
Abstract
Primary spinal peripheral primitive neuroectodermal tumors (pPNETs) are extremely rare entities that predominantly occur in children and young adults. Few studies have reported more than three cases. There are no current optimum treatment strategies due to the paucity of data. Here, we present 13 patients (nine females and four males) with primary intraspinal pPNETs who were surgically treated from April 2008 to February 2014. Histopathologic findings revealed the expression of CD99 in all cases. Limb weakness was the most common initial symptom (11/13, 85 %). The tumors were located mainly at the cervical level (6/13, 46 %) and in the epidural space (10/13, 77 %). The radiological diagnosis was neurinoma or meningioma in most cases (10/13, 77 %). Gross total resection was achieved in 77 % (10/13) of patients. During a mean follow-up of 25.5 months, local relapse occurred in 8 (61.5 %) patients and distant metastases occurred in 8 (61.5 %) patients. The overall 1-year survival rate was 77 % (10/13), and the overall 2-year survival rate was 54 % (7/13). The 2-year survival rate was 57.1 % in patients with adjuvant chemotherapy and 50 % in those without chemotherapy. Gross total resection and adjuvant radiotherapy with or without chemotherapy demonstrated a longer survival period (1-year survival rate: 100 %; 2-year survival rate: 86 %). Our data showed that primary spinal pPNETs are extremely rare and aggressive tumors with a poor prognosis. Radical resection is advocated. Gross total resection combined with adjuvant radiation may help to significantly improve patient survival period. Chemotherapy may also help to slightly prolong patient life.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Primary spinal primitive neuroectodermal tumors (PNETs) are poorly differentiated malignant tumors that usually occur in children and adolescents. PNETs include two categories: CNS PNETs or central PNETs and Ewing sarcoma/PNETs or peripheral PNETs. In recent classification by WHO, central PNETs (cPNETs) and medulloblastomas were subdivided under embryonic tumors [1]. Unlike cPNETs, which are referred to as tumors arising in the central nervous system (CNS) [2], peripheral PNETs (pPNETs) are non-CNS tumors derived from the neural crest [3], belonging to the Ewing sarcoma family of tumors (ESFTs) [4]. Primary spinal pPNETs are extremely rare entities. In the literature, slightly more than 100 cases of primary spinal pPNETs have been reported. Due to the limited reports, the epidemiology of primary spinal PNETs is unclear and the prognosis is very poor. Although multidisciplinary treatment has been well established as the standard treatment strategy for intracranial PNETs, there is no current standard treatment protocol for primary spinal pPNETs. Here, we review our series of 13 patients and discuss the clinical course, treatment and outcome of primary spinal pPNETs.
Methods
Patient population
Between April 2008 and February 2014, 13 patients with a definite pathological diagnosis of primary spinal pPNETs were treated at the Department of Neurosurgery in Beijing Tiantan Hospital and Beijing Jishuitan Hospital. This study included nine female and four male patients. The age at presentation ranged from 4 to 47 years (mean 19.8 years). The details of the patients’ baseline clinical characteristics are presented in Table 1. The inclusion criteria were primary spinal PNETs with the expression of CD99. The exclusion criteria were those with CNS PNETs, unavailable pathological data, incomplete follow-up, dissemination from the intracranial locations or any evidence of metastatic disease as revealed by brain MRI,chest CT, bone scan, bone marrow biopsy and CSF cytology. The natural history was based on the time span from the initial onset of the disease until surgery (Table 1).
Surgical treatment
Under general anesthesia, a dorsal approach was made through laminectomy in all patients. Neurophysiological monitoring was used. The tumors were located in the epidural space in 10 cases and in the intradural-extramedullary space in three cases. For those with epidural tumors, the dura mater was not opened. For those with intra- and extra-spinal tumors, the incision was extended according to tumor location. For those with intFradural tumors, prior to opening the dura mater, the location of the tumor was confirmed using an intraoperative ultrasonography device, which guided the extent of bone removal. At surgery, the tumors appeared as reddish or reddish-gray and soft lesions with abundant blood supply. The tumor margin appeared to be well demarcated. Intraoperative frozen sections showed undifferentiated small round cells. Under microsurgical conditions, gross total resection of the tumors was achieved in ten patients. Spinal fixation was performed through synthetic vertebral fixation devices in six patients to reduce vertebral mobility and avoid possible damage to the spinal cord and spinal roots. None of the patients experienced notable immediate-postoperative complications.
Adjuvant therapy
Two to four weeks after the initial surgery, the patients were referred to clinical oncologists. However, the use of chemotherapy or radiotherapy was determined by patient age, Karnofsky performance status scale (KPS) and patient preference. The pre- and post-operative KPS scores of the patients are listed in Table 1. Patients with a KPS ≥ 70 after surgery were suggested to undergo chemotherapy. The vincristine, doxorubicin, cyclophosphamide (VAC) or vincristine, ifosfamide, doxorubicin, etoposide (VIDE) protocol was recommended for chemotherapy. Radiotherapy was initiated 2 weeks after surgery for those who did not receive chemotherapy. For those with chemotherapy, radiotherapy was initiated concurrently or at a later stage, according to patient tolerance and compliance. The patients’ complete blood count and liver and kidney function were examined regularly to determine further adjuvant therapy.
Follow-up data
All patients underwent clinical and spinal MR imaging follow-up. Positron emission tomography-computed tomography (PET/CT) was used in some cases to reveal distant metastasis. The long-term treatment outcome was based on the follow-up from the time of surgery to the most recent medical information. The follow-up period ranged from 9 to 86 months (mean 25.5 months).
Results
Initial clinical presentation
The clinical presentation showed considerable variability (Table 1). The time interval between symptomatic onset and initial surgery was within 3 months (range 1 day–3 months; mean 40 days). The most common symptom at presentation was weakness in the limbs (in 11 cases), followed by pain (in six cases) and incontinence (in six cases). One patient presented sudden quadriplegia and incontinence because of tumor apoplexy, which led to acute spinal cord compression (Case 7). All patients had localized lesions, and none had metastatic disease at initial presentation.
Magnetic resonance data
The pPNETs were located throughout the spinal axis, but mainly at the cervical (6 cases) and lumbosacral levels (three cases) (Table 1). The tumors were located solely in the spinal canal in six cases. Intra and extra spinal communicating tumors were found in six cases (including dumbbell tumors). One was located in the foraminal space. A brain MRI, chest CT and bone scan did not reveal evidence of metastatic disease. The preoperative diagnosis was neurinoma in eight patients, meningioma in two patients, neurofibroma in one patient, sarcoma in one patient and hematoma in one patient. As revealed by postoperative MR imaging, gross total resection of the tumors was achieved in ten patients (Fig. 1).

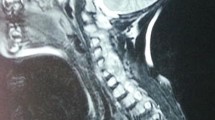
Case 8. a, b shows a moderate to prominent contrast-enhanced intradural lesion at the C3–5 level on sagittal and coronal T1-weighted MRI. 1 week after surgery, sagittal T1-weighted MRI (c) and T2-weighted MRI (d) show gross total resection of the tumor
Pathologic findings
The tumors presented uniform findings, showing well-circumscribed, reddish or reddish-gray soft tissues with an abundant blood supply. Because the primary preoperative diagnosis was mainly neurinoma, meningioma or neurofibroma, the histological findings were surprising. Light microscopic histological examination of hematoxylin-eosin-stained slides revealed highly cellular tumors consisting mainly of small round undifferentiated cells with hyperchromatic nuclei and a small cytoplasmatic wall. No well-defined Homer-Wright rosettes were found (Fig. 2a). High mitotic activity and necroses were presented. Immunohistochemistry using anti-MIB-1 (Ki 67) antibodies revealed a high proliferative index in most cases. Further staining revealed CD99 (MIC2) (+) in all patients (Fig. 2b). Vimentin was positive in 10 cases. Strong immunoreactivity for FLI-1 was detected in 8 patients. Fluorescence in situ hybridization and chromosomal study were not performed in this series.

Case 8. a The light microscopic photograph shows a typical highly cellular pPNET tumor consisting of undifferentiated small round cells with frequent mitoses. b Immunohistochemistry shows CD 99 immunoreactivity in pPNET tumor cells
Treatment modalities and long-term outcome
As listed in Table 2, the treatment modalities were gross total resection with adjuvant chemotherapy and radiotherapy in five patients, partial resection with adjuvant chemotherapy and radiotherapy in one patient, gross total resection with adjuvant chemotherapy in one patient, gross total resection with adjuvant radiotherapy in two patients, partial resection with adjuvant radiotherapy in two patients and gross total resection without adjuvant therapy in two patients. During follow-up, eight patients had local recurrence (Table 2; Fig. 3) and four patients underwent a second or third operation. The distant metastatic sites was the lung in five patients, sternum in two patients, rib in one patient, mediastinum in one patient and lumbar spinal canal in one patient (Table 2; Fig. 3). The overall 1-year survival rate was 77 % (10/13) and the 2-year survival rate was 54 % (7/13). The 1- and 2-year survival rates were 71 and 57.1 %, respectively, in patients with adjuvant chemotherapy, while they were 83 and 50 %, respectively, in patients without chemotherapy. Gross total resection and adjuvant radiotherapy with or without chemotherapy showed a relatively longer survival period (1-year survival rate: 100 %; 2-year survival rate: 86 %). However, whether adjuvant chemotherapy or radiotherapy or both were applied, partial resection of the tumors resulted in the rapid progress of local recurrence and distant metastasis, leading to a short survival period. Patients with early distant metastasis after surgery had a relatively shorter survival period. Due to the limited number of patients, statistical analysis was not used in this study.

MRI of Case 8 2 years after surgery. a, b Local recurrence of the tumor on sagittal and coronal T1-weighted contrast MRI. c, d Tumor dissemination at the L5 level on sagittal and coronal T1-weighted contrast MRI
Discussion
Concept and classification of PNETs
Primary spinal PNETs are very rare soft tissue neoplasms in both children and young adults [5]. In 1973,Hart and Earle formally introduced the concept and diagnostic criteria of PNETs, proposing that PNETs are undifferentiated malignant tumors of intracranial origin [2]. The World Health Organization classification in 1993 included cerebral medulloblastoma-like tumors as supratentorial PNETs [6]. In the third edition of the World Health Organization classification, PNETs were defined as embryonic tumors composed of undifferentiated or poorly differentiated neuroepithelial cells that have the capacity for or display divergent differentiation along neuronal astrocytic, ependymal, muscular or melanotic lines [7]. In recent classification by WHO, central PNETs (cPNETs) and medulloblastomas were subdivided under embryonic tumors [1]. Central PNET are referred to as neoplasms arising in the CNS but located outside the posterior fossa. pPNETs are non-CNS tumors derived from the neural crest [3], belonging to the ESFTs [4]. Primary intraspinal PNETs are extremely rare. These tumors may originate at all levels of the spine and can be intramedullary, intra- and extramedullary, extramedullary, or epidural. Because PNETs frequently disseminate or seed throughout the CNS, most cases of PNETs involving the spinal cord are “drop” metastases from primary intracranial tumors. For the correct diagnosis of spinal PNETs, the “drop” metastases of intracranial origin must be excluded [8, 9].
Epidemiology of primary spinal pPNETs
Primary spinal pPNETs are rare entities. Slightly more than 100 cases have been reported in the literature. Most were reports in the past 15 years with one or two cases and only four studies reported more than three cases [10–13]. It is unreliable to have an accurate epidemiology from these case reports. Engelhard et al. reported that spinal PNETs account for <1 % of the primary tumors in their 430 cases [14]. From 2008 to 2014, 3012 patients with primary spinal tumors were admitted to our department. We conclude that primary spinal pPNETs account for approximately 0.4 % of the primary spinal tumors treated at our department. In the recent two literature reviews of primary spinal pPNETs, a nearly 2:1 male sex preponderance was found [15, 16]. However, in our series, 69 % (9/13) were female and 31 % (4/13) were male, which is just contrary to their findings. Previous studies have asserted that primary spinal PNETs are most prevalent in children and young adults [4, 15–18]. Our study is consistent with those results. The mean age at presentation was 19.8 years. Additionally 77 % (10/13) of the patients were under the age of 20 years and 23 % (3/13) were in their 40s.
Clinical presentation
The clinical presentation of primary spinal pPNETs varied according to tumor locations and involved structures. In a recent meta-analysis of reported cases in the literature, muscle weakness is the most common symptom. Sensory symptoms, local pain, and radiculopathy are also common presentations [16]. In our series, 85 % (11/13) of the patients presented with varied degrees of limb weakness. Incontinence was found in 46 % (6/13) patients, mainly due to the location of the tumors. Patients may present with neck and shoulder pain mimicking cervical spondylosis (Case 1) or lumbago and scelalgia mimicking lumbar disc herniation (Case 6), particularly in adults. Additionally these patients would not be aware of their disease until a later stage, such as Case 1 and Case 6, who are in their 40s. The mean duration of symptoms before surgery was 40 days, much less than previously reported [16]. In 2011, Ellis et al. [15] reported the first case with intratumoral hemorrhage that resulted in acute spinal cord compression and neurological decline. Case 7 in our series presented with sudden quadriplegia and incontinence, which is the second reported case of intratumoral hemorrhage.
Radiological features
Ellis et al. [15] reported that spinal PNETs are typically hypo—to isointense on T1-weighted MR imaging and iso—to hyperintense on T2-weighted imaging. Our findings are consistent with those results. Their study showed that there was often minimal contrast enhancement [15]. Tsutsumi et al. [19] reviewed the literature and found that varying enhancement was noted. Moderate to prominent contrast enhancement were found in most of our cases. A recent meta-analysis showed that approximately half of the primary intraspinal pPNET cases occur in the lumbar spine [16]. Tsutsumi et al. [19] also reported that the thoracic spine was the most frequently affected area. In our series, 55 % (6/11) of the tumors were located at the cervical level and 27 % (3/11) at L5/S1 level, which is contrary to previous findings. Also in the recent meta-analysis, the author reported their case is the first female and the sixth patient with sacral epidural primary intraspinal pPNET [16]. Case 12 in our series is the sixth male and seventh patient in this category, and the patient is currently in good condition now. The radiographic findings of primary spinal pPNETs are usually not helpful in diagnosis and differential diagnosis, as was stated by previous studies [15, 19]. Radiological diagnosis may be neurinoma, meningioma, neurofibroma, especially neurinoma. Other diagnoses may be ependymoma or astrocytoma [15]. Most primary spinal pPNETs are epidural lesions and may often occupy the intraspinal and extraspinal space. Intradural locations are less frequent. Intramedullary location is extremely rare.
Diagnosis
PNETs are poorly differentiated small round blue cell tumors with hyperchromatic nuclei and features of neural differentiation, rarely with typical Homer–Wright rosettes [20]. cPNETs and pPNETs are often distinguishable according to their morphological features. Correct diagnosis depends on immunohistochemistry and cytogenetic analysis. In the literature, nearly half of the reports on primary spinal pPNETs did not make a histopathologic distinction between cPNET and pPNET [16]. Kampman et al. recently summarized the distinguishing features between cPNET and pPNET [4]. Immunocytochemical staining can distinguish between pPNET and cPNET by demonstrating the expression of MIC2 glycoprotein (CD99) in pPNET and showing the presence of the specific chimeric gene of EWS-FLI1 in pPNET [4]. The membrane protein FLI1 (Friend leukemia virus integration 1) are also commonly expressed in pPNETs [21–24]. Immunohistochemical staining showed that the tumor cells expressed CD99 in all 13 patients in our series, consistent with the diagnosis of pPNETs. FLI-1 was positive in 62 % (8/13) of the patients, which further confirmed the diagnosis of pPNETs. Cytogenetic analysis has shown that chromosomal translocation (11;22) (q24;q12) presents in more than 90 % of cases [8, 25, 26]. The presence of an (11;22) (q24;q12) translocation may be the strongest diagnostic tool for pPNETs [15]. However, chromosomal studies were performed in only a small portion (approximately 1/7) of the reported cases in the literature [16].
Treatment options
There are no current standard therapeutic strategies for primary spinal pPNETs. However, surgical removal of the tumors is the cornerstone of therapy [20]. Although tumor relapses and metastasis might occur even with gross total resection, radical resection is still recommended to prevent further neurological decline, improve quality of life, allow early mobilization and achieve sufficient volume reduction for further oncological management [18]. Gross total resection was achieved in 77 % (10/13) of patients in our series without causing further neurological damage. A standard laminectomy with preservation of the facet joints should be performed [15]. Intraoperative neurophysiological monitoring should be used. An ultrasonography device is advocated to guide the extent of bone removal and tumor resection. The tumor margins are usually well demarcated. Frozen sections should also be advocated during surgery. Spinal fixation should be performed in selected patients to reduce vertebral mobility and avoid possible damage to adjacent essential structures.
Due to the paucity of data, the benefit of adjuvant therapy remains unclear. A recent meta-analysis showed that adjuvant chemotherapy improved the 1- and 2-year survival rates after total or subtotal tumor resection [16]. The VAC or VIDE protocol was used in our patients with adjuvant chemotherapy. Although the 1-year survival rate in patients with adjuvant chemotherapy was slightly lower than that in those without chemotherapy (71 vs 83 %), the 2-year survival rate was relatively higher in patients with chemotherapy (57.1 vs 50 %). Gross total resection and adjuvant radiotherapy with or without chemotherapy had a relatively longer survival period (1-year survival rate: 100 %; 2-year survival rate: 86 %). Our experience showed that radiotherapy was relatively more important than adjuvant chemotherapy in prolonging the survival period. However, gross total resection should be the prerequisite. Patients with partial resection of tumors may survive for a period of <1 year even with adjuvant chemotherapy and/or radiotherapy. Radiation strategies varied in different reports. Controversies exist regarding the optimal radiation strategy, the indications of full neural axis radiation and dosing in the treatment of primary spinal PNETs [27]. In the literature, total doses ranged from 30 to 60 Gy [15]. In our series, the total doses employed ranged from 30 to 50 Gy. None of our patients received the combination of high-dose chemotherapy and autologous stem cell rescue as a supplement of surgery and radiotherapy. However, this treatment modality has shown promising results in previous reports [28, 29].
Prognosis
Primary spinal pPNETs are aggressive malignant tumors that lead to a lower 2-year survival rate [16]. Due to the limited reports in the literature, the prognostic factors are unclear. Regarding our series, partial resection, local recurrence and distant metastasis may predict a shorter survival period. However, gross total resection and radiation can significantly improve the survival rate of the patients. Adjuvant chemotherapy combined with gross total resection can also slightly prolong patient life. Patients with this tumor entity have a low 3-year survival rate (40 % in our series) and seldom survive 5 years after diagnosis (two patients in our series). Three patients were alive at our last follow-up. Two patients were uneventful and led a normal life (Case 6 and 12). Case 8 has experienced local recurrence and lumbar spinal canal dissemination of the tumor (Fig. 3). And this patient will undergo surgery at our department or another hospital. We will continue to follow up these three patients.
Conclusions
Primary spinal pPNETs are extremely rare and aggressive tumors with a poor prognosis. Clinical presentation varies from patient to patient. Radiological features may lead to misdiagnosis. Diagnosis of this disease depends on histopathologic findings. There is no standard treatment protocol due to the rarity of this disease. Microsurgical resection of the tumor is the mainstay of treatment. Gross total resection combined with adjuvant radiation may help to improve the survival period of the patients. Chemotherapy may also help to slightly prolong patient life.
References
Louis DN, Ohgaki H, Wiestler OD, Cavenee WK (2007) WHO classification of tumors of the central nervous system, 4th edn. IARC, Lyon
Hart MN, Earle KM (1973) Primitive neuroectodermal tumors of the brain in children. Cancer 32:890–897
Shimada H, Brodeur GM (1998) Tumors of peripheral neuroblastomas and ganglion cells. In: Bigner DD et al (eds) Russel and Rubinstein’s pathology of tumors of the nervous system, 6th edn. Arnold, London, pp 493–533
Kampman WA, Kros JM, De Jong TH, Lequin MH (2006) Primitive neuroectodermal tumors (PNETs) located in the spinal canal: the relevance of classification as central or peripheral PNET: case report of a primary spinal PNET occurrence with a critical literature review. J Neurooncol 77:65–72
Albrecht CF, Weiss E, Schultz-Schaeffer WJ, Albrecht T, Fauser S, Wickboldt J, Hess CF (2003) Primary intraspinal primitive neuroectodermal tumor: report of two cases and review of the literature. J Neurooncol 61:113–120
Kleihues P, Burger PC, Scheithauer BW (1993) The new WHO classification of brain tumors. Brain Pathol 3:255–268
Rorke LB, Hart MN, McLendon RE (2000) Supratentorial primitive neuroectodermal tumor (PNET). In: Kleihues P, Cavennee WK (eds) Pathology and genetics of tumors of the central nervous system. IARC Press, Lyon, pp 141–144
Deme S, Ang LC, Skaf G, Rowed DW (1997) Primary intramedullary primitive neuroectodermal tumour of the spinal cord: case report and review of the literature. Neurosurgery 41:1417–1420
Dorfmuller G, Wurtz FG, Umschaden HW, Kleinert R, Ambros PF (1999) Intraspinal primitive neuroectodermal tumour: report of two cases and review of the literature. Acta Neurochir (Wien) 141:1169–1175
Mukhopadhyay P, Gairola M, Sharma M, Thulkar S, Julka P, Rath G (2001) Primary spinal epidural extraosseous Ewing’s sarcoma: report of five cases and literature review. Australas Radiol 45:372–379
Harimaya K, Oda Y, Matsuda S, Tanaka K, Chuman H, Iwamoto Y (2003) Primitive neuroectodermal tumor and extraskeletal Ewing sarcoma arising primarily around the spinal column: report of four cases and a review of the literature. Spine (Phila Pa 1976) 28:E408–E412
Jingyu C, Jinning S, Hui M, Hua F (2009) Intraspinal primitive neuroectodermal tumors: report of four cases and review of the literature. Neurol India 57:661–668
Duan XH, Ban XH, Liu B, Zhong XM, Guo RM, Zhang F, Liang BL, Shen J (2011) Intraspinal primitive neuroectodermal tumor: imaging findings in six cases. Eur J Radiol 80:426–431
Engelhard HH, Villano JL, Porter KR, Stewart AK, Barua M, Barker FG, Newton HB (2010) Clinical presentation, histology, and treatment in 430 patients with primary tumors of the spinal cord, spinal meninges, or cauda equina. Clinical article. J Neurosurg Spine 13:67–77
Ellis JA, Rothrock RJ, Moise G, McCormick PC 2nd, Tanji K, Canoll P, Kaiser MG, McCormick PC (2011) Primitive neuroectodermal tumors of the spine: a comprehensive review with illustrative clinical cases. Neurosurg Focus 30:E1
Saeedinia S, Nouri M, Alimohammadi M, Moradi H, Amirjamshidi A (2012) Primary spinal extradural Ewing’s sarcoma (primitive neuroectodermal tumor): report of a case and meta-analysis of the reported cases in the literature. Surg Neurol Int 3:55
Jain A, Jalali R, Nadkarni TD, Sharma S (2006) Primary intramedullary primitive neuroectodermal tumor of the cervical spinal cord. Case report. J Neurosurg Spine 4:497–502
Hrabálek L, Kalita O, Svebisova H, Ehrmann J Jr, Hajduch M, Trojanec R, Kala M (2009) Dumbbell-shaped peripheral primitive neuroectodermal tumor of the spine—case report and review of the literature. J Neurooncol 92:211–217
Tsutsumi S, Yasumoto Y, Manabe A, Ogino I, Arai H, Ito M (2013) Magnetic resonance imaging appearance of primary spinal extradural Ewing’s sarcoma: case report and literature review. Clin Neuroradiol 23:81–85
Perry R, Gonzales I, Finlay J, Zacharoulis S (2007) Primary peripheral primitive neuroectodermal tumors of the spinal cord: report of two cases and review of the literature. J Neurooncol 81:259–264
Folpe AL, Goldblum JR, Rubin BP, Shehata BM, Liu W, Dei Tos AP, Weiss SW (2005) Morphologic and immunophenotypic diversity in Ewing family tumors: a study of 66 genetically confirmed cases. Am J Surg Pathol 29:1025–1033
Folpe AL, Hill CE, Parham DM, O’Shea PA, Weiss SW (2000) Immunohistochemical detection of FLI-1 protein expression: a study of 132 round cell tumors with emphasis on CD99-positive mimics of Ewing’s sarcoma/primitive neuroectodermal tumor. Am J Surg Pathol 24:1657–1662
Llombart-Bosch A, Navarro S (2001) Immunohistochemical detection of EWS and FLI-1 proteins in Ewing sarcoma and primitive neuroectodermal tumors: comparative analysis with CD99 (MIC-2) expression. Appl Immunohistochem Mol Morphol 9:255–260
Mhawech-Fauceglia P, Herrmann F, Penetrante R, Beck A, Sait S, Block AM, Odunsi K, Fisher J, Balos L, Cheneyet RT (2006) Diagnostic utility of FLI-1 monoclonal antibody and dual-colour, break-apart probe fluorescence in situ (FISH) analysis in Ewing’s sarcoma/primitive neuroectodermal tumour (EWS/PNET). A comparative study with CD99 and FLI-1 polyclonal antibodies. Histopathology 49:569–575
Bailly RA, Bosselut R, Zucman J, Cormier F, Delattre O, Roussel M, Thomas G, Ghysdael J (1994) DNA-binding and transcriptional activation properties of the EWS-FLI-1 fusion protein resulting from the t(11;22) translocation in Ewing sarcoma. Mol Cell Biol 14:3230–3241
Delattre O, Zucman J, Melot T, Garau XS, Zucker JM, Lenoir GM, Ambros PF, Sheer D, Turc-Carel C, Triche TJ, Aurias A, Thomas G (1994) The Ewing family of tumors—a subgroup of smallround-cell tumors defined by specific chimeric transcripts. N Engl J Med 331:294–299
Weber DC, Rutz HP, Lomax AJ, Schneider U, Lombriser N, Zenhausern R, Goitein G (2004) First spinal axis segment irradiation with spot-scanning proton beam delivered in the treatment of a lumbar primitive neuroectodermal tumour. Case report and review of the literature. Clin Oncol (R Coll Radiol) 16:326–331
Bohn Sarmiento U, Aguiar Bujanda D, Camacho Galán R, Rivero Vera JC, Aguiar Morales J (2005) Lumbar region intra-spinal primitive neuroectodermal tumour (PNET) combined with neurofibromatosis type 1. Clin Transl Oncol 7:464–467
Nutman A, Postovsky S, Zaidman I, Elhasid R, Vlodavsky E, Kreiss Y, Ben Arush MW (2007) Primary intraspinal primitive neuroectodermal tumor treated with autologous stem cell transplantation: case report and review of the literature. Pediatr Hematol Oncol 24:53–61
Acknowledgments
We are grateful for the help of Professor Luo Lin, chief of the Pathologic Department at the Beijing Neurosurgical Institute, and Professor Huang Xiaoyuan, chief of the Pathologic Department at Beijing Jishuitan Hospital.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
The authors declare that they have no conflict of interest.
Rights and permissions
About this article

Cite this article
Tong, X., Deng, X., Yang, T. et al. Clinical presentation and long-term outcome of primary spinal peripheral primitive neuroectodermal tumors. J Neurooncol 124, 455–463 (2015). https://doi.org/10.1007/s11060-015-1859-1
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s11060-015-1859-1