guidelines for the care and use of animals were followed. All procedures performed in studies involving animals were in accordance with the ethical standards of the institution or practice at which the studies were conducted.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
About this article

Cite this article
Albatany, M., Ostapchenko, V.G., Meakin, S. et al. Brain tumor acidification using drugs simultaneously targeting multiple pH regulatory mechanisms. J Neurooncol 144, 453–462 (2019). https://doi.org/10.1007/s11060-019-03251-7
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s11060-019-03251-7