
Abstract
Purpose
Both IDH1-mutated and wild-type gliomas abundantly display aberrant CpG island hypermethylation. However, the potential role of hypermethylation in promoting gliomas, especially the most aggressive form, glioblastoma (GBM), remains poorly understood.
Methods
We analyzed RRBS-generated methylation profiles for 11 IDH1WT gliomas (including 7 GBMs), 24 IDH1MUT gliomas (including 6 GBMs), and 5 normal brain samples and employed TCGA GBM methylation profiles as a validation set. Upon classification of differentially methylated CpG islands by IDH1 status, we used integrated analysis of methylation and gene expression to identify SPINT2 as a top cancer related gene. To explore functional consequences of SPINT2 methylation in GBM, we validated SPINT2 methylation status using targeted bisulfite sequencing in a large cohort of GBM samples. We assessed DNA methylation-mediated SPINT2 gene regulation using 5-aza-2′-deoxycytidine treatment, DNMT1 knockdown and luciferase reporter assays. We conducted functional analyses of SPINT2 in GBM cell lines in vitro and in vivo.
Results
We identified SPINT2 as a candidate tumor-suppressor gene within a group of CpG islands (designated GT-CMG) that are hypermethylated in both IDH1MUT and IDH1WT gliomas but not in normal brain. We established that SPINT2 downregulation results from promoter hypermethylation, and that restoration of SPINT2 expression reduces c-Met activation and tumorigenic properties of GBM cells.
Conclusions
We defined a previously under-recognized group of coordinately methylated CpG islands common to both IDH1WT and IDH1MUT gliomas (GT-CMG). Within GT-CMG, we identified SPINT2 as a top cancer-related candidate and demonstrated that SPINT2 suppressed GBM via down-regulation of c-Met activation.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Glioblastoma multiforme (GBM) is the most prevalent and most lethal form of brain cancer [1, 2], affecting 15,000 new patients yearly in the United States. Median survival with this type of cancer is 14.6 months, with only 3–5% of patients surviving over 5 years post-diagnosis [3]. This poor prognosis likely results from genetic and epigenetic influences, which vary between discrete subsets of patients [4, 5].
The silencing of endogenous tumor-suppressor genes by methylation of discrete CpG islands located within their promoter regions has been identified as a key process in gliomagenesis [1, 6, 7]. Following the recent discovery that somatic mutations to the isocitrate dehydrogenase 1 and 2 (IDH1/2) genes are present in a number of human cancers [8,9,10,11,12,13,14] and a majority of secondary glioblastomas [1, 15], genome-wide DNA methylation profiling of GBM and lower grade gliomas has identified several distinct methylation patient clusters, most notably the IDHMUT-associated glioma CpG island methylator phenotype (G-CIMP) [5, 6, 16,17,18].
Enhanced c-Met activation via HGF has been reported to promote growth, angiogenesis, invasion, and stem cell survival in GBM [19,20,21,22,23,24]. Serine Protease Inhibitor, Kunitz Type 2 (SPINT2) is a major inhibitor of hepatocyte growth factor activator (HGFA). HGFA is the primary enzyme catalyzing the conversion of pro-HGF to the active c-Met ligand HGF [25, 26]. While SPINT2 hypermethylation has been previously reported in several cancers [27,28,29,30], reports of SPINT2 hypermethylation in GBM have been limited [30, 31].
By performing methylation profiling of patient glioma samples, we confirmed a large set of CpG islands coordinately methylated in both IDH1WT and IDH1MUT gliomas (abbreviated as GT-CMG), which was potentially recognizable in other published methylomic datasets [5, 16, 17] but had yet to be clearly delineated. By applying unbiased bioinformatic criteria to GT-CMG, we identified SPINT2 as one of the top candidate tumor-suppressor genes that was hypermethylated and downregulated in IDH1WT GBMs. Furthermore, we confirmed that CpG island promoter methylation silenced SPINT2, and restoration of SPINT2 suppressed growth and migration of GBM cells by downregulating c-Met activation. Thus, our data supports a clinically relevant model for c-Met activation in GBM, in which SPINT2 methylation/downregulation releases the suppression of serine proteases such as HGFA on pro-HGF conversion and enables overactive c-Met activation.
Materials and methods
Details regarding cell cultures and pharmacological treatments, patient glioma specimens, methylation and expression data, in vitro and in vivo protocols and all data analyses are detailed in Online Resource 1_Supplemental Materials and Methods.
Results
GT-CMG: a group of CpG islands coordinately methylated in both IDH1 WT and IDH1 MUT gliomas
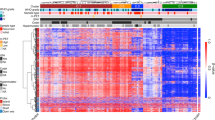
In order to classify groups of hypermethylated islands in terms of IDH1 genotype, we used our reduced representation bisulfite sequencing (RRBS) data to identify differentially methylated CpG islands depicted in a heatmap (Online Resource 2_Suppl. Figure 1a). First, as expected, gliomas demonstrated abundant hypermethylation as compared to normal brain. Instead of looking for methylation patient clusters (or CIMPs), we observed three sets of differentially methylated CpG islands based on whether they were methylated in IDH1MUT, IDH1WT, or both (Online Resource 2_Suppl. Figure 1a). We defined these groups as a Coordinately Methylated Group (CMG) of CpG islands in order to distinguish them from a CIMP. Thus, we designated: (1) Glioma-tumor-CMG (GT-CMG) as the set containing CpG islands methylated in both IDH1WT and IDH1MUT gliomas; (2) Glioma-IDH1MUT-CMG (GM-CMG) as the set containing CpG islands methylated in IDH1MUT gliomas only; and (3) Glioma-IDH1WT-CMG (GW-CMG) as the set containing CpG islands methylated in IDH1WT gliomas only (Online Resource 3_Suppl. Table 1). GT-CMG consisted of 1743 CpG islands exhibiting hypermethylation across both IDH1WT and IDH1MUT gliomas. GM-CMG exhibited hypermethylation in only IDH1MUT samples and consisted of 1421 CpG islands, which as expected exhibited high overlap with G-CIMP in IDH1MUT versus IDH1WT GBMs, with 84.4% overlap (Online Resource 3_Suppl. Tables 2, 3). Representing a much smaller group, GW-CMG consisted of 137 CpG islands hypermethylated in only IDH1WT samples (Online Resource 3_Suppl. Table 1).
In order to validate the CMG modules observed in our RRBS data in an independent dataset, methylation array data for 422 GBM samples (282 IDH1WT, 27 IDH1MUT, 113 unknown IDH1) obtained directly from TCGA (https://portal.gdc.cancer.gov/projects/TCGA-GBM) were similarly analyzed. This included 282 and 140 samples, generated via the Illumina Infinium Human Methylation 27 and 450 arrays, respectively. Because the TCGA GBM methylation data lacked normal brain samples, Illumina Infinium Human Methylation 450 platform data for 12 normal tissue samples were obtained from previously published work by Nardone et al. [32]. Similar to our RRBS results, comparisons of IDH1WT GBM, IDH1MUT GBM, and normal samples resulted in 3 distinct groups of CpG islands: GT-CMG, with 3115 CpG islands; GM-CMG, with 293 CpG islands; and GW-CMG, with 210 CpG islands (Online Resource 2_Suppl. Figure 1b; Online Resource 3_Suppl. Table 4). In addition to validating the presence of the three groups observed in the RRBS data, we also observed a small group of CpG islands that were hypomethylated in tumors versus normal.
We further validated our CMG classification by selecting 9 GT-CMG and 2 GW-CMG genes/CpG islands and performed targeted bisulfite sequencing (BiSeq) on patient GBM samples (Online Resource 4_Suppl. Table 5).
Identification of candidate tumor-suppressor genes within GT-CMG by integrated analysis of expression and methylation
In order to identify candidate tumor suppressors within GT-CMG, we applied bioinformatic filtering based on CpG island position within the gene and gene expression. Using genome annotation data downloaded directly from the UCSC genome browser (https://genome.ucsc.edu), we found 496 of 1743 GT-CMG CpG islands overlapping with promoter regions of known RefSeq genes. Differential gene expression between 573 GBM samples and 10 normal brain samples was determined using TCGA GBM gene expression array data (Online Resource 2_Suppl. Figure 1b; Online Resource 4_Suppl. Table 6). Of the 496 promoter-associated CpG islands, this filter yielded 58 corresponding genes exhibiting down-regulation in a tumor versus normal comparison, with a minimum threefold change and p < 1.0 × 10− 5. The top 10 genes, ranked by the significance of differential methylation, are shown in (Online Resource 4_Suppl. Table 7).
SPINT2 is one of the top cancer-related GT-CMG candidates with down-regulated gene expression in GBM patient tumors and cell lines
Identified within the top 10 GT-CMG genes (Online Resource 4_Suppl. Table 7), SPINT2 is an upstream regulator of the HGF/c-Met pathway [33] whose silencing may result in overactive c-Met activation by HGF. The SPINT2-associated CpG island revealed differential methylation of 0.38 (p = 8.3 × 10− 14) in gliomas as compared to normal samples in our RRBS screening (Online Resource 4_Suppl. Table 7). This is confirmed by inspection of RRBS data for SPINT2 CpG islands for gliomas (Online Resource 2_Suppl. Figure 2) including GBMs (Fig. 1a). We further validated tumor hypermethylation of the SPINT2 promoter associated CpG island using BiSeq, where we found hypermethylation in 47/74 IDH1WT GBM, 8/8 IDH1MUT GBM and 0/12 normal samples (Online Resource 4_Suppl. Table 5). As found from our filter, TCGA array data for 145 GBM (115 IDH1WT, 30 IDH1MUT) and 10 normal samples revealed 5.03-fold downregulation (p = 6.5 × 10− 10) of SPINT2 gene expression in GBMs (Online Resource 4_Suppl. Table 7), and the downregulation was associated with DNA hypermethylation (Fig. 1b). To confirm SPINT2 down-regulation in GBMs, we measured SPINT2 gene expression by qPCR in 10 normal brain tissues and 22 IDH1WT GBM samples and found SPINT2 gene expression was significantly lower in the hypermethylated GBM samples, compared to unmethylated GBM samples (Fig. 1c). We did not test IDH1MUT samples since they all appear to be methylated. Interestingly, even unmethylated GBMs had lower expression of SPINT2 compared to normal brain (Fig. 1b, c).

SPINT2 is hypermethylated and downregulated in GBMs compared to normal brain tissues. a Methylation profile of the SPINT2 associated CpG island via RRBS. Upper: map of the SPINT2 promoter region, showing position of CpG island, transcription start site (arrow), exon 1 (shaded box) and BiSeq primers (double headed arrows, Chr 19: 38,754,739–38,755,328); Lower: representative CpG site methylation pattern of IDH1WT gliomas or IDH1MUT GBMs and normal brain determined by RRBS. b Analysis of TCGA data demonstrates that SPINT2 expression was down-regulated in SPINT2 hypermethylated versus unmethylated GBMs, p = 0.026. c In our set of GBM samples, SPINT2 expression measured by qPCR was also down-regulated in SPINT2 hypermethylated versus unmethylated GBMs as determined by targeted BiSeq. p = 4.19 × 10−4
Silenced SPINT2 can be re-expressed by pharmacological and genetic disruption of DNA methyltransferases
We determined methylation status and gene expression of SPINT2 by BiSeq and qRT-PCR, respectively, in 6 GBM and 2 non-neoplastic cell lines (hTERT-immortalized astrocytes and HEK-293T cells). We found that SPINT2 was hypermethylated in all tested GBM cell lines and unmethylated in the 2 non-malignant cell lines. As expected, SPINT2 was highly expressed in the 2 non-malignant cell lines but significantly downregulated in GBM cell lines. In addition, SPINT2 was hypermethylated in 7 patient-derived GBM neurosphere lines and all 7 lines demonstrated silenced SPINT2 gene expression (Fig. 2a).

SPINT2 expression is regulated by promoter methylation. aSPINT2 was downregulated in promoter-methylated GBM cell lines and GBM neurosphere lines measured by qPCR. NHA and 293T are non-malignant cell lines; D54, U87, U251, U373, LN18 and T98 are GBM cell lines; HK207, HK250, HK261, HK308, HK211, HK213 and HK217 are GBM patient-derived neurospheres. bSPINT2 expression was upregulated in hypermethylated cell lines by treatment with demethylating agent 5-aza-CdR. SPINT2 expression was measured by qPCR in 293T, NHA, U251, T98G and LN18 cells treated with DMSO or 5-aza-CdR for 72 h (n = 3, mean ± SEM). c SPINT2 protein was upregulated in LN18 cells treated with 5-aza-CdR for 72 h (n = 3, representative figure shown). dDNMT1-siRNA transfection, three times every 2 days, achieved 80% downregulation of DNMT1, while SPINT2 gene expression was upregulated around threefold in LN18 cells (n = 2, mean ± SEM). eSPINT2 promoter activity was decreased by DNA methylation modification by HhaI in NHA cells. SV40 promoter, lacking “GCGC” sites was used as a negative control. HSV TK promoter, containing multiple “GCGC” sites, was used as a positive control. The Renilla luciferase vector was used as an internal control. SPINT2 promoter activity was measured as the mean value of Firefly/Renilla luciferase activity (n = 3, mean ± SEM). *p < 0.05; **p < 0.01; ***p < 0.001
To investigate whether methylation was responsible for silenced SPINT2 expression, we treated GBM cell lines (U251, T98G, LN18) harboring SPINT2 hypermethylation with 5-aza-2′-deoxycytidine (5-aza-CdR), a pharmacological demethylating agent. The treatment resulted in a substantial increase (> 100-fold change) in SPINT2 mRNA expression as compared to non-treated cells (Fig. 2b). Interestingly, NHA cells also showed a modest increase in expression. Moreover, we found that increased SPINT2 expression was dose-dependent (Online Resource 2_Suppl. Figure 3a). As expected, SPINT2 protein levels were also upregulated in a dose-dependent manner after 3 days of treatment with 5-aza-CdR (Fig. 2c). To provide further evidence, we treated U251 cells with a low dose of 5-aza-CdR for 9 days, and found that SPINT2 expression was greatly increased (Online Resource 2_Suppl. Figure 3b), and that this increase was associated with demethylation of the SPINT2 promoter (Online Resource 2_Suppl. Figure 3c). In addition to pharmacological treatment, we conducted siRNA-based knockdown of DNA (Cytosine-5)-Methyltransferase 1 (DNMT1) in LN18 cells and observed a threefold increase in the expression of SPINT2 as compared to nonspecific siRNA-treated cells (Fig. 2d). Taken together, our in vitro data combined with clinical sample data strongly suggests that SPINT2 gene expression is regulated by promoter CpG island methylation.
SPINT2 promoter activity is regulated by DNA methylation
To directly demonstrate regulation of SPINT2 promoter activity by methylation, we generated 4 reporter constructs by inserting the SPINT2 promoter and 3 control promoters (CMV, SV40, HSV TK) into the promoter free pGL4.17 reporter plasmid. The luciferase assay showed that SPINT2 promoter activity was comparable to the control promoters, indicating that the SPINT2 promoter was highly active in driving transcription (Online Resource 2_Suppl. Figure 4a). Transfection of NHA cells also demonstrated that the SPINT2 promoter was highly active (Online Resource 2_Suppl. Figure 4b).
To determine the role of methylation in regulation of SPINT2 promoter activity, we treated the three plasmid constructs in vitro with HhaI methyltransferase, which specifically methylates the first cytosine of GCGC DNA sites. We used the SV40 promoter construct, lacking GCGC sites, as a negative control for methylation-dependent expression regulation; we used the TK promoter construct, containing multiple GCGC sites, as a positive control (Online Resource 2_Suppl. Figure 5a). Treated constructs were then transfected into 293T and NHA cells. As expected, HhaI methylation modification did not alter SV40 promoter activity. In contrast, HhaI methylation dramatically reduced SPINT2 and TK promoter activities in NHA and 293T cells (Fig. 2e and Online Resource 2_Suppl. Figure 5b). These results demonstrated direct regulation of SPINT2 transcription via promoter methylation.
SPINT2 exerts tumor-suppressive properties in GBM cell lines
To determine whether SPINT2 could exert tumor suppression, we achieved stable SPINT2 overexpression by retroviral infection (pLPCX, Clontech) in LN18 and U87 cells. These cell lines were selected because both demonstrated simultaneous expression of HGF and c-Met (Online Resource 2_Suppl. Figure 6). We confirmed SPINT2 overexpression by Western blot (Fig. 3a). As compared to pLPCX-control cells, Transwell invasion assays (Fig. 3b) showed that SPINT2 overexpression strongly arrested cell invasion of LN18 cells. In addition, SPINT2 overexpression clearly inhibited cell migration as measured by the wound healing assay in LN18 cells (Fig. 3c). SPINT2 overexpression significantly reduced cell proliferation as measured by the MTT assay both in LN18 (Fig. 3d) and U87 cells (Fig. 3e). SPINT2 overexpression also significantly reduced cell colony formation as measured by the colony growth assay in LN18 (Fig. 3f) and U87 cells (Fig. 3g), and anchorage-independent growth as demonstrated by soft agar growth assays in LN18 (Fig. 3h) and U87 cells (Fig. 3i). By showing reduced cell invasion, migration, and proliferation, these assays demonstrate the tumor-suppressive properties of SPINT2 overexpression in vitro.

SPINT2 overexpression blocks glioma cell invasion, migration and proliferation. a Stable overexpression of SPINT2 in LN18 and U87 cell lines was achieved by retroviral infection and puromycin selection. bSPINT2 overexpression reduced LN18 cell invasion in the Transwell migration assay (n = 6, mean ± SEM). cSPINT2 overexpression inhibited the migration of LN18 cells in the wound healing assay (n = 6, mean ± SEM). d, eSPINT2 overexpression reduced cell proliferation (MTT assay) in LN18 (n = 8) and U87 (n = 3) cells (mean ± SEM). f, gSPINT2 overexpression reduced colony formation in LN18 (n = 7) and U87 (n = 3) cells (mean ± SEM). h, iSPINT2 overexpression reduced anchorage-independent growth in LN18 (n = 3) and U87 (n = 4) cells (mean ± SEM). *p < 0.05; **p < 0.01; ***p < 0.001
Conditioned medium from SPINT2-overexpressing cells (SPINT2-CM) and recombinant SPINT2 (rSPINT2) application reduce c-Met phosphorylation and growth in GBM cell lines
Based on previous reports that secreted SPINT2 can inhibit HGFA-mediated conversion of pro-HGF to active HGF [33, 34], we tested whether GBM cells shed SPINT2 proteins extracellularly by harvesting starvation-derived conditioned medium (CM) from LN18 cells (FBS-free, 24 h starvation). Western blot analysis of concentrated proteins (molecular weight ≥ 10 kD) demonstrated the presence of SPINT2 protein (~ 30 kD) in the conditioned medium (Fig. 4a). In comparison to protein extracts of LN18 cells, concentrated CM proteins, as expected, did not contain α-tubulin protein, excluding the possibility that conditioned medium was contaminated with cell-derived SPINT2 (Fig. 4a). By applying starvation-derived CM from LN18-SPINT2 cells (SPINT2-CM) to LN18-pLPCX cells, we found that SPINT2-CM inhibited c-Met phosphorylation in LN18-pLPCX cells in the presence of FBS/HGF (Fig. 4b) (note that serum provides bovine HGFA [26, 28]). SPINT2-CM treated LN18-pLPCX cells (Fig. 4c) and U87-pLPCX cells (Fig. 4d) showed reduced growth compared with pLPCX-CM treated cells. SPINT2-CM treated LN18-pLPCX cells also showed reduced cell invasion in the Transwell migration assay (Fig. 4e). Similarly, recombinant SPINT2 (rSPINT2) was found to reduce c-Met phosphoryation compared with vehicle to a level approaching that of the c-Met receptor inhibitor, iMet (Fig. 4f, upper panel). As expected, treatment with rSPINT2 also significantly reduced LN18 cell growth compared with vehicle-treated cells, and comparably with iMet (Fig. 4f, lower panel). A reduction in c-Met activation was also noted in SPINT2-CM treated SPINT2-overexpressing LN18 cells (Online Resource 2_Suppl. Figure 7).

SPINT2 protein diminishes c-Met activation, cell proliferation and invasion in LN18 cells. a Starvation-derived conditioned medium of LN18-SPINT2 cells contained SPINT2 protein indicating shedding of soluble SPINT2 into medium by glioma cells (n = 4, representative figure shown). b Conditioned medium from LN18-SPINT2 cells suppressed c-Met activation in LN18-pLPCX cells. pLPCX-CM, serum-starvation-conditioned medium from LN18-pLPCX cells; SPINT2-CM, serum-starvation-conditioned medium from LN18-SPINT2 cells (n = 3, representative figure shown). c Conditioned medium from LN18-SPINT2 cells suppressed cell proliferation of LN18-pLPCX cells (n = 3, mean ± SEM). d Conditioned medium from LN18-SPINT2 cells suppressed cell proliferation of U87-pLPCX cells (n = 4, mean ± SEM). e Conditioned medium from LN18-SPINT2 cells suppressed cell invasion of LN18-pLPCX cells in the Transwell migration assay (n = 3, mean ± SEM). f Treatment with rSPINT2 suppressed c-Met activation in LN18 cells (upper panel; n = 3, representative figure shown). Treatment with rSPINT2 suppressed cell proliferation in LN18 cells measured by MTT assay (lower panel; n = 3, mean ± SEM). *p < 0.05; **p < 0.01; ***p < 0.001
SPINT2 overexpression inhibits tumor growth in vivo
To determine whether SPINT2 overexpression affects GBM growth and influences survival in vivo, we xenografted U87 cells intracranially into 12 NSG mice (6 U87-SPINT2, 6 U87-pLPCX) and assessed tumor burden using overall survival (OS) as the primary endpoint. Using Kaplan–Meier analysis, we found that the median OS of mice injected with U87-SPINT2 cells (20 days) exceeded those of mice injected with U87-pLPCX cells (15.5 days; log-rank, p = 0.0016; Fig. 5a). Intracranially xenografted LN-18 cells failed to generate tumors (data not shown). We then xenografted 107 LN18 cells into the flank of 10 NSG mice (5 LN18-SPINT2, 5 LN18-pLPCX), and found that 2 out of 5 mice from the LN18-SPINT2 group grew tumors as compared to the 5 out of 5 mice from control LN18-pLPCX group (data not shown). LN18-SPINT2 cells formed smaller tumors (n = 5) as compared to the control mice as assessed over 76 days post-transplantation (Fig. 5b). The two LN18-SPINT2 tumors also displayed a reduced rate of growth (data not shown).

SPINT2 overexpression diminishes tumor growth in vivo and prolonged survival of mice with intracranial transplantation. aSPINT2 overexpression prolonged the survival of NSG mice. U87 cells were intracranially transplanted into NSG mice. Tumor burden was assessed by overall survival (OS) analysis (Kaplan–Meier analysis). The median OS of mice injected with U87-pLPCX control cells was 15.5 days, the median OS of mice injected with U87-SPINT2 cells was 20 days, p = 0.0016 (log-rank; n = 6). bSPINT2 overexpression inhibited tumor formation and growth in mice transplanted subcutaneously with LN18 cells. Tumor size was measured with a caliper (n = 5). c Schematic model summarizing the impact of SPINT2 methylation on the regulation of c-Met in GBM. SPINT2 promoter methylation downregulates SPINT2 expression. As less SPINT2 is available to inhibit HGFA, HGFA maximally converts pro-HGF into HGF which then activates c-Met in GBM cells. *p < 0.05; **p < 0.01; ***p < 0.001
Conclusions
In this study, we defined and confirmed a group of coordinately methylated CpG islands (GT-CMG) common to both IDH1WT and IDH1MUT gliomas including GBMs. Within GT-CMG, we identified SPINT2 as a top cancer-related candidate and demonstrated that SPINT2 exerts tumor-suppressive properties in GBM via inhibition of c-Met activation. The value of defining GT-CMG is to acknowledge a group of genes that has been overlooked in the G-CIMP paradigm. Recognition of GT-CMG contributes an orthogonal view of the CpG island methylation landscape of gliomas [5, 16,17,18, 35], ultimately to enable a better understanding of the generation of aberrant methylation and its subsequent contribution to glioma formation and progression. For example, it has recently been noted that a subset of IDH1MUT tumors more closely resemble the methylation profile of IDH1WT at recurrence [36]. The existence of GT-CMG strongly suggests that aberrant hypermethylation in gliomas can occur via multiple mechanisms, possibly separated into IDH1MUT-dependent and -independent mechanisms. Comparison of methylation at specific CpG sites for GT-CMG CpG islands in the context of IDH1WT and IDH1MUT gliomas may provide important insight into these mechanisms.
Through multiple lines of evidence, we have demonstrated that SPINT2 gene expression is suppressed by methylation of its associated CpG island. Moreover, through a series of in vitro and in vivo experiments, we show that overexpression of SPINT2 exerts tumor suppression in GBM cell lines. These results corroborate similar findings previously reported in GBM cells [30, 31]. In our study, we demonstrated that cell surface or soluble SPINT2 suppressed the proliferation and invasion of tumor cells via downregulation of c-Met activation.
SPINT2 has been found to be hypermethylated in patient tumors from several other cancer types [27, 29, 37]. Evidence for SPINT2 hypermethylation is more limited in the context of GBM patient tumors [30, 31]. Kongkham and colleagues [37] identified SPINT2 hypermethylation and associated reductions in SPINT2 mRNA expression in medulloblastoma cell lines, and demonstrated reductions in cell growth and motility following forced SPINT2 re-expression in these cell lines. They also demonstrated increased overall survival in SPINT2-overexpressing murine medulloblastoma xenografts. Lee and colleagues [31] examined SPINT2 promoter hypermethylation in GBMs and GSC lines and observed associated reductions in SPINT2 mRNA expression in GSC lines; they similarly demonstrated reduced cell growth and migration following forced SPINT2 expression in U87 cells. A study published as this manuscript was being prepared also identified hypermethylation of the SPINT2 gene in human glioma-derived cell lines and high-grade glioma tissue samples [38].
The overall clinical significance of our findings is in the apparent intersection with the HGF/c-Met pathway. In GBM, although c-Met mutations only occur in 1.6% of GBM patients [5], HGF and c-Met are frequently overexpressed, and one-third of patients simultaneously overexpress both HGF and c-Met. This provides further evidence corroborating other recent work suggesting that HGF/c-Met signaling is important in GBM [16, 17, 20, 39,40,41]. The downstream effectors of the c-Met pathway have been well-characterized in prior studies [42,43,44,45]. In preclinical models, inhibition of c-Met or downregulation of HGF and c-Met can suppress GBM growth [22, 46], and targeted c-Met therapies have been tested in three phase II clinical trials for adult GBM [47, 48]. However, none of these trials has demonstrated clear clinical benefit, possibly due to the inability to predict those patients most likely to benefit from c-Met targeted therapy. In this regard, our data suggest that SPINT2 methylation may be associated with c-Met activation and serve as a biomarker indicative of c-Met activation in GBM.
In conclusion, this study defines a set of hypermethylated genes, GT-CMG, that contains a potential therapeutic target, SPINT2. Based on our data, we formulate a model in which SPINT2 downregulation may promote GBM progression via unregulated c-Met activation by HGF (Fig. 5c). This model suggests that targeting c-Met activation via blocking HGFA-mediated activation of HGF is potentially an effective strategy to treat GBM with hypermethylated SPINT2.
References
Parsons DW, Jones S, Zhang X, Lin JC, Leary RJ, Angenendt P, Mankoo P, Carter H, Siu IM, Gallia GL, Olivi A, McLendon R, Rasheed BA, Keir S, Nikolskaya T, Nikolsky Y, Busam DA, Tekleab H, Diaz LA Jr, Hartigan J, Smith DR, Strausberg RL, Marie SK, Shinjo SM, Yan H, Riggins GJ, Bigner DD, Karchin R, Papadopoulos N, Parmigiani G, Vogelstein B, Velculescu VE, Kinzler KW (2008) An integrated genomic analysis of human glioblastoma multiforme. Science 321:1807–1812. https://doi.org/10.1126/science.1164382
Bleeker FE, Molenaar RJ, Leenstra S (2012) Recent advances in the molecular understanding of glioblastoma. J Neuro Oncol 108:11–27. https://doi.org/10.1007/s11060-011-0793-0
Stewart BW, Wild C, International agency for research on cancer, World Health Organization World cancer report 2014
Cloughesy TF, Cavenee WK, Mischel PS (2014) Glioblastoma: from molecular pathology to targeted treatment. Ann Rev Pathol Mech Dis 9: null. doi. https://doi.org/10.1146/annurev-pathol-011110-130324
Brennan CW, Verhaak RG, McKenna A, Campos B, Noushmehr H, Salama SR, Zheng S, Chakravarty D, Sanborn JZ, Berman SH, Beroukhim R, Bernard B, Wu CJ, Genovese G, Shmulevich I, Barnholtz-Sloan J, Zou L, Vegesna R, Shukla SA, Ciriello G, Yung WK, Zhang W, Sougnez C, Mikkelsen T, Aldape K, Bigner DD, Van Meir EG, Prados M, Sloan A, Black KL, Eschbacher J, Finocchiaro G, Friedman W, Andrews DW, Guha A, Iacocca M, O’Neill BP, Foltz G, Myers J, Weisenberger DJ, Penny R, Kucherlapati R, Perou CM, Hayes DN, Gibbs R, Marra M, Mills GB, Lander E, Spellman P, Wilson R, Sander C, Weinstein J, Meyerson M, Gabriel S, Laird PW, Haussler D, Getz G, Chin L, Network TR (2013) The somatic genomic landscape of glioblastoma. Cell 155:462–477. https://doi.org/10.1016/j.cell.2013.09.034
Noushmehr H, Weisenberger DJ, Diefes K, Phillips HS, Pujara K, Berman BP, Pan F, Pelloski CE, Sulman EP, Bhat KP, Verhaak RG, Hoadley KA, Hayes DN, Perou CM, Schmidt HK, Ding L, Wilson RK, Van Den Berg D, Shen H, Bengtsson H, Neuvial P, Cope LM, Buckley J, Herman JG, Baylin SB, Laird PW, Aldape K, Cancer Genome Atlas Research N (2010) Identification of a CpG island methylator phenotype that defines a distinct subgroup of glioma. Cancer Cell 17:510–522. https://doi.org/10.1016/j.ccr.2010.03.017
Issa JP (2004) CpG island methylator phenotype in cancer. Nat Rev Cancer 4:988–993. https://doi.org/10.1038/nrc1507
Sjoblom T, Jones S, Wood LD, Parsons DW, Lin J, Barber TD, Mandelker D, Leary RJ, Ptak J, Silliman N, Szabo S, Buckhaults P, Farrell C, Meeh P, Markowitz SD, Willis J, Dawson D, Willson JK, Gazdar AF, Hartigan J, Wu L, Liu C, Parmigiani G, Park BH, Bachman KE, Papadopoulos N, Vogelstein B, Kinzler KW, Velculescu VE (2006) The consensus coding sequences of human breast and colorectal cancers. Science 314:268–274. https://doi.org/10.1126/science.1133427
Mardis ER, Ding L, Dooling DJ, Larson DE, McLellan MD, Chen K, Koboldt DC, Fulton RS, Delehaunty KD, McGrath SD, Fulton LA, Locke DP, Magrini VJ, Abbott RM, Vickery TL, Reed JS, Robinson JS, Wylie T, Smith SM, Carmichael L, Eldred JM, Harris CC, Walker J, Peck JB, Du F, Dukes AF, Sanderson GE, Brummett AM, Clark E, McMichael JF, Meyer RJ, Schindler JK, Pohl CS, Wallis JW, Shi X, Lin L, Schmidt H, Tang Y, Haipek C, Wiechert ME, Ivy JV, Kalicki J, Elliott G, Ries RE, Payton JE, Westervelt P, Tomasson MH, Watson MA, Baty J, Heath S, Shannon WD, Nagarajan R, Link DC, Walter MJ, Graubert TA, DiPersio JF, Wilson RK, Ley TJ (2009) Recurring mutations found by sequencing an acute myeloid leukemia genome. N Engl J Med 361:1058–1066. https://doi.org/10.1056/NEJMoa0903840
Hemerly JP, Bastos AU, Cerutti JM (2010) Identification of several novel non-p.R132 IDH1 variants in thyroid carcinomas. Eur J Endocrinol 163:747–755. https://doi.org/10.1530/EJE-10-0473
Amary MF, Bacsi K, Maggiani F, Damato S, Halai D, Berisha F, Pollock R, O’Donnell P, Grigoriadis A, Diss T, Eskandarpour M, Presneau N, Hogendoorn PC, Futreal A, Tirabosco R, Flanagan AM (2011) IDH1 and IDH2 mutations are frequent events in central chondrosarcoma and central and periosteal chondromas but not in other mesenchymal tumours. J Pathol 224:334–343. https://doi.org/10.1002/path.2913
Borger DR, Tanabe KK, Fan KC, Lopez HU, Fantin VR, Straley KS, Schenkein DP, Hezel AF, Ancukiewicz M, Liebman HM, Kwak EL, Clark JW, Ryan DP, Deshpande V, Dias-Santagata D, Ellisen LW, Zhu AX, Iafrate AJ (2012) Frequent mutation of isocitrate dehydrogenase (IDH)1 and IDH2 in cholangiocarcinoma identified through broad-based tumor genotyping. Oncologist 17:72–79. https://doi.org/10.1634/theoncologist.2011-0386
Cairns RA, Iqbal J, Lemonnier F, Kucuk C, de Leval L, Jais JP, Parrens M, Martin A, Xerri L, Brousset P, Chan LC, Chan WC, Gaulard P, Mak TW (2012) IDH2 mutations are frequent in angioimmunoblastic T-cell lymphoma. Blood 119:1901–1903. https://doi.org/10.1182/blood-2011-11-391748
Wang RX, Wu DP, Chen SN, He J, Xu Y, Wang XL, Yin J, Tian H, Tian XP, Sun AN (2013) Clinical characteristics in adult acute myeloid leukemia with isocitrate dehydrogenase gene mutation. Zhonghua yi xue za zhi 93:751–755
Ichimura K, Pearson DM, Kocialkowski S, Backlund LM, Chan R, Jones DT, Collins VP (2009) IDH1 mutations are present in the majority of common adult gliomas but rare in primary glioblastomas. Neuro Oncol 11:341–347. https://doi.org/10.1215/15228517-2009-025
Goodwin CR, Rath P, Oyinlade O, Lopez H, Mughal S, Xia S, Li Y, Kaur H, Zhou X, Ahmed AK, Ho S, Olivi A, Lal B (2018) Crizotinib and erlotinib inhibits growth of c-Met(+)/EGFRvIII(+) primary human glioblastoma xenografts. Clin Neurol Neurosurg 171:26–33. https://doi.org/10.1016/j.clineuro.2018.02.041
Tasaki T, Fujita M, Okuda T, Yoneshige A, Nakata S, Yamashita K, Yoshioka H, Izumoto S, Kato A (2016) MET expressed in glioma stem cells is a potent therapeutic target for glioblastoma multiforme. Anticancer Res 36:3571–3577
Sturm D, Witt H, Hovestadt V, Khuong-Quang DA, Jones DT, Konermann C, Pfaff E, Tonjes M, Sill M, Bender S, Kool M, Zapatka M, Becker N, Zucknick M, Hielscher T, Liu XY, Fontebasso AM, Ryzhova M, Albrecht S, Jacob K, Wolter M, Ebinger M, Schuhmann MU, van Meter T, Fruhwald MC, Hauch H, Pekrun A, Radlwimmer B, Niehues T, von Komorowski G, Durken M, Kulozik AE, Madden J, Donson A, Foreman NK, Drissi R, Fouladi M, Scheurlen W, von Deimling A, Monoranu C, Roggendorf W, Herold-Mende C, Unterberg A, Kramm CM, Felsberg J, Hartmann C, Wiestler B, Wick W, Milde T, Witt O, Lindroth AM, Schwartzentruber J, Faury D, Fleming A, Zakrzewska M, Liberski PP, Zakrzewski K, Hauser P, Garami M, Klekner A, Bognar L, Morrissy S, Cavalli F, Taylor MD, van Sluis P, Koster J, Versteeg R, Volckmann R, Mikkelsen T, Aldape K, Reifenberger G, Collins VP, Majewski J, Korshunov A, Lichter P, Plass C, Jabado N, Pfister SM (2012) Hotspot mutations in H3F3A and IDH1 define distinct epigenetic and biological subgroups of glioblastoma. Cancer Cell 22:425–437. https://doi.org/10.1016/j.ccr.2012.08.024
Li Y, Li A, Glas M, Lal B, Ying M, Sang Y, Xia S, Trageser D, Guerrero-Cazares H, Eberhart CG, Quinones-Hinojosa A, Scheffler B, Laterra J (2011) c-Met signaling induces a reprogramming network and supports the glioblastoma stem-like phenotype. Proc Natl Acad Sci USA 108:9951–9956. https://doi.org/10.1073/pnas.1016912108
Xie Q, Bradley R, Kang L, Koeman J, Ascierto ML, Worschech A, De Giorgi V, Wang E, Kefene L, Su Y, Essenburg C, Kaufman DW, DeKoning T, Enter MA, O’Rourke TJ, Marincola FM, Vande Woude GF (2012) Hepatocyte growth factor (HGF) autocrine activation predicts sensitivity to MET inhibition in glioblastoma. Proc Natl Acad Sci USA 109:570–575. https://doi.org/10.1073/pnas.1119059109
Joo KM, Jin J, Kim E, Ho Kim K, Kim Y, Gu Kang B, Kang YJ, Lathia JD, Cheong KH, Song PH, Kim H, Seol HJ, Kong DS, Lee JI, Rich JN, Lee J, Nam DH (2012) MET signaling regulates glioblastoma stem cells. Cancer Res 72:3828–3838. https://doi.org/10.1158/0008-5472.CAN-11-3760
Martens T, Schmidt NO, Eckerich C, Fillbrandt R, Merchant M, Schwall R, Westphal M, Lamszus K (2006) A novel one-armed anti-c-Met antibody inhibits glioblastoma growth in vivo. Clin Cancer Res 12:6144–6152. https://doi.org/10.1158/1078-0432.CCR-05-1418
Uchinokura S, Miyata S, Fukushima T, Itoh H, Nakano S, Wakisaka S, Kataoka H (2006) Role of hepatocyte growth factor activator (HGF activator) in invasive growth of human glioblastoma cells in vivo. Int J Cancer 118:583–592. https://doi.org/10.1002/ijc.21362
Abounader R, Laterra J (2005) Scatter factor/hepatocyte growth factor in brain tumor growth and angiogenesis. Neuro Oncol 7:436–451. https://doi.org/10.1215/S1152851705000050
Kitajima Y, Ide T, Ohtsuka T, Miyazaki K (2008) Induction of hepatocyte growth factor activator gene expression under hypoxia activates the hepatocyte growth factor/c-Met system via hypoxia inducible factor-1 in pancreatic cancer. Cancer Sci 99:1341–1347. https://doi.org/10.1111/j.1349-7006.2008.00828.x
Shimomura T, Ochiai M, Kondo J, Morimoto Y (1992) A novel protease obtained from FBS-containing culture supernatant, that processes single chain form hepatocyte growth factor to two chain form in serum-free culture. Cytotechnology 8:219–229
Dong W, Chen X, Xie J, Sun P, Wu Y (2010) Epigenetic inactivation and tumor suppressor activity of HAI-2/SPINT2 in gastric cancer. Int J Cancer 127:1526–1534. https://doi.org/10.1002/ijc.25161
Hwang S, Kim HE, Min M, Raghunathan R, Panova IP, Munshi R, Ryu B (2015) Epigenetic silencing of SPINT2 promotes cancer cell motility via HGF-MET pathway activation in melanoma. J Investig Dermatol 135:2283–2291. https://doi.org/10.1038/jid.2015.160
Tung EK, Wong CM, Yau TO, Lee JM, Ching YP, Ng IO (2009) HAI-2 is epigenetically downregulated in human hepatocellular carcinoma, and its Kunitz domain type 1 is critical for anti-invasive functions. Int J Cancer 124:1811–1819. https://doi.org/10.1002/ijc.24115
Hamasuna R, Kataoka H, Meng JY, Itoh H, Moriyama T, Wakisaka S, Koono M (2001) Reduced expression of hepatocyte growth factor activator inhibitor type-2/placental bikunin (HAI-2/PB) in human glioblastomas: implication for anti-invasive role of HAI-2/PB in glioblastoma cells. Int J Cancer 93:339–345
Lee EJ, Rath P, Liu J, Ryu D, Pei L, Noonepalle SK, Shull AY, Feng Q, Litofsky NS, Miller DC, Anthony DC, Kirk MD, Laterra J, Deng L, Xin HB, Wang X, Choi JH, Shi H (2015) Identification of global DNA methylation signatures in glioblastoma-derived cancer stem cells. J Genet Genomics 42:355–371. https://doi.org/10.1016/j.jgg.2015.06.003
Nardone S, Sams DS, Reuveni E, Getselter D, Oron O, Karpuj M, Elliott E (2014) DNA methylation analysis of the autistic brain reveals multiple dysregulated biological pathways. Transl Psychiatry 4:e433. https://doi.org/10.1038/tp.2014.70
Kawaguchi T, Qin L, Shimomura T, Kondo J, Matsumoto K, Denda K, Kitamura N (1997) Purification and cloning of hepatocyte growth factor activator inhibitor type 2, a Kunitz-type serine protease inhibitor. J Biol Chem 272:27558–27564
Shimomura T, Denda K, Kitamura A, Kawaguchi T, Kito M, Kondo J, Kagaya S, Qin L, Takata H, Miyazawa K, Kitamura N (1997) Hepatocyte growth factor activator inhibitor, a novel Kunitz-type serine protease inhibitor. J Biol Chem 272:6370–6376
Brat DJ, Verhaak RG, Aldape KD, Yung WK, Salama SR, Cooper LA, Rheinbay E, Miller CR, Vitucci M, Morozova O, Robertson AG, Noushmehr H, Laird PW, Cherniack AD, Akbani R, Huse JT, Ciriello G, Poisson LM, Barnholtz-Sloan JS, Berger MS, Brennan C, Colen RR, Colman H, Flanders AE, Giannini C, Grifford M, Iavarone A, Jain R, Joseph I, Kim J, Kasaian K, Mikkelsen T, Murray BA, O’Neill BP, Pachter L, Parsons DW, Sougnez C, Sulman EP, Vandenberg SR, Van Meir EG, von Deimling A, Zhang H, Crain D, Lau K, Mallery D, Morris S, Paulauskis J, Penny R, Shelton T, Sherman M, Yena P, Black A, Bowen J, Dicostanzo K, Gastier-Foster J, Leraas KM, Lichtenberg TM, Pierson CR, Ramirez NC, Taylor C, Weaver S, Wise L, Zmuda E, Davidsen T, Demchok JA, Eley G, Ferguson ML, Hutter CM, Mills Shaw KR, Ozenberger BA, Sheth M, Sofia HJ, Tarnuzzer R, Wang Z, Yang L, Zenklusen JC, Ayala B, Baboud J, Chudamani S, Jensen MA, Liu J, Pihl T, Raman R, Wan Y, Wu Y, Ally A, Auman JT, Balasundaram M, Balu S, Baylin SB, Beroukhim R, Bootwalla MS, Bowlby R, Bristow CA, Brooks D, Butterfield Y, Carlsen R, Carter S, Chin L, Chu A, Chuah E, Cibulskis K, Clarke A, Coetzee SG, Dhalla N, Fennell T, Fisher S, Gabriel S, Getz G, Gibbs R, Guin R, Hadjipanayis A, Hayes DN, Hinoue T, Hoadley K, Holt RA,Hoyle AP, Jefferys SR, Jones S, Jones CD, Kucherlapati R, Lai PH, Lander E, Lee S, Lichtenstein L, Ma Y, Maglinte DT, Mahadeshwar HS, Marra MA, Mayo M, Meng S, Meyerson ML, Mieczkowski PA, Moore RA, Mose LE, Mungall AJ, Pantazi A, Parfenov M, Park PJ,Parker JS, Perou CM, Protopopov A, Ren X, Roach J, Sabedot TS, Schein J, Schumacher SE, Seidman JG, Seth S, Shen H, Simons JV, Sipahimalani P, Soloway MG, Song X, Sun H, Tabak B, Tam A, Tan D, Tang J, Thiessen N, Triche T, Jr., Van Den Berg DJ, Veluvolu U, Waring S, Weisenberger DJ, Wilkerson MD, Wong T, Wu J, Xi L, Xu AW, Zack TI, Zhang J, Aksoy BA, Arachchi H, Benz C, Bernard B, Carlin D, Cho J, DiCara D, Frazer S, Fuller GN, Gao J, Gehlenborg N, Haussler D, Heiman DI, Iype L, Jacobsen A, Ju Z, Katzman S, Kim H, Knijnenburg T, Kreisberg RB, Lawrence MS, Lee W, Leinonen K, Lin P, Ling S, Liu W, Liu Y, Lu Y, Mills G, Ng S, Noble MS, Paull E, Rao A, Reynolds S, Saksena G, Sanborn Z, Sander C, Schultz N, Senbabaoglu Y, Shen R, Shmulevich I, Sinha R, Stuart J, Sumer SO, Sun Y, Tasman N, Taylor BS, Voet D, Weinhold N, Weinstein JN, Yang D, Yoshihara K, Zheng S, Zhang W, Zou L, Abel T, Sadeghi S, Cohen ML, Eschbacher J, Hattab EM, Raghunathan A, Schniederjan MJ, Aziz D, Barnett G, Barrett W, Bigner DD, Boice L, Brewer C, Calatozzolo C, Campos B, Carlotti CG, Jr., Chan TA, Cuppini L, Curley E, Cuzzubbo S, Devine K, DiMeco F, Duell R, Elder JB, Fehrenbach A, Finocchiaro G,Friedman W, Fulop J, Gardner J, Hermes B, Herold-Mende C, Jungk C, Kendler A, Lehman NL, Lipp E, Liu O, Mandt R, McGraw M, McLendon R, McPherson C, Neder L, Nguyen P,Noss A, Nunziata R, Ostrom QT, Palmer C, Perin A, Pollo B, Potapov A, Potapova O, Rathmell WK, Rotin D, Scarpace L, Schilero C, Senecal K, Shimmel K, Shurkhay V, Sifri S, Singh R, Sloan AE, Smolenski K, Staugaitis SM, Steele R, Thorne L, Tirapelli DP, Unterberg A, Vallurupalli M, Wang Y, Warnick R, Williams F, Wolinsky Y, Bell S, Rosenberg M, Stewart C, Huang F, Grimsby JL, Radenbaugh AJ (2015) Comprehensive, integrative genomic analysis of diffuse lower-grade gliomas. N Engl J Med 372: 2481–2498. https://doi.org/10.1056/NEJMoa1402121
de Souza CF, Sabedot TS, Malta TM, Stetson L, Morozova O, Sokolov A, Laird PW, Wiznerowicz M, Iavarone A, Snyder J, deCarvalho A, Sanborn Z, McDonald KL, Friedman WA, Tirapelli D, Poisson L, Mikkelsen T, Carlotti CG Jr, Kalkanis S, Zenklusen J, Salama SR, Barnholtz-Sloan JS, Noushmehr H (2018) A distinct DNA methylation shift in a subset of glioma CpG island methylator phenotypes during tumor recurrence. Cell Rep 23:637–651. https://doi.org/10.1016/j.celrep.2018.03.107
Kongkham PN, Northcott PA, Ra YS, Nakahara Y, Mainprize TG, Croul SE, Smith CA, Taylor MD, Rutka JT (2008) An epigenetic genome-wide screen identifies SPINT2 as a novel tumor suppressor gene in pediatric medulloblastoma. Cancer Res 68:9945–9953. https://doi.org/10.1158/0008-5472.CAN-08-2169
Fukushima T, Kawaguchi M, Yamamoto K, Yamashita F, Izumi A, Kaieda T, Takezaki Y, Itoh H, Takeshima H, Kataoka H (2018) Aberrant methylation and silencing of the SPINT2 gene in high-grade gliomas. Cancer Sci. https://doi.org/10.1111/cas.13732
Koochekpour S, Jeffers M, Rulong S, Taylor G, Klineberg E, Hudson EA, Resau JH, Vande Woude GF (1997) Met and hepatocyte growth factor/scatter factor expression in human gliomas. Cancer Res 57:5391–5398
Zhang Y, Farenholtz KE, Yang Y, Guessous F, Dipierro CG, Calvert VS, Deng J, Schiff D, Xin W, Lee JK, Purow B, Christensen J, Petricoin E, Abounader R (2013) Hepatocyte growth factor sensitizes brain tumors to c-MET kinase inhibition. Clin Cancer Res 19:1433–1444. https://doi.org/10.1158/1078-0432.CCR-12-2832
Petterson SA, Dahlrot RH, Hermansen SK, Gundesen SKAM, Wohlleben MT, Rasmussen H, Beier T, Hansen CP, Kristensen S BW (2015) High levels of c-Met is associated with poor prognosis in glioblastoma. J Neuro Oncol 122:517–527. https://doi.org/10.1007/s11060-015-1723-3
Szabo R, Rasmussen AL, Moyer AB, Kosa P, Schafer JM, Molinolo AA, Gutkind JS, Bugge TH (2011) c-Met-induced epithelial carcinogenesis is initiated by the serine protease matriptase. Oncogene 30:2003–2016. https://doi.org/10.1038/onc.2010.586
Matsumoto K, Umitsu M, De Silva DM, Roy A, Bottaro DP (2017) Hepatocyte growth factor/MET in cancer progression and biomarker discovery. Cancer Sci 108:296–307. https://doi.org/10.1111/cas.13156
Xiang C, Chen J, Fu P (2017) HGF/Met signaling in cancer invasion: the impact on cytoskeleton remodeling. Cancers 9:44. https://doi.org/10.3390/cancers9050044
Kataoka H, Kawaguchi M, Fukushima T, Shimomura T (2018) Hepatocyte growth factor activator inhibitors (HAI-1 and HAI-2): emerging key players in epithelial integrity and cancer. Pathol Int 68:145–158. https://doi.org/10.1111/pin.12647
Abounader R, Ranganathan S, Lal B, Fielding K, Book A, Dietz H, Burger P, Laterra J (1999) Reversion of human glioblastoma malignancy by U1 small nuclear RNA/ribozyme targeting of scatter factor/hepatocyte growth factor and c-met expression. J Natl Cancer Inst 91:1548–1556
Wen PY, Schiff D, Cloughesy TF, Raizer JJ, Laterra J, Smitt M, Wolf M, Oliner KS, Anderson A, Zhu M, Loh E, Reardon DA (2011) A phase II study evaluating the efficacy and safety of AMG 102 (rilotumumab) in patients with recurrent glioblastoma. Neuro Oncol 13:437–446. https://doi.org/10.1093/neuonc/noq198
Awad AJ, Burns TC, Zhang Y, Abounader R (2014) Targeting MET for glioma therapy. Neurosurg focus 37:E10. https://doi.org/10.3171/2014.9.FOCUS14520
Acknowledgements
The authors thank the support of UCLA Jonsson Comprehensive Cancer Center (JCCC); Dr. Paul S Mischel for glioma cell lines; and UCLA Brain Tumor Translational Resource for logistical and technical support.
Funding
Art of the Brain; Bradley Zankel Foundation; National Institutes of Health/National Cancer Institute [R01CA179071]; UCLA SPORE in Brain Cancer [P50 CA211015].
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
Author Byram Ozer has received an honorarium for a one-time consultation with Neurocore, 2015. Author Timothy Cloughesy reports personal fees from Pfizer, Tocagen, Roche, Novocure, Nektar, VBL, ABBVIE, Upshire Smith, Notable Labs, Oxigene, NewGen, Agios, Cortice, MedQia, PRoNai, Wellcome Trust, Merck, Insys, Human Longevity, Sunovion, Boston Biomedical, Alexion, Novogen, VBI, BMS, GW Pharma, Bioclinica, Amgen outside the submitted work. Dr. Cloughesy is a board member of the Global Coalition for Adaptive Research 501c3 and the PI for GBM Agile. Dr. Cloughesy has stock options for Notable Labs. Author Albert Lai has received Honoraria from Merck, Genentech, Abbvie, and Optune.
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
About this article

Cite this article
Liu, F., Cox, C.D., Chowdhury, R. et al. SPINT2 is hypermethylated in both IDH1 mutated and wild-type glioblastomas, and exerts tumor suppression via reduction of c-Met activation. J Neurooncol 142, 423–434 (2019). https://doi.org/10.1007/s11060-019-03126-x
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s11060-019-03126-x