
Abstract
Optic pathway glioma (OPG) represents the most common central nervous system tumor in children with Neurofibromatosis type-1 (NF1). Although overall survival is usually good, no clear prognostic factors have been identified so far. We assessed the natural history of OPG in a cohort of unselected patients affected by NF1. We retrospectively evaluated 414 consecutive patients affected by NF1 and referred to our NF1 clinic before age 6. Average follow-up was 11.9 years: 52 out of 414 patients had OPG with a total cumulative incidence of 15.4% at age 15 (Kaplan–Meier estimate) and a statistically significant difference according to sex. Brain and orbit MRI was performed in 44.7% of patients: 34.6% for screening purposes and 65.4% because of the presence of neurological, ocular or other symptoms. OPG was diagnosed in 12.5% of cases in the first group, whereas in 36.4% in the latter group (p = 0.001). Clinical management was conservative in most patients, while 8 of them underwent therapy mainly because of visual deterioration. OPG was diagnosed earlier in treated patients, but the difference was not statistically significant. Conversely, all patients who underwent screening MRI had normal visual outcome. In conclusion, OPG location does not correlate with need for treatment; female patients were more frequently affected by OPG but not more frequently treated. OPG diagnosis by screening MRI does not affect the natural history of the tumor.
Similar content being viewed by others
Avoid common mistakes on your manuscript.
Introduction
Neurofibromatosis type 1 (NF1) is one of the most common autosomal dominant conditions with an incidence of about 1:3500 [1]. It is caused by loss-of-function mutations of NF1 [2], encoding neurofibromin that acts as a tumor suppressor by negatively regulating the GTPase activity of p21RAS and by controlling the serine threonine kinase MTOR [3]. About 50% of patients carry a de novo mutation.
NF1 is a multisystem disease with heterogeneous age-dependent clinical manifestations [4, 5]: NF1 patients are prone to develop benign and malignant tumors, with optic pathway glioma (OPG) being the most common NF1-associated central nervous system tumor that affects about 15% of NF1 patients [6, 7] and usually arises during the first decade of life, nevertheless later onset is well documented [8, 9].
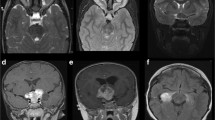
Although OPG in NF1 usually shows a more indolent course compared to sporadic cases [10], it may present a hazardous evolution with severe impairment of visual function and potentially life-threatening behavior [11]. OPG in NF1 are classified according to their anatomical location: the pre-chiasmatic tracts of the optic nerves, the chiasmatic-hypothalamic region and/or the posterior optic pathways [12]. Some studies have reported that posterior involvement of the optic pathway is associated with a worse visual outcome [11, 13,14,15], however other studies did not confirm this finding [16, 17]. No other prognostic factors of OPG behavior have been clearly proven and tumor evolution remains unpredictable so far [18, 19].
Surgery or radiotherapy are generally avoided in symptomatic OPG in NF1 patients and treatment options are limited to a classical chemotherapeutic regimen (Carboplatine and Vincristine) that often shows poor outcome concerning visual outcome [20, 21]; currently, prospective multicenter clinical trials are warranted to test the efficacy of novel biological drugs. However, many NF1 patients with OPG will never experience clinical progression and the decision to undertake a treatment is mostly based on clinical and ophthalmological assessment [13, 15, 18], whereas radiological progression alone, at least at the initial follow-up MRI, does not justify treatment.
The utility of systematic brain MRI for screening purposes in NF1 asymptomatic children is still a matter of debate: some studies recommend it for the ability to detect earlier the presence of an OPG [13, 15], and others exclude a clear benefit of such an approach [9, 18].
The aim of our study was to assess the natural history of OPG and possible prognostic factors for OPG progression in a cohort of unselected patients affected by NF1 who developed the tumor during the follow-up.
Materials and methods
We retrospectively evaluated the clinical records and imaging data in a cohort of 599 consecutive patients referred to our NF1 Clinic for a clinical diagnosis or suspicion of NF1 between January 1980 and June 2016. To avoid bias in patients’ selection, only patients who were first evaluated before the age of 6 were included. The analysis was performed in those individuals who fulfilled the clinical diagnosis of NF1, according to the established international criteria, during our first evaluation or the follow-up [22]. Exclusion criteria include: segmental NF1 defined as the presence of diagnostic criteria limited to one body area without crossing the midline [23], or a diagnosis of OPG before our first evaluation. Standard care for NF1 patients consists of annual detailed clinical evaluations and complete ophthalmologic examinations, including fundus oculi analysis, visual acuity measurement, visual field assessment and since 2012 also ocular computerized tomography (OCT) [24]. Visual acuity impairment was defined using visual acuity age-based norms data and the International Classification of Impairments, Disabilities and Handicaps (WHO) [25, 26]. Moderate visual acuity impairment was defined as visual acuity inferior to age-based norms data [25]. Severe visual acuity impairment was defined as visual acuity inferior to 0.12 logMAR [26]. Brain and orbit MRI scans are performed only on clinical or ophthalmologic indications; however, during the follow-up a proportion of NF1 patients were evaluated in other centers and sixty-four of them underwent brain and orbit MRI for screening purposes.
OPG patients were divided into two groups: the first one included those who presented with symptoms suggestive of OPG (decreased visual acuity, ophtalmoscopic or OCT alterations, proptosis, precocious puberty); the second one included asymptomatic individuals or patients who underwent MRI because of symptoms unrelated to OPG (macrocephaly, developmental delay, headache, seizures, plexiform/orbital neurofibromas of the face/neck, brain ischemia).
Statistical analyses
Descriptive statistics for the main variables of interest were calculated. The Student t test was used to analyze differences between independent continuous variables; the Chi square test or the Fisher’s exact test were used to assess differences between categorical variables. Kaplan–Meier survival curves were used to estimate the cumulative incidence of OPG. The log-rank test was used to evaluate whether the incidence of OPG was significantly different according to sex, the presence of congenital plexiform neurofibromas, a positive family history for NF1. All p values were calculated at a 95% CI.
Results
A total of 414 patients affected by NF1 fulfilled the inclusion criteria and were selected for this study. All these subjects were evaluated for the first time in our clinic before age 6 and before a diagnosis of OPG was established; 26 children were excluded since they had been diagnosed with OPG before they were referred to our clinic and the remaining 159 because of a diagnosis of segmental NF1 or the presence of cafè-au-lait spots not related to NF1.
The mean age at the first evaluation was 2.5 years (SD 1.6) and the follow-up duration was, on average, 11.9 years (SD 8.5) without statistically significant differences between patients with or without OPG.
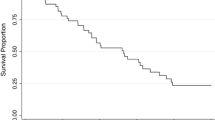
The main characteristics of our population are shown in Table 1. Among the 414 patients, 52 (12.6%) developed OPG during their follow-up (29 females and 23 males). Cumulative incidence of OPG at 15 years was of 15.4% [standard error (SE) 2.0%, Kaplan–Meier estimate], and we observed a statistically significant difference according to sex [females 19.9% (SE 3.4%), males 12.1% (SE 2.4%), p value 0.035], that was not dependent on differences in average follow-up (Fig. 1).

a Total cumulative incidence of OPG (Kaplan–Meier estimate), b cumulative incidence of OPG according to sex (p value 0.035)
We could not observe any correlation between the presence of OPG and both congenital plexiform neurofibromas and NF1 family history. OPG was diagnosed at an average age of 5.4 years (SD 3.1) and the mean follow-up duration was 11.2 years (SD 6.9). Follow-up was slightly longer in patients who did not manifest OPG than patients with this tumor, but the difference was not statistically significant (11.2 years vs. 12 years; Table 1). Mean age at last evaluation in OPG patients was 13.3 years (range 3.4–32.4).
A total of 185 out of 414 NF1 patients underwent brain/orbit MRI examination: 64 (34.6%) for screening purposes, whereas the remaining 121 (65.4%) because of a specific clinical indication. OPG was diagnosed more frequently in the second group (36.4%) compared to the first one (12.5%) with a statistically significant difference (p value 0.001). In particular, among the 52 OPG patients, brain/orbit MRI was performed in 43 subjects for a clinical indication: twenty-five patients (48.1%) manifested symptoms highly suggestive of an OPG, whereas in 18 cases (34.6%), the clinical problems requiring brain imaging were not indicative of the presence of an OPG. In one patient the indication for MRI was an unspecified clinical symptom, while in the remaining 8 individuals (15.4%) OPG was identified by screening MRI performed in other NF1 clinics. Indications for brain and orbit MRI in patients with OPG are reported in Table 1.
OPG were diagnosed more frequently in the group of patients with symptoms highly suggestive of OPG (25/55, 45.5% vs. 26/129, 20.2%, p value 0.001) compared to patients who underwent brain/orbit MRI for screening purpose or symptoms unrelated to the presence of an OPG; in the first group the diagnosis was made at a younger age than in the second one, but the difference was not statistically significant (Table 2).
According to the modified Dodge OPG classification [12], 32.7% (17/52) of our patients presented a class Ia OPG, involving a single optic nerve, 15.4% (8/52) had Ib OPG, involving bilaterally the optic nerves, 23.1% (12/52) had 2a or 2b OPG, affecting symmetrically or asymmetrically the optic chiasma, 5.8% (3/52) a class 4 OPG involving the optic tracts and 23.1% (12/52) a 2a or 2b H + OPG involving also the hypothalamic region.
Among all the NF1 patients who underwent a brain/orbit MRI, other neuroradiological findings were documented (which are reported in Online Resource 1). Unidentified bright objects (UBOs) were not considered since their high frequency among NF1 populations and the absence of correlations with OPG [27].
Most of OPG patients in our cohort (44/52, 84.6%) did not require any treatment and a “wait and see” approach was adopted with frequent complete ophthalmological evaluations and serial brain/orbit MRI.
Only eight patients required therapy (in seven case for a clinical deterioration, in one case for a radiological progression). OPG were diagnosed earlier in treated patients [mean age 4.5 years (SD 3.1) vs. 5.6 years (SD 3.1)], but the difference was not statistically significant. Average age at the beginning of treatment was 6.4 years (SD 3.5) with a mean interval between OPG diagnosis and treatment of 1.5 years (S.D 1.4). Six patients were treated with chemotherapy (requiring a surgical decompression in one case) and two subjects underwent radiotherapy (treatment was performed before 2000 in both cases, before the radiotherapy-associated long-term risk for secondary malignancies had become apparent) [28].
A total of five patients with OPG manifested precocious puberty (at the moment of OPG diagnosis or after) and of those subjects three suffered from concurrent hydrocephalus; in three cases OPG was localized to the chiasma, in one case involved both optic nerves and in the remaining case affected one optic nerve. All these individuals underwent chemotherapy because of visual decline and/or visual field alterations, with the exception of the patient with the unilateral optic nerve glioma.
Five out of 52 patients with OPG were subsequently lost at follow-up and there is no detailed information regarding tumor evolution. Among the 47 patients with follow-up data, the frequency of stable tumors was comparable between individuals who performed MRI for symptoms suggestive of OPG (“Symptomatic” group) and those who performed MRI for screening purpose or symptoms unrelated to OPG (“Asymptomatic” group) (65.2 vs. 70.8%). Tumors with a clinical evolution were more frequently observed in the “symptomatic” group; conversely, radiological progression without an associated clinical decline was more frequent in the “asymptomatic” group (Table 3). Among the seven OPG patients who manifested clinical progression there were three males and four females, the average age at diagnosis was 5.6 years (min–max: 2.1–8.8); in three patients OPG involved the optic nerve bilaterally, in three cases both the optic nerve and the chiasma, and in one patient the chiasma and the posterior pathway.
The mean age at the last ophthalmological evaluation was 12.9 years (SD 6.2). Most patients with OPG had a normal visual outcome (39/47; 83%). In the remaining eight patients who presented a decline in visual function, two patients showed a mild deficit and six patients manifested a severe deterioration.
In the “asymptomatic” group, a normal visual outcome was present in 22/24 of patients (91.6%), whereas a visual decline was documented in two patients (8.4%): OPG was incidentally found during a MRI performed for a plexiform neurofibroma of the neck or for headache, respectively, but only the second patient required treatment. In the “symptomatic” group, 73.9% had a normal visual outcome, with around a quarter (26.1%) manifesting a visual deficit (severe in five cases). In our cohort, no significant differences in the visual outcome were observed between the two groups (Table 3).
Finally, we evaluated the presence of possible differences between the groups of treated and untreated patients according to sex, age at diagnosis of OPG, indications for the first brain/orbit MRI, tumor localization and progression. Results are summarized in Table 4. Male patients underwent therapy more frequently than females, but the difference is not statistically significant. Most untreated patients were older than 3 years at OPG diagnosis (72.5%), whereas in 57.1% of treated patients, OPG was diagnosed before age 3 (p value 0.188). Concerning visual outcome, patients with a precocious diagnosis of OPG (before age 3) had a significantly worse visual outcome compared to later diagnosed OPG.
The rates of patients with OPG requiring treatment were not statistically different among groups with different OPG classes according to the modified Dodge classification [12]. To avoid bias due to the small number of patients undergoing therapy, we also classified OPG into two main categories (OPG involving or not the chiasmatic region) according to the previous OPG classification [29]; again, no significant difference in the rates of patients who underwent treatment was observed between the two groups.
Treatment requirement was more frequent in patients who underwent brain/orbit MRI because of the onset of symptoms, but there was no statistical significance. No significant difference was observed neither considering the different onset manifestations. As expected, 80% of untreated patients showed stable lesions, with evidence of a radiological progression (not associated with a decline in visual function) only in 17.5%.
Most treated patients showed a visual decline, whereas 95% of untreated patients maintained a normal visual function.
We also compared the group of patients selected for the present study and those excluded because the diagnosis of OPG was made before our first evaluation (Online Resource 2); the latter group comprises children with diagnosis of OPG established at a younger age than in the study cohort but with a comparable follow-up duration. There was a higher rate of males in the group of patients who were excluded from the study, but the difference was not statistically significant.
No statistically significant differences were observed between the two groups regarding the rate of children who underwent brain/orbit MRI because of OPG symptoms and the frequency of OPG involving the chiasma.
Overall, there was a higher rate of children needing OPG therapy in the group of patients who were excluded from the study, and particularly among those who presented symptoms at diagnosis. On the other hand, the number of asymptomatic individuals at OPG diagnosis who required treatment for tumor progression during the follow-up was comparable between the two groups. Finally, the visual outcome was significantly worse in the group of patients excluded from the study who manifested symptoms at OPG diagnosis.
Discussion
The cohort of patients included in our study was selected among individuals with a clinically confirmed NF1 who came to our attention before the diagnosis of OPG and first evaluated before the age of 6. These criteria were applied to avoid the selection bias due to the inclusion of patients referred to our NF1 Clinic for the presence of OPG. The incidence of OPG in children affected by NF1 reproduces previously reported data [8], confirming a significant higher incidence in females [6, 30]. In agreement with previous works, we did not find specific factors predisposing to OPG development, in particular concerning a positive family history of NF1 [31] or the presence of plexiform neurofibromas [19].
Mean age of OPG diagnosis in our cohort was 5.4 years, in agreement with what described in other large retrospective studies in NF1 patients not undergoing systematic screening MRI [32]; conversely, MRI in asymptomatic NF1 children allows to anticipate the diagnosis of around 2 years [11, 15, 18, 33].
Currently, the usefulness of baseline brain MRI in NF1 pediatric patients is still controversial since many NF1 clinics are against the use of systematic brain imaging in these patients [18, 34, 35], whereas others claim the opposite [15, 33, 36], being these contrasting results mainly due to differences in patients’ selection.
There is still no definitive evidence that an earlier chemotherapic treatment may prevent visual deterioration in these patients. In previous works [15, 33], neuroradiological screening has been advocated for its ability to improve the visual outcome in OPG patients, but in both cohorts all patients who maintain a good visual function had been treated for a radiological progression and had not manifested visual problems at the beginning of the therapy. Therefore, it is possible that the same outcome would have been achieved without any treatment. In fact there are well documented examples of stabilization or even regression of such tumors in the absence of therapy [37, 38]. Standardized guidelines are warranted for these patients [5, 9] and prospective studies are required to define prognostic factors for OPG progression.
We confirmed that about half of NF1 patients with OPG presents clinical symptoms or signs at the diagnosis or during the follow-up [13, 15], including mainly a decrease in visual acuity, ophtalmoscopic or OCT alterations and, in five patients, precocious puberty, which however, as previously observed, is not always associated with a hypothalamic involvement and it is not an indication to undertake a chemotherapic treatment, even though it is often associated with obstructive hydrocephalus and the potential need for derivation [19]. We did neither observe significant differences in the visual outcome, in the need for treatment nor in the tumor location between patients who underwent brain/orbit MRI for screening purposes or for specific clinical indications. Therefore the present work further supports previous findings regarding the lack of indication of baseline MRI in children with NF1 [19]. However, we observed a greater proportion of treated patients among those who were referred to our clinic after the diagnosis of OPG; in particular, the rate was higher in the group of patients who underwent MRI because of the presence of symptoms related to OPG. This group included subjects with OPG diagnosis at a younger age, suggesting that the early onset of OPG symptoms is likely a negative prognostic factor. Our data confirm the well known heterogeneous distribution of OPG location and show that it does not correlate with treatment requirement, in agreement with the results of previous works [9, 10, 12], but it may be secondary to sample selection criteria and the indication to perform screening MRI. In fact, among patients attending neuro-oncological or ophthalmological Clinics, it is more likely to find a higher number of patients with OPG involving multiple tracts of the optic pathway, that requires a closer follow-up [9]. On the other hand, performing screening MRI may reveal the presence of indolent asymptomatic OPG that may not be diagnosed otherwise [12]. The difficulties in selecting an unbiased population may explain the inconsistency of the prognostic factors so far investigated [9, 15].
In our cohort we found that female patients presented more frequently OPG. Although this finding confirms previous works, the number of patients of the present study is limited and further studies are warranted to support the influence of the female gender on NF1 manifestations and in particular on OPG onset; moreover, we found a higher rate of male patients in the group of OPG patients excluded from the study.
Another main difficulty in comparing data about the natural history of OPG in NF1 patients is the lack of a consensus on the indications for treatment, which however is mainly based on clinical progression. In fact, the absence of clear correlations between radiological and clinical outcomes have been previously confirmed [8, 13]. In our cohort, 8 out of 52 patients underwent a therapy, in seven cases for visual deterioration and just in one case for a radiological progression (who however was treated before 2000).
Significantly, we found that patients with an OPG diagnosed by screening MRI only rarely need treatment compared to those with an OPG diagnosed because of the presence of symptoms and the overall visual outcome after therapy is not improved by screening MRI in the asymptomatic group. These findings corroborate the previous indications of NF1 management, which do not include screening brain/orbit MRIs but recommend adequate ophthalmological follow-up, since a precocious diagnosis of asymptomatic OPG does not appear to change tumour prognosis [11, 21]. Interestingly, no difference in the necessity of treatment is present neither if the onset of subclinical signs, such as fundus oculi abnormalities or OCT alterations, are considered. Prospective studies aimed at evaluating the usefulness of OCT as a precocious marker of neuronal damage and its prognostic significance in patients with OPG are warranted [24]. Our data on visual examination are limited by the retrospective design of the study and by the poor compliance to test in younger patients.
We confirmed that most OPGs identified through screening MRI remain stable and, when evolutive, they generally show only radiological progression.
It should be noted that indications for brain imaging were not homogeneous in our cohort; in fact, although OPG screening in our clinic includes only clinical and ophthalmological examinations and not MRI, some patients underwent neuroradiological studies in other centers.
Another limitation of our study is the small number of cases showing clinical progression, which is a consequence of the strict inclusion criteria. On the other hand, our work is based on prospectively collected data and provides unbiased information compared to previous studies regarding OPG incidence, symptoms, treatment rate and clinical outcome.
In conclusion, although our study presents some limitations that could explain few differences in results compared to previous works, our findings show that decreased visual function at diagnosis appears to be the main prognostic factors in OPG patients and, considering the current outcome after chemotherapy, do not support the role of systematic brain MRI in children affected by NF1. Further prospective studies on larger populations and the identification of potential subclinical signs may allow to clarify the prognostic factors relevant for OPG evolution and to improve the clinical outcome.
References
Clementi M, Barbujani G, Turolla L, Tenconi R (1990) Neurofibromatosis-1: a maximum likelihood estimation of mutation rate. Hum Genet 84(2):116–118
Wallace MR, Marchuk DA, Andersen LB, Letcher R, Odeh HM, Saulino AM, Fountain JW, Brereton A, Nicholson J, Mitchell AL, Brownstein BH, Collins FS (1990) Type-1 Neurofibromatosis gene—identification of a large transcript disrupted in 3 Nf1 patients. Science 249(4965):181–186. doi:10.1126/science.2134734
Ratner N, Miller SJ (2015) A RASopathy gene commonly mutated in cancer: the Neurofibromatosis type 1 tumour suppressor. Nat Rev Cancer 15(5):290–301. doi:10.1038/nrc3911
DeBella K, Szudek J, Friedman JM (2000) Use of the National Institutes of Health Criteria for diagnosis of Neurofibromatosis 1 in children. Pediatrics 105(3):608–614. doi:10.1542/peds.105.3.608
Ferner RE, Huson SM, Thomas N, Moss C, Willshaw H, Evans DG, Upadhyaya M, Towers R, Gleeson M, Steiger C, Kirby A (2007) Guidelines for the diagnosis and management of individuals with Neurofibromatosis 1. J Med Genet 44(2):81–88. doi:10.1136/jmg.2006.045906
Listernick R, Louis DN, Packer RJ, Gutmann DH (1997) Optic pathway gliomas in children with Neurofibromatosis 0.1. Consensus statement from the NF1 optic pathway glioma task force. Ann Neurol 41(2):143–149. doi:10.1002/ana.410410204
Shamji MF, Benoit BG (2007) Syndromic and sporadic pediatric optic pathway gliomas: review of clinical and histopathological differences and treatment implications. Neurosurg Focus 23(5):E3. doi:10.3171/foc.2007.23.5.4
Listernick R, Ferner RE, Piersall L, Sharif S, Gutmann DH, Charrow J (2004) Late-onset optic pathway tumors in children with neurofibromatosis 1. Neurology 63(10):1944–1946
Listernick R, Ferner RE, Liu GT, Gutmann DH (2007) Optic pathway gliomas in neurofibromatosis-1: controversies and recommendations. Ann Neurol 61(3):189–198. doi:10.1002/ana.21107
Helfferich J, Nijmeijer R, Brouwer OF, Boon M, Fock A, Hoving EW, Meijer L, den Dunnen WF, de Bont ES (2016) Neurofibromatosis type 1 associated low grade gliomas: a comparison with sporadic low grade gliomas. Crit Rev Oncol Hematol 104:30–41. doi:10.1016/j.critrevonc.2016.05.008
Balcer LJ, Liu GT, Heller G, Bilaniuk L, Volpe NJ, Galetta SL, Molloy PT, Phillips PC, Janss AJ, Vaughn S, Maguire MG (2001) Visual loss in children with neurofibromatosis type 1 and optic pathway gliomas: relation to tumor location by magnetic resonance imaging. Am J Ophthalmol 131(4):442–445
Taylor T, Jaspan T, Milano G, Gregson R, Parker T, Ritzmann T, Benson C, Walker D, Group PS (2008) Radiological classification of optic pathway gliomas: experience of a modified functional classification system. Br J Radiol 81(970):761–766. doi:10.1259/bjr/65246351
Fisher MJ, Loguidice M, Gutmann DH, Listernick R, Ferner RE, Ullrich NJ, Packer RJ, Tabori U, Hoffman RO, Ardern-Holmes SL, Hummel TR, Hargrave DR, Bouffet E, Charrow J, Bilaniuk LT, Balcer LJ, Liu GT (2012) Visual outcomes in children with neurofibromatosis type 1-associated optic pathway glioma following chemotherapy: a multicenter retrospective analysis. Neuro Oncol 14(6):790–797. doi:10.1093/neuonc/nos076
Tow SL, Chandela S, Miller NR, Avellino AM (2003) Long-term outcome in children with gliomas of the anterior visual pathway. Pediatr Neurol 28(4):262–270
Prada CE, Hufnagel RB, Hummel TR, Lovell AM, Hopkin RJ, Saal HM, Schorry EK (2015) The use of magnetic resonance imaging screening for optic pathway gliomas in children with Neurofibromatosis type 1. J Pediatr 167(4):851–856 e851. doi:10.1016/j.jpeds.2015.07.001
Opocher E, Kremer LC, Da Dalt L, van de Wetering MD, Viscardi E, Caron HN, Perilongo G (2006) Prognostic factors for progression of childhood optic pathway glioma: a systematic review. Eur J Cancer 42(12):1807–1816. doi:10.1016/j.ejca.2006.02.022
Segal L, Darvish-Zargar M, Dilenge ME, Ortenberg J, Polomeno RC (2010) Optic pathway gliomas in patients with neurofibromatosis type 1: follow-up of 44 patients. J AAPOS 14(2):155–158. doi:10.1016/j.jaapos.2009.11.020
Blanchard G, Lafforgue MP, Lion-Francois L, Kemlin I, Rodriguez D, Castelnau P, Carneiro M, Meyer P, Rivier F, Barbarot S, Chaix Y, network NFF (2016) Systematic MRI in NF1 children under six years of age for the diagnosis of optic pathway gliomas. Study and outcome of a French cohort. Eur J Paediatr Neurol 20(2):275–281. doi:10.1016/j.ejpn.2015.12.002
King A, Listernick R, Charrow J, Piersall L, Gutmann DH (2003) Optic pathway gliomas in neurofibromatosis type 1: the effect of presenting symptoms on outcome. Am J Med Genet A 122A(2):95–99. doi:10.1002/ajmg.a.20211
Dalla Via P, Opocher E, Pinello ML, Calderone M, Viscardi E, Clementi M, Battistella PA, Laverda AM, Da Dalt L, Perilongo G (2007) Visual outcome of a cohort of children with Neurofibromatosis type 1 and optic pathway glioma followed by a pediatric neuro-oncology program. Neuro Oncol 9(4):430–437. doi:10.1215/15228517-2007-031
Kalin-Hajdu E, Decarie JC, Marzouki M, Carret AS, Ospina LH (2014) Visual acuity of children treated with chemotherapy for optic pathway gliomas. Pediatr Blood Cancer 61(2):223–227. doi:10.1002/pbc.24726
Neurofibromatosis. Conference statement. National Institutes of Health Consensus Development Conference (1988) Arch Neurol 45(5):575–578
Listernick R, Mancini AJ, Charrow J (2003) Segmental neurofibromatosis in childhood. Am J Med Genet A 121A(2):132–135. doi:10.1002/ajmg.a.20183
Parrozzani R, Clementi M, Kotsafti O, Miglionico G, Trevisson E, Orlando G, Pilotto E, Midena E (2013) Optical coherence tomography in the diagnosis of optic pathway gliomas. Invest Ophthalmol Vis Sci 54(13):8112–8118. doi:10.1167/iovs.13-13093
Pan Y, Tarczy-Hornoch K, Cotter SA, Wen G, Borchert MS, Azen SP, Varma R, Grp M (2009) Visual acuity norms in pre-school children: the multi-ethnic pediatric eye disease study. Optom Vis Sci 86 (6):607–612
International Classification of Impairments, Disabilities, and Handicaps. A manual of classification relating to the consequences of disease (1980). World Health Organization 1980 edn
Friedrich RE, Nuding MA (2016) Optic pathway glioma and cerebral focal abnormal signal intensity in patients with Neurofibromatosis type 1: characteristics, treatment choices and follow-up in 134 affected individuals and a brief review of the literature. Anticancer Res 36(8):4095–4121
Sharif S, Ferner R, Birch JM, Gillespie JE, Gattamaneni HR, Baser ME, Evans DG (2006) Second primary tumors in neurofibromatosis 1 patients treated for optic glioma: substantial risks after radiotherapy. J Clin Oncol 24(16):2570–2575. doi:10.1200/JCO.2005.03.8349
Dodge HW Jr, Love JG, Craig WM, Dockerty MB, Kearns TP, Holman CB, Hayles AB (1958) Gliomas of the optic nerves. AMA. Arch Neurol Psychiatry 79(6):607–621
Diggs-Andrews KA, Brown JA, Gianino SM, Rubin JB, Wozniak DF, Gutmann DH (2014) Sex Is a major determinant of neuronal dysfunction in neurofibromatosis type 1. Ann Neurol 75(2):309–316. doi:10.1002/ana.24093
Johnson KJ, Scheurer ME, Woehrer A, Wiemels J (2016) Evolving evidence on tumor and germline genetic classification of gliomas: implications for etiology and survival studies. Clin Neuropathol 35(1):31–38. doi:10.5414/NP300917
Thiagalingam S, Flaherty M, Billson F, North K (2004) Neurofibromatosis type 1 and optic pathway gliomas: follow-up of 54 patients. Ophthalmology 111(3):568–577. doi:10.1016/j.ophtha.2003.06.008
Blazo MA, Lewis RA, Chintagumpala MM, Frazier M, McCluggage C, Plon SE (2004) Outcomes of systematic screening for optic pathway tumors in children with Neurofibromatosis Type 1. Am J Med Genet A 127 A(3):224–229. doi:10.1002/ajmg.a.20650
Massry GG, Morgan CF, Chung SM (1997) Evidence of optic pathway gliomas after previously negative neuroimaging. Ophthalmology 104(6):930–935
Listernick R, Charrow J, Greenwald M (1992) Emergence of optic pathway gliomas in children with neurofibromatosis type 1 after normal neuroimaging results. J Pediatr 121(4):584–587
Doganis D, Pourtsidis A, Tsakiris K, Baka M, Kouri A, Bouhoutsou D, Varvoutsi M, Servitzoglou M, Dana H, Kosmidis H (2016) Optic pathway glioma in children: 10 years of experience in a single institution. Pediatr Hematol Oncol 33(2):102–108. doi:10.3109/08880018.2016.1155101
Gottschalk S, Tavakolian R, Buske A, Tinschert S, Lehmann R (1999) Spontaneous remission of chiasmatic/hypothalamic masses in Neurofibromatosis type 1: report of two cases. Neuroradiology 41(3):199–201
Parsa CF, Hoyt CS, Lesser RL, Weinstein JM, Strother CM, Muci-Mendoza R, Ramella M, Manor RS, Fletcher WA, Repka MX, Garrity JA, Ebner RN, Monteiro MLR, McFadzean RM, Rubtsova IV, Hoyt WF (2001) Spontaneous regression of optic gliomas—thirteen cases documented by serial neuroimaging. Arch Ophthalmol-Chic 119(4):516–529
Acknowledgements
Funding for this work was provided by the Italian NF patients’ association Linfa Onlus (www.associazionelinfa.it). The ophthalmological part of this research was financially supported by Ministry of Health and Fondazione Roma. We appreciate the kind cooperation of the patients and their families.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
The authors declare that they have no conflict of interest.
Ethical approval
For this type of study formal consent is not required.
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
About this article

Cite this article
Trevisson, E., Cassina, M., Opocher, E. et al. Natural history of optic pathway gliomas in a cohort of unselected patients affected by Neurofibromatosis 1. J Neurooncol 134, 279–287 (2017). https://doi.org/10.1007/s11060-017-2517-6
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s11060-017-2517-6