
Abstract
Salen and Salphen ligands have attracted important attention due to their easy synthesis and ability to form stable complexes with transition metals for main application in catalysis. MetalloSalen and metalloSalphen complexes exhibited good catalytic activity for many chemical transformations. But they suffer from some limitations including difficult separation from the reaction mixture and non-reusability. Immobilizing these complexes into solid supports could solve such issues and afford efficient and recyclable heterogeneous catalysts. Mesoporous silica materials (MSMs), such as SBA-15, MCM-41, and MCM-48, are ideal support materials due to their important features, such as large surface areas, highly ordered nanostructure, narrow pore size distribution, tunable porosity, easy to functionalize, and high chemical and hydrothermal stability. These properties allowed high dispersion and better catalytic activity of the reported heterogeneous catalysts. This review article provides an overview about the synthesis of metalloSalen/Salphen complexes, their immobilization into MSMs, and their utilization as heterogeneous catalysts for different types of reactions such as the oxidation and hydrogenation of hydrocarbons. Different techniques to incorporate these complexes into MSMs have been discussed, including grafting, co-condensation, periodic mesoporous organosilicas (PMOs), and coordination-assisted grafting. The developed heterogeneous catalysts combine the benefits of metalloSalen/Salphen complexes as efficient homogeneous catalysts and the advantageous features of MSMs as support. Compared to their homogeneous analogues, the MSM-supported complexes are recyclable and often exhibit higher catalytic activity due to the high active site’s distribution.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Salen and Salphen ligands and related metal complexes
Salen and related metal complexes
Simple Salen
The coupling of aldehyde group with primary amine forms imine bond which is called Schiff base [1]. For many years, Schiff base ligands have been used successfully as ligands in conjunction with different metals to create homogeneous catalysts which can catalyze a variety of chemical transformations [2, 3]. Schiff base reaction is a simple condensation reaction between a primary amine and an aldehyde or ketone, in the presence of an acid as a catalyst (Scheme 1) [4].

General Schiff base reaction mechanism [4]
In fact, the reaction begins with a nucleophilic attack of the nitrogen atom to the carbon atom of the carbonyl group, to form an unstable ammonium intermediate. This intermediate transforms to the imine (C = N) form after the removal of the hydroxyl group in form of water molecule as follows [4]:
There are many important factors that can affect the Schiff base condensation reaction to yield Schiff base products. One important parameter is the pH of the reaction medium. An acidic pH is necessary to accelerate multiple steps in the reaction mechanism. First of all, the amine needs to be protonated to an ammonium ion, because under very basic or neutral conditions, the amine cannot be protonated and nucleophilic enough to attack the carbonyl carbon [5]. This attack forms a carbinolamine intermediate. Then, acid also is required to protonate and remove the hydroxyl group from this intermediate, enabling formation of the carbonyl group and imine bond generation. The steric and electronic effects of the carbonyl and amine of the starting materials also significantly affect the Schiff base reaction. Aldehydes react more smoothly than ketones due to their carbonyl carbon appearing less sterically hindered, which would facilitate interaction with the amine group of the ligand [6]. Furthermore, the electron-withdrawing groups on aldehydes increase the carbonyl’s electrophilicity. However, large substituents like tert-butyl form steric hindrance that can slow the reaction, while such steric hindrance can present a challenge. The optimization of some parameters such as temperature, solvent, catalyst, and acid concentration can promote the condensation reaction even of highly substituted precursors. This increases the diversity of synthetically designed ligands [7, 8].
The “Salen” ligand is one of the most popular Schiff base ligands, which has undergone extensive study (Fig. 1) [2, 3].

Structure of the Salen (or H2Salen) ligand [2]
Pfeiffer et al. [2] published the first study on Salen-metal complexes in 1933. The word “Salen” is made up of two abbreviations, “sal” stands for salicylaldehyde and “en” stands for ethylenediamine [9]. Salen ligands are linear tetradentate, N2O2-coordinating ligand systems which are formed from the condensation reaction of two equivalents of a salicylaldehyde precursor with one equivalent of ethylenediamine as illustrated in Scheme 2 below [10].

Structure of the Salen (or H2Salen) ligand [9]
Usually, these reactions are simple and do not require any catalyst. However, often, the products undergo reversible hydrolysis. To avoid the hydrolysis of the Salen, dehydrating agents or molecular sieves can be used, so water molecules formed during the reaction can be absorbed. When a water-immiscible solvent is employed (such toluene or benzene), a Dean-Stark apparatus can be also used to trap water [9, 10]). Moreover, Salen ligands exhibit several properties that make them attractive to prepare metalloSalen catalysts. As flexible organic frameworks, they can favorably interact with orient substrates through non-covalent interactions Also, the electronic and steric characteristics of the ligands can be modified by varying the substituents on the phenolate and diamine units [10]. This enables a systematic tuning of some properties such as acidity, basicity, and lipophilicity through rational ligand design [11].
Furthermore, by using other diamine derivative, the analogues H2salmen, H2saltn, H2salchxn, and H2salben have been also reported (Fig. 2) [12,13,14,15]. The utilization of a different carbonyl moiety is also reported, as shown for H2hapen and H2acacen (Fig. 2) [12,13,14,15]. These Schiff bases have been probably used for coordinating almost all the metals of the periodic table of elements due to the hosting tetradentate pocket and applied in catalysis biomimetic systems and materials for their optical, electronic, and magnetic features [16,17,18,19,20].

The utilization of chiral diamine can allow accesses to chiral Salen (Fig. 3). This category of Salen ligands can be used to prepare enantioselective catalysts which are in the asymmetric synthesis [21].

Examples of chiral Salen ligands [21]
Non-symmetrical Salen is an important category of Salen ligands which can be distinguished by tunable electronic, steric, and catalytic properties compared to their symmetrical analogues [22]. The easiest pathway to synthesize a non-symmetrical Salen is direct two-step Schiff base synthesis. In the first step, the reaction between salicylaldehyde and ethylenediamine in 1:1 molar ratio affords a mono-keto-imine product followed by the reaction with a substituted salicylaldehyde in the second step (Scheme 3) [23].

Synthesis of non-symmetrical Salen ligand [23]
One of the most efficient techniques to synthesize Salen-type Schiff base ligands is the template synthesis [24]. In this method, a metal can be added in the reaction to form a temporary stable complex by chelation which will be removed in the end of the reaction via protonation, usually using concentrated hydrochloric acid, to afford the desired ligand. This method is often used to direct the synthesis towards the formation of the desired ligand with improved yield and avoid secondary reactions. A screening over the different transition metals (e.g., Cu, Ni, Fe, Mn, Co, Zn) able to show such a template effect has also been reported. In this process, usually, a metal-salicylaldehyde intermediate is first formed in situ to serve as the templating species. Then, an ethylenediamine is subsequently added, which coordinates to the metal center. This makes it easier for the condensation of an additional salicylaldehyde unit, which eventually results in the desirable metal-Salen chelate that is anchored around the metal ion (Scheme 4) [24]

Template syntheses of tridentate and tetradentate Salen-type Schiff base ligands and related complexes [24]
Template method provides an effective route for the synthesis of macrocyclic Salen ligands with well-defined ring sizes and geometries (Scheme 5) [25]. In this approach, a metal ion such as zinc first reacted with the aldehyde,then, imine was added to the mixture, bringing them into proximity. The bridging diamine ligand then chelates around the metal center, with the metal templating the correct orientation of the linkage formation between the aldehyde/imine moieties. This yields a stable cyclic intermediate with the metal ion at its core. Chelation of the organic ligands tightly around the metal exerts strict control over the macrocyclization process and self-assembly of the desired macrocycle topology. Finally, removal of the metal template via protonation using concentrated hydrochloric acid yields the pure organic macrocyclic ligand. This template method allows for facile preparation of macrocycles that may be difficult to access without the direction of the metal-mediated self-assembly.

Template synthesis for macrocyclic Salen ligand [25]
Metal complexes of Salen ligand derivatives
MetalloSalen complexes are a unique and exciting class of organometallic complexes, with exceptionally versatile applications ranging from laboratory reaction to mass scale industries level [26]. Interestingly, metal Salen complexes gained popularity because of their roles in multiple areas, especially in catalysis. MetalloSalen complexes have been substantially investigated as both homogeneous and heterogeneous catalysts for multiple uses [27]. The most attracting feature of metalloSalen catalysts is their high stability [28]. In addition, MetalloSalen complexes have some benefits such as the simplicity of varying steric and electronic features and the use of strategies that enable manipulation of various fragments of ligand system [29]. Therefore, many important reactions that were catalyzed by metalloSalen include Friedel–Crafts reactions, mixed-aldol condensation, Claisen rearrangements, Diels–Alder reactions, and ene reaction [30,31,32,33,34]. Interestingly, metalloSalen catalysts exhibited an important role in various oxidation reactions such as the epoxidation of alkene and the oxidation of heterocyclic compounds [35, 36]. In addition, metalloSalen complexes have been known to be effective catalysts for many asymmetric conversions including (ep)oxidations, epoxide ring-opening reactions, and stereo-selective polymerizations [37].
In fact, the donor sites N2O2 of Salen ligands allow metal ions to adopt different geometries such as square planar, tetrahedral, square pyramidal, octahedral, and with additional ligand(s) if needed. Many metal ions have been introduced to Salen and its analogue ligands to produce a variety of metalloSalen complexes [27]. Kokubo and Katsuki [38] reported the synthesis of a panoply of non-substituted and substituted metalloSalen complexes of different transition metals, such as Co(II), Cu(II), Ni(II), and Mn(II), using a direct reaction between the relevant metal acetate and Salen-type ligand in ethanol as a solvent (Fig. 4). The most obtained complexes were purified by recrystallization in ethanol [38,39,40]. The synthesis of Ni(II) and Cu(II) complexes was carried out in ambient air, while all Co(II) and Fe(II) complexes were prepared using standard Schlenk line methods under an argon atmosphere [8, 40]. In addition to the Salen complexes mentioned above, an attempt was made to synthesize Zn(Salen) complexes with extended alkyl side chains, using Zn acetate as metal precursor. However, the products were insoluble in the standard solvents. Zn(Salen) has been reported to be easier to isolate when pyridine was used as a solvent. Without pyridine, Co(Salen) complex forms a polymer where the cobalt of one molecule was connected to the oxygen atoms of neighboring molecules [41].

Synthesis of metalloSalen with various Salen analogues and different metal ions [38]
The most used Salen-based catalytic systems are the manganese (III) complexes of the Salen ligand bearing a chiral cyclohexyl bridging fragment, also known as Jacobsen’s epoxidation catalyst [11]. Furthermore, other chiral Salen systems based on substituted diamino scaffolds have also shown to be attractive for a variety of catalytic applications (Fig. 5) [42].

Structures of the most used chiral Salen ligands analogues [42]
According to the method described by Jacobsen et al. [43], the synthesis of Jacobsen’s catalyst involves a condensation reaction between two equivalents of 3,5-di-tert-butyl salicylaldehyde and one equivalent of 1,2-diaminocyclohexane in methanol (Scheme 6). The reaction was catalyzed using an acidic catalyst such as hydrochloric acid. This promoted the loss of two water molecules and formation of an imine bond between the aldehyde and amine functional groups. Under reflux, the intermediate was cyclized to yield the desired Salen ligand. Then, the obtained ligand was mixed with one equivalent of manganese (II) acetate tetrahydrate in the presence of lithium chloride in methanol under reflux to form the desired metalloSalen [43].

Synthesis of Jacobsen’s catalyst [43]
Homobinuclear bi-Salen complexes were also synthesized under argon using Schlenk line technique (Fig. 6) [44]. Reaction mixtures were heated at 70 °C ethanol for 20 h to guarantee full complexation. The formation of mononuclear metal complexes, as byproducts, occurred in all cases with short reaction durations. To isolate the binuclear complex from soluble impurities, such as unreacted ligand, metal acetate, and mononuclear complex, the reaction mixture was filtered at room temperature, and the product was washed with ethanol [44].

Synthesis of homobinuclear bi-Salen complexes 41–43 [44]
Bimetallic Salen derivatives can be also formed by creating a second metal cavity in the same Salen ligand. Coupling ethane-1,2-diamine with 2,3-dihydroxybenzaldehyde or 2-hydroxy-3-methoxybenzaldehyde form a Salen ligand derivative with O2O2 outer cavity. This outer cavity can also be filled with cationic metals, such as rare earth metal [45]. The resulting heterobimetallic system can exhibit an efficient activity in various types of cooperative asymmetric catalysts [46]. Matsunaga and Shibasaki reported the incorporation of some transition metal ions into the N2O2 inner cavity and oxophilic rare earth metals, with much larger ionic radius, into the O2O2 outer cavity (Fig. 7) [45].

Synthesis of heterobimetallic mono-Salphen complexes with cationic rare earth metals into the outer cavity [45]
In another work, Finelli et al. [47] reported the synthesis of a heterobimetallic Salen complex, named as LCuMn (Scheme 7). The H2Salen ligand was prepared by reacting ethane-1,2-diamine with 2-hydroxy-3-methoxybenzaldehyde. Then, LCu complex was prepared first followed by the addition of Mn(NO3)2∙4H2O in a mixture of acetonitrile/ethanol. The desired material was obtained as a red precipitate after overnight stirring at room temperature. The obtained bimetalloSalen complex LCuMn was used as a single-source precursor (SSP) to synthesize bimetallic metal oxides Cu1.5Mn1.5O4 at lower temperatures [47].

Example of heterobimetallic Salen complexes. (a) Synthetic route of Salen ligand and (b) coordination of both chelating sites by Cu(II) and Mn(II) [47]
Salphen ligand and related metal complexes
Salphen ligand

Structure of Salphen ligand [48]

Synthesis of derivative of Salphen ligand [24]
The Salphen structures have been ignored for a long time despite offering significant advantages over their Salen analogues [48]. Salphen ligands are \(\pi\)-conjugated systems with tunable photophysical properties. In addition, the rigid geometry introduced by the Salphen ligand surrounding the metal center can be used to manage properties such as the metal’s Lewis acid character, thus improving the reactivity of the resultant complexes [49]. These properties make Salphen systems ideal building blocks in a large range of fields, including catalysis [49]. MetalloSalphen derivatives have been used effectively in the catalysis of various important organic transformations, such as the oxidation, epoxidation, hydroamination, and hydroformylation of hydrocarbons [50]. Substituted analogue of Salphen, bis-Salphen, and also thiophene analogues are also reported (Fig. 9) [51,52,53].

Some Salphen derivatives [54]
Salphen-based tri [3 + 3] (31, Fig. 10), tetra [4 + 4], and hexa [6 + 6] macrocycles have also been synthesized using 1,2-phenylenediamine and 2,3-dihydroxybenzene-1,4-dicarbaldehyde [54,55,56]).

Synthesis of tri-Salphen macrocycle (31) [56]
More recently, Hui and MacLachlan and Jiang and MacLachlan reported also the development tetra-Salphen macrocyclic architectural types (32) (Fig. 11) [56, 57]. Remarkably, a stepwise strategy was used to build isomeric structures for the tetra-Salphen macrocyclic systems by exploiting the aldimine-ketimine reactivity difference to build up the macrocyclic ring gradually through repetitive condensation reactions. The obtained macrocycles can be accessible and fine-tuned, depending on the directional characteristics of the construction blocks. These macrocyclic Salphen ligands also have potential as biomimetic catalysts and reported also as a catalyst of the oxidation reactions involved in the breakdown of lignin biomass [56, 57]).

Metal complexes of Salphen
Kokubo and Katsuki [38] reported also the synthesis of a panoply of metalloSalphen complexes of different metals such as Co(II), Cu(II), Ni(II), and Mn(II) using a direct reaction between the relevant metal acetate and Salphen ligand (Fig. 12). Ethanol was often considered the suitable solvent. The obtained complexes were purified by recrystallization in ethanol [38,39,40]. The synthesis of Ni and Cu complexes (23–30) was carried out in ambient air, while all Co(II) and Fe(II) complexes were prepared using standard Schlenk line methods under an argon atmosphere [8, 40]. However, because they do not coordinate dioxygen as easily as their Salen analogues, Co(Salphen) complexes (19–22) were not sensitive to the used synthesis technique [8]. Fe(II) complexes (31–34) were synthesized by producing a ligand/sodium salt first, using NaH as a base and THF as a solvent. Then, FeCl2 was added to the mixture to form the desired complex at room temperature [58].

Synthesis of some metalloSalphen derivatives (19–34) [38]
Bis-zinc(II)Salphen with symmetrical (1) and unsymmetrical (2) Salphen and also bis-metalloSalphen with unsymmetrical (2) Salphen and different metals were reported by Castilla and co-workers (Scheme 9) [59]. These bis(metalloSalphen) complexes were the result of further condensation with a panoply of salicylaldehydes and different metals such Zn, Co, and Mn. In addition, this synthetic strategy allowed the creation of a variety of heterobimetallic bis-Salphen complexes, with two different metallic centers, which can be highly desirable in catalysis [22]. The same research team reported also the synthesis of bimetallic bis-Salphen complexes through metal-templation method to preferentially afford the heterobimetallic Salphen complex via a stepwise condensation/metalation sequence of 3,3′-diaminobenzidene (3) [22].

Synthesis of symmetrical and non-symmetrical bis(metalloSalphen) complexes derived from the 3,3′-diaminobenzidene scaffold [59]
The same research group developed a straightforward, metal-templated process for producing non-symmetrical bis-Salphen scaffolds with different patterns of substitution (Scheme 10) [60]. In the reported method, the first chelated metal was Zn(II), which is crucial because it has high Lewis acidity due to its ability to strongly withdraw electrons. This makes zinc very reactive towards nucleophiles like hydroxide (OH−). In the presence of OH− ions, zinc provokes a selective hydrolysis reaction where it will only react with one equivalent of OH− forming useful mono-imine phenolate salts that were easily converted into non-symmetrical mono- and bis(metalloSalphen) products [61].

Incorporation of Salen/Salphen complexes into mesoporous silica materials (MSMs) and application in oxidation of hydrocarbons
Mesoporous silica materials: MCM-41, SBA-15, and MCM-48
Overview
Mesoporous silica refers to a class of porous silicon materials that have pore sizes classified as mesoporous according to definitions set by (IUPAC). The pore sizes of mesoporous materials fall between those of macroporous materials, which have pores larger than 50 nm in diameter, and microporous materials with pores smaller than 2 nm [62]. In the early 1990s, researchers started paying more attention to mesoporous materials. The first synthesis of mesoporous silica was reported in 1992 by researchers in the Mobil Oil Corporation Company. They referred to these as MCM-X (Mobil Crystalline Materials) [63]. The researchers in this company were able to synthesize mesoporous silica with varying pore structures including MCM-41, which has a hexagonal arrangement of cylindrical pores,MCM-48, which exhibits a cubic porous structure; and MCM-50, which has a laminar structure [64, 65].
After this time, Yu et al. [66] reported the synthesis of a new type of mesoporous materials named as SBA-X (Santa Barbara Amorphous), where X refers to pore structure and surfactant used. For example, SBA-15 synthesized with P123 as a surfactant results in cylindrical pores that are ordered hexagonally, whereas SBA-16 is synthesized with F127 and has spherical pores that are arranged in a body-centered cubic structure [66,67,68].
In recent years, a variety of highly ordered mesoporous silica materials such as MSU, KIT, FDU, and AMS have also been successfully synthesized using anionic, neutral, or non-ionic surfactants, and the different assembly methods have been modified in order to optimize the synthesis process (Fig. 13) [66,67,68].

Different types of mesoporous silica nanoparticles [69]
In comparison to other silica, MCM-41 has a high ordered structure with uniform mesopores arranged into a hexagonal, honeycomb-like lattice as shown in Fig. 14a [70]. TEM image of MCM-41 shows details of its well-organized pore structures which are separated from each other by thin walls of amorphous silica, approximately 1–1.5 nm thick. The mesopores are not necessarily arranged in straight lines running directly through the silica matrix. Instead, they may display some slight curvature in their paths. However, despite this curvature, they still keep an overall hexagonal ordered configuration, as can be seen in Fig. 14b and c [71]. Moreover, as seen in its micrographs, MCM-41 has a large void fraction because of the presence of the mesopores and concomitantly a rather low density. Due to this, MCM-41 exhibits a large specific surface area of approximately 1000 m2 g−1 and pore volume around 1 cm3/g [72]. This property makes MCM-41 very promising to be utilized as a support for catalysts [73, 74]. In addition, since MCM-41 exclusively contains mesopores, it can both allow access to large molecules and mitigate diffusion issues, which are commonly observed in microporous materials such as zeolites [75, 76].

SBA-15 is another type of mesoporous silica which has cylindrical pores arranged in a hexagonal order as shown in Fig. 15 [77]. Its well-defined structure is produced during synthesis using the triblock copolymer P123 as a structure-directing agent [78]. The size of the pore channels can be tuned from 5 to 30 nm, and the pore volume around 1.1 cm3/g. This elongated, wormhole-like structure is due to the larger pore dimensions compared to other mesoporous silicas like MCM-41 [79].

The structure of SBA-15-type mesoporous silica and a TEM micrograph [77]
In comparison to MCM-41, SBA-15 has certain advantages in its pore structure and stability. MCM-41 has a well-ordered hexagonal structure of cylindrical pores that usually range between 2 and 10 nm in diameter [80]. On the other hand, SBA-15 contains a hexagonal arrangement of larger pores sized around 5–30 nm [81]. These larger pores in SBA-15 are separated by thicker walls around 3–6.5 nm. This contrasts with the thinner walls of only 2–3 nm for MCM-41 [82]. Due to these features, they enhance the hydrothermal and chemical stabilities of SBA-15 in comparison to MCM-41. Moreover, it has been reported that SBA-15 has pores with a curved nature, which enhances the adsorption capacity as molecules can easily diffuse into the framework [83,84,85]. The combination of these advantages described above makes SBA-15 a favored mesoporous support for use in various applications such as catalysis, adsorption, separations, and drug delivery [83,84,85].
MCM-48 is another type of mesoporous silica material with a three-dimensional cubic structure, as shown in Fig. 16 [86]. The pores in MCM-48 form an interconnecting system in three dimensions with segments that intersect uniformly to form a continuous porous network. The pore diameter of MCM-48 is usually in the range of 2–14 nm [87]. Compared to MCM-41 which has a hexagonal arrangement of cylindrical pores, the cubic structure of MCM-48 provides more accessibility and faster mass transport kinetics. This is due to its continuous, non-directional pore system which allows molecules to rapidly diffuse in and out through multiple pathways [88]. The synthesis of MCM-48 can be more complex and difficult to control compared to MCM-41 and SBA-15. Furthermore, the pore walls of MCM-48 tend to be thinner than SBA-15, making it less hydrothermally stable. But its unique three-dimensional structure provides a higher surface area, typically around 1000–1400 m2/g, and pore volume, around 0.7–2 cm3/g, making MCM-48 promising for applications that require fast diffusion such as catalysis and ion-exchange [89].

Synthesis of mesoporous silica MCM-41, SBA-15, and MCM-48
The mesoporous silica MCM-41 can be produced using a simple process that uses surfactant templating to guide the self-assembly of silica into its unique hexagonal pore structure [90]. The general preparation of MCM-41 involves the use of cetyltrimethylammonium bromide (CTAB) as surfactant which is commonly used due to its ability to self-assemble into ordered supramolecular architectures and tetraethyl orthosilicate (TEOS) as the silica precursor (Scheme 11) [74, 91]. The formation mechanism of MCM-41 was reported by Beck et al. [65]. They suggested that under strong alkaline condition, the surfactant initially forms micellar rods, which then stack and arrange in hexagonal arrays [65]. Upon addition of a silica precursor such as TEOS, driven by electrostatic interactions between the negatively charged silicate species and positively charged CTAB heads, this results in the hydrolysis and condensation of silanes. After the synthesis, the surfactant template can be removed either by calcination or solvent extraction (e.g., EtOH, HCl), forming the final mesoporous material MCM-41 [92].

Schematic representation of MCM-41 synthesis [92]
Mesoporous material SBA-15 is typically synthesized following similar procedure described above for MCM-14, but using pluronics P123 (PEO20PPO70PEO20) as a structure-directing agent under acidic conditions [93]. The synthesis of MCM-48 follows a similar process described for MCM-41, utilizing surfactant templating to guide the self-assembly of silica into its cubic pore structure. In the case of MCM-48, surfactants that form cubic micelle phases such as P123 or F127 are commonly used as surfactants [94]. Under alkaline medium, these tri-block copolymers self-assemble into cubic micellar phases. However, obtaining the precise cubic phase is more difficult than the hexagonal phase formed by MCM-41 surfactants [95]. After the addition of a silica precursor such as TEOS, silica polymerization occurs on the surface of the cubic micelle template. Factors such as surfactant/silica ratio, pH, temperature, and time may affect the mesostructure formation. Typically, a basic pH is maintained between 10 and 13 during the hydrothermal synthesis which takes place at 100–150 °C for 1–7 days. This allows the silica framework to continuously build up on the surface of the surfactant micelles [96]. However, controlling the reaction conditions to reliably synthesize MCM-48 with its well-defined three-dimensional cubic structure can be challenging due to the greater complexity compared to the linear channel structure of MCM-41. After synthesis, the surfactant is removed via solvent extraction or calcination, forming the three-dimensionally interconnected silica framework with an inverted replica of the original cubic micelle structure [88].
Methods to immobilize metallo(Salen/Salphen) complexes into MSMs
Metallo(Salen/Salphen) complexes are common examples of organometallic homogeneous catalysts that have some advantages such as high activity and selectivity. However, they also have limitations such as difficult recovery and recycling from the reaction mixtures. This can lead to loss of costly metal complexes and catalytic site deactivation over time [97]. To overcome these issues while maintaining the benefits of homogeneous systems, efforts have been made to heterogenize these homogeneous catalysts using solid support materials [98, 99]. MSMs are often employed as catalyst support due to their excellent physical and chemical properties, such as large surface area, tunable pore size, easy to functionalize, high chemical and thermal stability, and different pores network dimensions (1D, 2D and 3D).
Mesoporous materials have shown great potential in many industrially important processes such as refining, petrochemical production, and renewable energy technologies. Their ordered structure maximizes catalyst accessibility and utilization, leading to higher activity, selectivity, and stability [100,101,102]. In addition, the abundant silanol groups on the MSM surface allow easy functionalization and immobilization of active species [103]. The heterogenization of Salen/Salphen complexes onto MSM support combines advantages from both phases [104, 105]. The support facilitates easy recovery and reuse through simple filtration or centrifugation. The immobilization of Salen/Salphen complexes through MSM surface prevents the agglomeration and leaching of the active sites. This minimizes the deactivation of the catalyst and maintains its catalytic activity through multiple cycles [106]. Overall, the heterogenization addresses key limitations around recyclability and deactivation while retaining the favorable homogeneous catalyst characteristics.
There are several approaches to incorporate metallo(Salen/Salphen) complexes into MSMs. One technique involves their physical adsorption through weak van der Waals interactions [93]. However, this method relies only on transient and non-covalent bonding between the complex and MSM surface. This led often to leaching issues over time. A more robust alternative method is to form stronger bonds (e.g., covalent or coordination bonds) between the complex and MSM support using chemical methods [107]. The main chemical methods have been used to incorporate organic moieties (R) into MSMs including post-synthesis (grafting), co-condensation, and periodic mesoporous organosilicas (PMOs) (Scheme 12) [107]. Grafting is a post‐synthesis method, where the organic moiety is covalently attached to the silica surface [108]. Co-condensation involves introducing the organic material during the synthesis process of the silica through co-condensation between the silica source (e.g., TEOS) and the mono-, bi-, or multi-silylated organic moiety in the presence of a structure directing agent [109, 110], while, in the PMO method, the organosilica material is obtained by the condensation of 100% of bi- or multi-silylated organic precursor, without any other silica source, in the presence also of a structure directing agent [111]. In addition to these three methods, coordination bond between the central metal ion in metallo(Salen/Salphen) complex and a ligand grafted onto MSM surface can also allow the preparation of a stable metallo(Salen/Salphen)-MSMs catalysts [112]. The grafting-coordination method can be considered a simple and effective alternative to immobilize organometallic complexes onto MSM surface. All these four methods will be discussed with more detail in the following section [112].

Different methods to incorporate organic moieties onto mesoporous silica materials: 1 grafting, 2 co-condensation, 3 periodic mesoporous organosilica (PMO) [107]
Grafting
In this method, organosilanes RSi(OR′)3 react with the surface-silanol groups of a pre-prepared MSM (Fig. 17) [113]. Many organic and organometallic moieties, including Salen/Salphen derivatives, have been incorporated onto MSM surface using this method [113]. A key advantage of this approach is that the mesoporous structure of the original silica material is often preserved under the reaction conditions employed. However, the textural properties (surface area, pore size, and pore volume) of the obtained MSMs will be reduced [114]. Additionally, achieving a homogeneous distribution of the organic groups throughout the pores surface can be challenging. If the grafting reactants react preferentially at the pore openings through the initial step of the synthesis process, the diffusion of further molecules into the center of the pores can be hampered [115] (Scheme 13).

Mn(III)Salen complex immobilized on MCM-48 [116]

Grafting process of an organic moiety onto MSM pore walls [113]
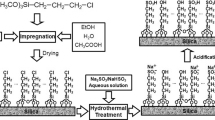
Among metallo(Salen/Salphen)-MSMs described in the literature, the Salen derivatives are the most reported. The metalloSalen complexes are immobilized onto MSM surface through different types of linkers. Yu et al. [116] reported the immobilization of Mn(III)Salen complex onto MCM-48 in the 5-position of the Salen ligand through an alkylamine linker (Fig. 17). The preparation of Mn(III)Salen complex involved a multi-step synthesis. First, MCM-48 silica was synthesized using cetyltrimethylammonium bromide (CTAB) as surfactant and TEOS as silica source at a molar ratio of 1:0.17:0.41:52 for TEOS to CTAB to NaOH to H2O. Then, CTAB was removed by calcination at 550 °C. Aminopropyltriethoxysilane (APTES) was then grafted into the calcined MCM-48 surface at a silica to APTES ratio of 1:0.15. Meanwhile, the chloromethyl group on the Mn(III)Salen complex reacted with the amine groups of APTES resulting in the preparation of Mn(III)Salen-MCM-48 material.
The immobilization of Mn(III)Salen complex onto MCM-48 surface was confirmed by IR, nitrogen sorption analysis, and inductively coupled plasma atomic emission spectroscopy ICP-AES. The adsorption–desorption isotherms indicated the reduction in the textural properties after modification. Specifically, the surface area and pore volume decreased significantly compared to MCM-48 (Table 1) [116]
ICP-AES analysis confirmed the Mn loading to be 0.13 mmol/g. The catalytic activity of these catalysts was evaluated in the epoxidation reaction of styrene. The epoxidation was carried out using tert-butyl hydroperoxide (THBP) as oxidant at 0 °C. For comparison purposes, the unsupported Mn(III)Salen complex was also evaluated under the same reaction conditions. Mn(III)Salen-MCM-48 exhibited up to 98% conversion of styrene within 8 h, indicating high activity compared to 40% for Mn(III)Salen. More importantly, an enantiomeric excess of 90% was obtained for the epoxide product using the heterogeneous catalyst compared to 75% Mn(III)Salen complex.
In another work, propylthiol groups were also used as linkers to immobilize Mn(III)Salen derivatives onto the MSM surface. Thiol groups are particularly suitable for this role due to their high nucleophilicity. Moreover, propyl spacers between the thiol and silica allow more flexibility and less steric hindrance for the Mn(III)Salen complex. The flexible propyl chains also help disperse the immobilized complexes and reduce possible diffusional limitations within the mesoporous framework. Ma et al. [117] reported the immobilization of Mn(III)Salen complexes onto the SAB-15 surface using the propylthiol group as a linker (Fig. 18). SBA-15 was prepared using TEOS or 1,2-bis(trimethoxysilyl)ethane (BTMSE) as silica source and P123 as the structure-directing surfactant. P123 was removed via calcination at 550 ℃. Then, the obtained silica was functionalized with propylthiol groups by reacting 3-mercaptopropyltriethoxysilane (MPTES) with the surface silanol groups. The Salen ligand was immobilized onto SBA-15 surface after forming covalent bonds between propylthiol groups and benzene rings of the Salen nucleus via nucleophilic substitution between the benzyl chloride and propylthiol group. Mn(III) ions were then introduced by stirring the obtained Salen-SBA-15 material in toluene with manganese acetate and LiCl to afford the desired catalyst Mn(III)Salen-SBA-15 [117].

Immobilization of Mn(III)Salen-SBA-15 via propylthiol linker for the oxidation of styrene [117]
The prepared catalyst was tested for the catalytic epoxidation of styrene by H2O2. The obtained results showed high conversion of styrene (> 90%) and good selectivity towards styrene epoxide (> 98%) under mild conditions [117]
Jones et al. [118] incorporated Ru(II)Salen bis-pyridine complex onto SBA-15 surface through the grafting method using the thiol group on the Salen complex as a linker (Fig. 19). SBA-15 was synthesized using P123 as a structure directing agent and TEOS as silica source. A trimethoxysilane-modified Salen ligand containing a terminal thiol group was synthesized and reacted with the hydroxylated SBA-15 via condensation reaction. Ruthenium was complexed to the immobilized Salen ligands by reaction with [Ru(cymene)2Cl2]2 and n-butyllithium, affording the Ru(II)Salen-SBA-15 catalyst [118].

Ru(II)Salen bis-pyridine complexes immobilized SBA-15 [118]
The structure of Ru(II)Salen-SBA-15 was confirmed by 13C and 29Si solid NMR. This catalyst was evaluated in the asymmetric cyclopropanation of styrene with ethyl diazoacetate, achieving high conversion (100%) and selectivity (96%). Furthermore, recycling experiments demonstrated high stability of the catalyst over three cycles.
In another work, Zhao et al. [119] reported the utilization of a bifunctional amine group as a dual connector to immobilize a Mn(III)Salen complex onto MCM-41 surface (Fig. 20). MCM-41 was prepared following a similar method described above. The linking unit was covalently bonded to the MCM-41 surface by condensation between the triethoxysilyl group and MCM-41 surface silanols. The Salen ligand was coupled to the linking using a Salen to silica ratio of 1:19. As expected, the textural properties of the silica were dramatically reduced after the immobilization of Mn(III)Salen complex (Table 2). ICP analysis indicated that the Mn loading in the obtained material was around 4.2%. The catalytic activity of the obtained Mn(III)Salen-MCM-41 catalyst was evaluated in the epoxidation reaction of styrene and methylstyrene, using m-CPBA as oxidants at room temperature for 2 h. The result showed good catalytic activity with 57% conversion and 97% selectivity and 50% conversion and 95% selectivity for styrene and methylstyrene epoxidation respectively. Recycling investigation performed for the methylstyrene epoxidation using m-CPBA showed good stability of the catalyst during 5 runs, with 0.8% Mn leaching [119].

Incorporation of a Mn(III)Salen complex onto MCM-41 surface via bifunctional amine group [119]
The TEM image of the fresh catalyst confirmed preservation of MCM-41 nanostructure order after the incorporation of Mn(III)Salen (Fig. 21). However, the TEM was not performed for the spent catalyst to investigate the stability of its nanostructure after use.

TEM images of Mn(III)Salen@MCM-41 [119]
More complicated linkers were also used to immobilize metalloSalen complexes into MSMs. Bigi et al. [120] reported the immobilization of Mn(III)Salen complex onto MCM-41 surface via a triazine-based linker (Fig. 22). In this method, MCM-41 was first synthesized using CTAB as a template and TEOS as a silica source at a molar ratio of 1:0.15:0.41:52 for TEOS to CTAB to NaOH to H2O. The surfactant template was removed in the end of the synthesis by calcination process at 500 °C [120].

Immobilization of Mn(III)Salen complex into MCM-41 surface via a triazine-based linker [120]
A triazine-based linker containing trimethoxysilane and Salen moieties was then reacted with the hydroxylated MCM-41 surface via condensation reaction between the trimethoxysilane group and surface silanol groups. The Salen to silica ratio was 1:19. Manganese was then added to the immobilized Salen ligands by reaction with Mn(OAc)2 and LiCl, producing MCM-41-supported Mn(III)Salen catalyst. According to ICP analysis, manganese loading was 14%. The obtained catalyst exhibited a good catalytic activity towards the asymmetric epoxidation of 1-phenylcyclohexene with H2O2/AcOH, using 4 mol% catalyst at 0 °C for 4 h to produce the epoxide with high enantioselectivity (82% ee). Recycling experiments showed high stability of the catalyst after over three runs [120].
Chiral metalloSalphen complexes have been also immobilized onto MSM surface via grafting method. Kim and Shin reported a multi-step grafting method to immobilize chiral Mn(III)Salen complexes into MCM-41 through an alkylamine linker (Scheme 14) [121]. In this work, (3-aminopropyl)trimethoxysilane (APTMS) was grafted onto MCM-41 surface, followed by condensation with 2,6-diformyl-4-tert-butylphenol to form the chiral half unit. Then, the obtained precursor was reacted with various salicylaldehyde derivatives and Mn(OAc)2·4H2O to prepare chiral Mn(III)Salen-MCM-41 catalysts [121].

Immobilization of chiral Mn(III)Salen complexes onto MCM-41 surface using a multi-step grafting method [121]
ICP analysis revealed 5% of Mn loading. The obtained catalyst was evaluated asymmetric in the epoxidation of styrene and α-methylstyrene. Up to 98% conversion and 89% enantioselectivity of styrene were obtained using m-CPBA as an oxidant (Table 3). Furthermore, no significant loss in activity was observed after four successive cycles [121]
However, only few examples of metalloSalphen analogues have been immobilized onto MSM surface via grafting method. Yu et al. [122] grafted a Mn(III)Salphen complex onto pre-prepared MCM-48 surface through a multi-step grafting process (Scheme 15) [122]. MCM-48 was synthesized using a gel composition of TEOS, CTAB surfactant in basic pH. CTAB was removed from the as-made silica by solvent extraction method. Then, chiral Mn(II)salphen complexes were immobilized onto the surface of the prepared MCM-48 via a multi-step synthesis. (3-Aminopropyl)triethoxysilane was grafted onto silica surface. Subsequently, 2,6-diformyl-4-tert-butylphenol was condensed to form surface-bound chiral half units. Reaction of these half-units with salicylaldehyde formed Mn(III)Salphen-MCM-48 catalyst. The Mn(III)Salphen content was ca. 0.3 mmol/g according to ICP-AES analysis. The obtained heterogeneous catalyst exhibited higher activity and enantioselectivity comparable to the unsupported Mn(III)Salphen for the asymmetric epoxidation of some challenging olefin substrates, such as α-methylstyrene and indene, using m-CPBA as an oxidant. In addition, enantiomeric excess ee% exceeded 89% (Table 4). Moreover, the obtained catalysts exhibited high stability during four runs [122]

Mn catalyst immobilization on a type MCM-48 support [122]
Co-condensation method
This one-pot method involves the condensation of tetraalkoxysilanes Si(OR)4, TEOS, or tetramethyl orthosilicate (TMOS), with one or more organoalkoxysilanes containing terminal organic functional groups RSi(OR′)3 (Scheme 12) [107]. The reaction takes place in a single vessel containing the tetraalkoxysilane and organoalkoxysilane precursors, along with a structure-directing agent to guide the formation of the mesoporous organosilica (MOS) material (Scheme 16) [113]. The templates such as CTAB and P123 are commonly used as structure-directing agents for the synthesis of MOS materials through co-condensation method. Among the advantages of this approach is that the organic functionalities become an inherent part of the silica framework from the beginning of the reaction. Therefore, the formed pores within the structure are not at risk of becoming blocked later during the synthesis [123]. In addition, the organic components tend to be more homogeneously distributed across the co-condensation materials compared to those prepared via grafting method [113].

Co-condensation method for the organic modification of ordered MSMs with organosilanes [113]
However, this method also has some disadvantages, for example, the level of mesopores ordering among the products also tends to decrease as the concentration of RSi(OR′)3 in the reaction mixture increases, which eventually leads to totally disordered products. As a result, the amount of organic functional groups that could be incorporated into MOS via co-condensation method is generally limited to below 40 mol% [124]. Furthermore, the quantity of terminal organic groups that are immobilized into the pore-wall network is normally less than expected based on the starting concentrations of the components in the reaction mixture [113]. These observations can be attributed to the fact that an increasing proportion of (R’O)3SiR in the reaction mixture promotes homocondensation reactions over cross-linking condensation reactions with the silica sources [113]. One drawback of this method is that different precursors have different hydrolysis and condensation rates. The ones that react faster will condense with each other first before the slower ones fully react. This leads to a lack of homogeneous incorporation of all precursors [125]. Additionally, increasing the loading of the organic groups can lead to reduction of the pore diameter, pore volume, and specific surface area of the final material. So a high organic content may compromise the porous network structure [125]. Moreover, special care must be taken when removing the surfactant template to avoid the degradation of sensitive organic functionality. The solvent extraction method is typically the only option employed, instead of the calcination method, that could compromise the incorporated organic moieties in most synthesized materials [126].
Recently, Barker et al. [127] developed a co-condensation sol–gel route to immobilize both symmetrical and unsymmetrical Salen/Salphen ligands into MSMs (Scheme 17). For symmetrical ligands, the ligand (Salen or salophen) was mixed with TEOS in a ratio ranging from 1:5 to 1:20 of ligand to TEOS, and this mixture was subjected to hydrolysis and condensation. For unsymmetrical ligands, the silica immobilization precursor was first prepared by reacting the aldehyde precursor (2-hydroxy-5-(3-triethoxysilylpropyl)benzaldehyde) with TEOS in a 1:5 ratio, which formed silica-supported aldehyde. This was then reacted with an excess of a diamine to obtain the silica-supported amine, which was further reacted with salicylaldehyde to obtain the desired silica supported unsymmetrical ligand. The reactants were dissolved in ethanol and water with acetic acid and heated at 60 °C for 1 h to initiate the polycondensation via formation of silicon-oxygen-silicon bonds between the sol–gel network and the triethoxysilyl moiety on the ligand. The structures of the prepared in situ prepared metalloSalen/Salphen were confirmed by solid 1H and 13C NMR. While ICP-MS showed ligand loadings ranging from 0.13 to 0.2 mmol/g in silica, with surface areas between 300 and 470 m2/g as determined by N2 adsorption/desorption technique. The obtained heterogeneous catalysts were able to catalyze cyclic carbonate synthesis from terminal epoxides and CO2 comparable to homogeneous catalysts, with ease of separation and recycling afforded by the silica support [127].

Synthesis of silica-supported metal complexes [127]
Amarasekara et al. [128] reported the immobilization of Co(III)Salphen complex into silica through polycondensation of the complex with 10 equivalents of tetraethyl orthosilicate (Scheme 18). First, Claisen rearrangement of the 2-allyloxybenzaldehyde (1) yielded 3-allyl-2-hydroxybenzaldehyde (2) in 85% yield. Compound (2) then underwent hydrosilylation with triethoxysilane utilizing PtO2 as a catalyst. Then, the desired precursor 2-hydroxy-3-(triethoxysilylpropyl)benzaldehyde (3) was obtained with high yield after addition of HSi(OEt)3 to the double bond of the allyl group. Subsequently, compound (3) was reacted with 1,2-phenylenediamine then followed by addition of CoCl2 in ethanol to form the Co(III)Salen complex, which was immediately utilized in the co-condensation reaction with TEOS in a 10:1 molar ratio to the ligand with water and ammonium hydroxide to afford the catalyst (4). This initiated the sol–gel polycondensation process. Through hydrolysis and condensation reactions, the triethoxysilylpropyl bearing Co(III)Salen complex became covalently linked to the growing silica network.

a 190 ℃, 24 h. b HSi(OEt)3 (1 eq.), PtO2 (0.01 eq.), 85 ℃, 20 h (c) (i) p-phenylenediamine (0.5 eq.), EtOH, 60 ℃, 30 min. (ii) CoCl2 6H2O (1 eq.), 60 ℃, 15 min. (iii) Si(OEt)4 (10 eq.), H2O, NH4OH, r.t., 5 days then 110 ℃, 2 days. (d) (i) p-Phenylenediamine (0.5 eq.), EtOH, 60 ℃ 30 min (ii) Si(OEt)4 (10 eq.), H2O, NH4OH, r.t., 5 days then 110 ℃, 2 days [128]
The successful immobilization of Co(III)Salen into silica was confirmed using TGA, IR, and UV–Vis spectroscopy. The obtained catalyst was tested in selective oxidation of some alkane and alkene, using O2 as oxidant. The obtained results showed high catalytic activity and stability of this catalyst [128].
Later, same research group reported the synthesis of Jacobsen–Katsuki-type chiral Mn(III)Salen catalyst immobilized in silica by sol–gel using similar approach (Scheme 19) [129]. A salicylaldehyde ligand with a triethoxysilylpropyl group was synthesized and involved in a condensation reaction with o-phenylenediamine to form the Salphen scaffold. Then, 1 eq of Mn(OAc)2·H2O was added to form the desired Mn(III)Salen complex. Then, TEOS was added with water and ammonium hydroxide to initiate the sol–gel co-condensation reaction. This allowed one pot formation and immobilization of Mn(III)Salen complex into the growing silica network via covalent bonds [129].

Synthesis of immobilized Jacobsen–Katsuki-type Mn(III)Salen catalyst 3 [129]
The structure and thermal stability of the obtained catalyst (3) were confirmed by FT-IR, XRD, TGA, and elemental analysis. However, TEM and BET are not performed to investigate the order of the nanostructure and porosity of the obtained material. The prepared catalyst exhibited good catalytic activity and recyclability for the epoxidation reaction of styrene and methylstyrene (Table 5), using sodium hypochlorite as oxidant [129].
Periodic mesoporous organosilicas (PMOs)
This approach involves the condensation of 100% of bi- or multi-silylated organic precursors R[Si(OR′)3]n (n ≥ 2) (R: organic bridge, R′: ethyl or methyl group) in the presence of a template (e.g., CTAB, P123), following (often) the MCM-41 or SBA-15 protocol [113]. These precursors are capable of undergoing cross-linking reactions and simultaneously carry the organic functionality R as described in (Scheme 20). In contrast to the organically functionalized silica phases, which are obtained by grafting and co-condensation methods, the organic components in this technique are built into the three-dimensional structure of the silica network. They are immobilized through two or more covalent linkages, resulting in the homogenous incorporation of these units across the pore wall structure [113]. However, the pore systems typically often demonstrate a wide array of pore radii rather than a narrow distribution [130].

General synthetic pathway of PMOs. R: organic bridge, R′: Me, Et, or iPr [113]
Unlike amorphous aerogels and xerogels, PMOs exhibit a periodically arranged pores and narrow pore distribution. The first PMO material was synthesized independently by three research groups in 1999 [131,132,133]. PMO materials show great potential in numerous technical applications such as catalysis, adsorption, and chromatography [134,135,136].
Significant progress has been made in the diversity of organic precursors that can be used to prepare PMO materials [125]. Figure 23 presents examples of organic moieties which have been successfully employed as organic bridge to prepare new PMOs [109]. This allowed various chemical functionalities to be structurally incorporated into the final mesoporous silica architecture, such as short-chained saturated aliphatic groups like alkylenes (1) and unsaturated alkenylene chains (2). Other implemented organic moieties included cyclic (3) and branched (4) multi-silylated compounds, aromatic residues like phenyl (5,6), heterocyclic (7,8), and polycyclic (9) structures, and aromatic compounds with tunable substituents (10,11). Even ether (12) and conjugated π-system (13) containing linkers have been developed to template the formation of PMO networks with their intended properties encoded [125, 130, 137]. This diversity of tailored organic precursors guides the sol–gel co-assembly of mesoporous silicas with increasingly complex compositions [138]. PMO materials exhibit unique molecular-scale periodicity compared to other mesoporous organosilica forms produced via grafting methods or co-condensation sol–gel processes. Conventional amorphous organosilica frameworks modified with organic groups often lack ordered internal structure [139]. In contrast, PMOs produced using a single aromatic organic precursor can induce the formation of crystal-like layered architectures within the framework [140]. Specifically, PMOs containing bridging aromatic organic groups self-assemble into frameworks possessing repeated units of alternating organic and silica layers [141]. As shown in Fig. 24, the layered structures exhibit distinct molecular-scale periodicity, with distances between layers ranging from 5.6 to 11.9 Å depending on the size of the incorporated organic bridge [142].

Examples of bis- and multi-organosilica precursors that have been converted into PMOs. Si = Si(OR)3 with R = Me, Et, or iPr [125]

A list of important aromatic organosilane precursors (a) and their periodic arrangement in the mesopore walls of the PMOs (b), with a TEM image of a 2,6-naphthylene-bridged PMO (c) and its crystal-like framework (d) [142]
Moreover, PMO materials with chiral centers were also attracting the interest of researchers due to their potential applications such as asymmetric catalysis and separation [111]. A variety of organosilica precursors with chiral centers, which have excellent asymmetric catalytic properties, have been reported (Fig. 25) [111, 143].

Examples of chiral PMOs [111]
Additionally, organometallic complexes have been also incorporated in PMOs for main application in heterogeneous catalysis (Fig. 26). Metal centers can be introduced into PMOs by two different ways. They may chelate to the PMO precursor (ligand) before polymerization or can be added to pre-synthesized PMOs [111].

Examples of organometallic complexes used to prepare PMO materials for main application in heterogeneous catalysis [111]
However, only few examples reported the incorporation of metalloSalen into MSMs as PMO materials. Baleizão et al. [144] developed a novel approach to immobilize the VO(Salen) complex into PMO (Scheme 21). First, vanadyl Salen ligands containing biphenyl units were synthesized and functionalized with two terminal trimethoxysilyl groups. This was achieved by reacting the Salen complexes with 3-mercaptopropyltrimethoxysilane using azobisisobutyronitrile (AIBN) as an initiator to graft the trimethoxysilyl moieties onto the ligands. This affords catalytically active vanadyl Salen structures covalently attached to trimethoxysilyl anchors. These precursors containing both the Salen complexes and trimethoxysilyl groups were then used along with CTAB as the structure-directing agent in the PMO sol–gel process. During this process, the trimethoxysilyl groups of vanadyl Salen complex underwent hydrolysis and condensation reactions, acting as both the silica source and structure directing agent to form a siloxane network with the vanadyl Salen complexes integrated into the inorganic–organic hybrid framework. After synthesis, the CTAB surfactant was removed by extraction with acidic ethanol resulted in PMO material containing covalently immobilized VO(Salen) complexes [144].

Preparation of VOSalen-PMO and VOSalen-ChiMO(ChiMO = Chiral PMO ((a) 3-mercaptopropyltrimethoxysilane, AIBN, CHCl3 (degassed), 70 ◦C, 20 h; (b) TEOS, NH3, CTAB, H2O, EtOH, 90 ◦C, 4 days) [144]
The structure of VO(Salen)-PMO was confirmed by XRD, nitrogen adsorption, and 29Si MAS-NMR spectroscopy. XRD revealed that the material possesses high long-range structural periodicity, characteristic of an ordered mesoporous structure as shown in Fig. 27. N2 adsorption determined a high specific surface area of 900 m2/g, indicating a porous framework. Pore size analysis demonstrated a narrow monomodal distribution centered around 42 Å, suggesting uniform mesopores. Finally, 29Si solid-state NMR spectroscopy (Fig. 28) showed peaks assigned to T3 silicon atoms (CH2–Si(OSi≡)3), providing evidence functionalization of VO(Salen)complex into silica through covalent Si–C bonds. The catalytic performance of the immobilized of VO(Salen)-PMO was tested for cyanosilylation of aldehydes using trimethylsilyl cyanide as reagent in chloroform at room temperature. The result showed high conversion around (82%) with complete selectivity to the silylated cyanohydrins. The reusability of the immobilized VO(Salen)-PMO catalyst was demonstrated over multiple reaction cycles. In four consecutive runs catalyzing the same cyanosilylation reaction, the catalyst maintained high activity with only a minor decrease in conversion around (79%) which was observed during the final run [144].

X-ray diffraction of VO(Salen)@PMO before extraction of the template (a) and after extraction the template (b) (1), and X-ray diffraction of VO(Salen)@ChiMO before extraction of the template (a) and after extraction the template (b) (2) [144]

MAS.29Si NMR spectra of VO(Salen)@ChiMO after CTABr removal [144]
Incorporation of metallo(Salen/Salphen) complexes in mesoporous silica via coordination bonds
One straightforward method to incorporate organometallic complexes into MSMs involves coordination bonding between the silica surface and metal center of the complex. This approach consists of the functionalization of MSM surface first by a ligand, then the addition of the organometallic complex. Usually 3-aminopropyltriethoxysilane (APTES) is used to incorporate propylamine (-CH2CH2-NH2) into MSM surface (NH2-MSMs) which plays a ligand role for metal centers (Scheme 22) [145]. NH2-MSMs can be also prepared by co-condensation method between TEOS and APTES in the presence of a template (e.g., CTAB, P123) [146]. This facile route maintains the native coordination geometry of the organometallic complexes, preserving their catalytic properties and allowing their reusability. Additionally, coordination interactions allow homogeneous introduction and tunable activation of each metal site via coordination variations. Due to these key advantages, immobilization of organometallic complexes onto silica supports through coordination bonding has become widespread. This approach has been used to immobilize different organometallic complexes into MSMs for various applications, including heterogeneous catalysis, sensing, and biomedical applications that benefit from tunable surface coordination environments [125, 145, 147].

Grafting method of APTES onto MSM surface
Metallo(Salen/Salphen) complexes have been also immobilized onto MSM surface using coordination approach. Liu et al. [148] reported the immobilization of Mn(II)Salen complex into SBA-15 via coordination method and used it to oxidize styrene to styrene oxide (Scheme 23). In the first step, the SBA-15 surface was functionalized with aminopropyl groups via grafting reaction of APTES to afford the corresponding NH2-SBA-15. In the second step, Mn(II)Salen complex was added to NH2-SBA-15 in order to immobilize Mn(II)Salen onto SBA-15 via coordination between the amino groups and Mn(II) center, yielding the heterogeneous catalyst denoted as Mn(II)Salen-NH-SBA-15 [148]

Preparation of the catalyst Mn(II)Salen-NH-SBA-15 via coordination method and its evaluation in the oxidation of styrene to produce styrene epoxide using 4-phenylpyridine N-oxide (PPNO) as an axial ligand and NaClO as oxidant [148]
The high dispersion of the Mn(II)Salen complex through the SBA-15 surface was evaluated by XRD (Fig. 29) and TEM (Fig. 30). The absence of reflection peaks in Mn(II)Salen-NH-SBA-15 in XRD curve indicates the absence of crystalline phases of Mn(II) Salen and the high dispersion of the complex through the silica surface [148]

XRD patterns of SBA-15, NH2-SBA-15, and Mn(II)Salen-NH-SBA-15 [148]

Transmission electron micrograph (TEM) images of Mn(II)Salen-NH-SBA-15 [148]
Moreover, TEM images showed the preservation of the honeycomb-like highly ordered mesostructure of SBA-15 after the modification reactions (Fig. 30). The Mn(Salen)-NH-SBA-15 material exhibited hexagonal lattice fringes with uniform parallel cylindrical pore network. The obtained catalyst was evaluated in the epoxidation reaction of styrene using NaClO as oxidant. The obtained results revealed high conversion of styrene (88.9%) and excellent selectivity (100%) towards styrene oxide. This catalyst also demonstrated a good reusability over multiple cycles [148].
In another work, Saikia et al. [149] reported the immobilization of Mn(II)SalenCl complex onto SBA-15 surface using different types of linkers including APTES (Scheme 24). In this work the pre-prepared SBA-15 was functionalized by 3-aminopropyltrimethoxysilane (APTMS) and 3-mercaptopropyltrimethoxysilane (MPTMS), to obtain SBA-15-NH2 and SBA-15-SH materials, respectively. The sulfonic acid functionalized SBA-15 (SBA-15-SO3H) was obtained by oxidizing the thiol groups in SBA-15-SH using hydrogen peroxide. Then, Mn(II)SalenCl was added to each modified silica to afford SBA-15-NH2-Mn(II)SalenCl, SBA-15-SH-Mn(II)SalenCl, and SBA-15-SO3H-Mn(II)SalenCl materials.

Immobilization of Mn(II)SalenCl onto NH2-SBA-15, SH-SBA-15, and SO3H-SBA-15 surface [149]
Atomic absorption spectrometry (AAS) was used to determine the Mn loading in all materials, and the surface area of the prepared materials was measured by N2 adsorption–desorption analysis. The obtained results are presented in (Table 6). Mn loading was affected by linker type, and the surface area of SBA-15 decreased after functionalization and was affected also by the size of the linker and the amount of Mn(II)Salen [149]
The TEM images showed the original hexagonal structure and ordered pores of the SBA-15 material were maintained even after functionalizing the surface and introducing the Mn(II)Salen complexes (Fig. 31). The obtained catalysts were then tested for the selective aerial oxidation of R-( +)-limonene using molecular oxygen as the oxidant. It was found that Mn(II)SalenCl immobilized on propylthiol-functionalized SBA-15 (SBA-15-pr-SH Mn(II)Salen yielded the 1,2-limonene epoxide product with 100% chemo- and regioselectivity [149].

TEM images of a SBA-15-pr-NH2-Mn(II)Salen, b SBA-15-pr-SH- Mn(II)Salen, and c SBA-15-pr-SO3H-Mn(II)Salen catalysts [149]
Imidazole-propyl linker was also used to immobilize Mn(III)SalenCl derivatives into the MSMs. Lou et al. [150] synthesized an imidazole-functionalized organosilica precursor and used as ligand for a chiral Mn(III)SalenCl complex (Scheme 25) [150]. TEOS and N-(3-triethoxysilylpropyl)imidazole (TESPI) were used as the co-condensing silica precursors, with TEOS providing the inorganic silica framework and TESPI introducing imidazole functionality, using CTAB as surfactant. The used molar ratios were 1 mol TEOS, 0.12 mol CTAB, and 0.12–0.14 mol TESPI. Subsequently, the surfactant was removed by extraction method using acidic ethanol. The prepared imidazole-functionalized MCM-41 support was utilized to immobilize the chiral Mn(III)SalenCl complex via reflux in toluene. The imidazole groups on the surface of MCM-41 were coordinated directly to the central Mn(III) metal ion of the Salen complex. This coordination interaction robustly tethered the complex within the mesoporous silica pores to afford Mn(III)SalenCl-Imd-MCM-41 material [150].

Immobilization of a chiral Mn(III)SalenCl derivative via coordination to imidazole-functionalized Mn(III)SalenCl-Imd-MCM-41) [150]
The Mn content in the obtained material was 0.14 mmol/g as determined by inductively coupled plasma atomic emission spectroscopy (ICP-AES). N2 adsorption–desorption isotherms showed decrease in BET surface area, pore volume, and pore size of the prepared material indicating the successful immobilization of Mn(III)SalenCl complex into MCM-41 pores surface. Moreover, TEM images showed regular hexagonal array of the mesochannels with uniform pore size (Fig. 32) which confirmed the preservation of the MCM-41 mesoporosity and high order of the nanostructure after the immobilization of Mn(III)SalenCl. The obtained heterogeneous catalyst exhibited an excellent activity and enantioselectivity in the asymmetric epoxidation of styrene, using only 0.6 mol% of the catalyst loading and during a short reaction time. The obtained results revealed quantitate conversion of styrene (100%) with ee 49% and high TOF (up to 165 h−1) [150].

TEM images of Mn(III)SalenCl-pr-Imd-MCM-41material [150]
In another work, Mandal et al. reported the immobilization of chiral metalloSalen derivatives (complex 1 and complex 2) into MSMs via coordination method using a pyridine derivative linker (Fig. 33) [151]. The pyridine linker was grafted onto MCM-41 surface (MCM-41-Py) via multi-step synthesis. Then, Mn(II)Salen complexes were immobilized onto MCM-41 surface via coordination bonds between the nitrogen atom of the pyridine and Mn ions to afford Mn(II)Salen-Py-MCM-41 material [151].

Immobilization of Mn(II)Salen derivatives onto MCM-41 surface via coordination bonding between Mn(II) centers and pyridine linker to form Mn(II)Salen-Py-MCM-41 [151]
The amount of Mn loading determined by ICP was 10.82 mg/100 mg and 11.35 mg/100 mg for complex 1 and complex 2 in MCM-41, respectively. As expected, the surface area, pore size, and pore volume of MCM-41 were decreased progressively after grafting the pyridine linker and the immobilization of Mn(II)Salen (Table 7).
Both MCM-41 supported and unsupported complexes 1 and 2 were tested as catalysts for the asymmetric epoxidation of styrene, using 30% hydrogen peroxide as oxidant. The obtained results for unsupported complex 1 revealed 56% styrene conversion and 58% enantiomeric excess (ee) for the styrene oxide product. While unsupported complex 2 achieved 65% styrene conversion and 70% enantioselectivity under the same conditions. However, MCM-41 supported complexes 1 and 2 showed better activity, with 75% and 80% styrene conversion respectively. Importantly, the immobilized complexes maintained high catalytic activity even after five successive cycles [151].
MetalloSalen derivatives were also immobilized onto PMO surface via coordination method. Ru(II)Salen complex was immobilized via coordination bonds onto a prepared Phenyl-PMO (Ph-PMO) through three steps using methylpiperazine as a linker (Scheme 26) [152]. First 1,4-bis(triethoxysilyl)benzene (BTEB) was used as the bridging organosilane precursor, and P123 triblock copolymer was used as a template in acidic medium following SBA-15 method to prepare Ph-PMO. The template P123 was removed by solvent extraction using an ethanol solution containing 5% HCl. The bridging benzene units of Ph-PMO were then sequentially functionalized by CH2Cl after treatment with formaldehyde and HCl. Then, the CH2Cl group was transformed to CH2-piperazine. In the final step, Ru(II)Salen complex was immobilized into the obtained aminated Ph-PMO via coordination bond between Ru center and piperazine ligand to afford the desired material Ru(II)Salen-Ph-PMOs [152].

Immobilization of Ru(II)Salen onto Ph-PMO surface via coordination bond using piperazine as a linker [152]
The successful immobilization of Ru(II)Salen onto Ph-PMO surface was confirmed by 13C CP/MAS NMR (Fig. 34). While TEM images indicated the preservation of the typical hexagonal nanostructure of Ph-PMOs after the immobilization of Ru(II)Salen complex (Fig. 35) [152].


TEM images of A Ph-PMOs and B Ru(II)Salen-Ph-PMOs [152]
The obtained catalyst Ru(II)Salen-Ph-PMOs showed good activity for the oxidation of cyclohexene with H2O2 as oxygen donor agent. The highest conversion achieved with this catalyst was 35.9%, which is higher than that obtained with Ru(II)Salen grafted onto SBA-15 [152].
Silica-coated magnetic nanoparticles are also used as support of metalloSalen derivatives using coordination methods. In this approach, iron oxide (Fe3O4) magnetic nanoparticles were used as a core coated with a silica shell. Fe3O4 nanoparticles have important properties such as strong magnetism, chemical inertness, and easy separation using an external magnet.
Rayati et al. [153] reported the immobilization of Mn(III)Salen complex onto silica-coated Fe3O4 magnetic nanoparticles via direct and simple coordination bonding between silica-surface silanol groups and Mn metal centers (without linker) (Scheme 27) [153].

Illustration of Mn(III)Salen complex supported onto silica-coated Fe3O4 magnetic nanoparticles via coordination bonds [153]
The pre-synthesized Mn(III)Salen complex immobilized on Fe3O4@SiO2 nanoparticles via axial coordination. The TEM investigation of the obtained material showed the preservation of the spherical shape of the nanoparticles after the immobilization of Mn(III)Salen complex. TEM images depicted the well-defined core–shell structure of the final material, with an average shell (silica) thickness of 30.4 nm [153].
The immobilized Mn(III)Salen-Fe3O4@SiO2 catalyst was evaluated for the epoxidation of various organic substrates, including cyclohexene, styrene, and methyl phenyl sulfide, using H2O2 as oxidation agent, at room temperature. Styrene oxidation showed complete conversion and excellent selectivity within 90 s, with a turnover frequency of 17,750 h−1. Similar results were obtained with cyclohexene and methylphenyl sulfide, with slight decrease in the conversion. The obtained results are displayed in Table 8.
However, only few examples reported the immobilization of metalloSalphen onto MSM surface via coordination method. Wu et al. [154] reported the immobilization of Cr(III)Salphen complex onto MCM-41 surface via coordination bonding, using APTES as a linker (Fig. 36). APTES was first grafted onto MCM-41 surface followed by addition of Cr(III)Salen complex. The desired material MCM-41-NH-Cr(III)SalphenSO3H-NH-MCM-41 was obtained after eventual coordination bonding between the Cr ion center and nitrogen atom of the propylamine group [154].

Immobilization of Cr(III)Salphen onto MCM-41 surface via APTES linker using the coordination method [154]
ICP analysis revealed that Cr loading in MCM-41-NH-Cr(II)SalphenSO3H was around 2%. TEM (Fig. 37) images and N2 adsorption–desorption isotherms confirmed the preservation of the highly ordered mesoporous nanostructure of MCM-41. Furthermore, TEM images exhibited high dispersion of Cr(II)Salphen through the MCM-41 surface [154].

TEM images of Cr(II)SalphenSO3H-NH2-MCM-41 [154]
The prepared material Cr(II)SalphenSO3H-NH2-MCM-41 was tested as a catalyst for the oxidation of benzyl alcohol to benzaldehyde using hydrogen peroxide as an oxidant. Compared to the homogeneous catalyst Cr(II)SalphenSO3H, the heterogeneous system demonstrated higher catalytic activity under identical conditions, with benzyl alcohol conversion of 60.3%, and 100% selectivity towards benzaldehyde. Moreover, the prepared catalyst exhibited good stability during five successive cycles [154].
In another example of metalloSalphen, Wang and coworker reported also the immobilization of Zr(II)SalphenCl2 complex onto MCM-41 surface via APTES linkers using the coordination method (Scheme 28) [155]. In this work, amino-functionalized Zr(II)Salphen complex (NH2-Zr(Salphen)) was first synthesized then grafted onto MCM-41 surface.

a Synthetic route of Zr(II) SalphenCl2. b Synthetic pathway of Zr(II)Cl2Salphen-(NH2)2-MCM-41 [155]
As expected, N2 adsorption–desorption showed decrease in the surface area, pore size, and pore volume of MCM-41 after the immobilization of Zr(II) Salphen Cl2. TEM images of the prepared material showed the preservation of the long-range order and uniform pore structure of MCM-41 after the modification (Fig. 38). Furthermore, the absence of metal aggregation (black spots) indicates high dispersion of Zr(II)SalphenCl2 through the silica surface. The obtained material was evaluated as a catalyst for the transfer hydrogenation of ethyl levulinate to gamma-valerolactone using isopropanol as hydrogen source. The prepared heterogeneous catalyst showed excellent activity with GVL yield of up to 90% under the optimized conditions compared to unsupported Zr(II)SalphenCl2. Moreover, Zr(II)Salphen-(NH2)2-MCM-41was easily separable, recyclable, and reusable over four successive cycles [155].

TEM images of the prepared Zr(II)SalphenCl2-(NH2)2 MCM-41 [155]
Conclusion
In conclusion, immobilizing metalloSalen and metalloSalphen complexes into mesoporous silica supports has successfully addressed many drawbacks associated with their utilization as homogeneous catalysts for the oxidation and hydrogenation reactions of hydrocarbons. The heterogenization of these complexes maintains their catalytic activity while enabling their facile separation from reaction mixtures and allows their recyclability and reusability. Incorporation methods such as grafting, co-condensation, periodic mesoporous organosilicas (PMOs), and coordination have proven effective for dispersing the catalysts within high surface area frameworks such as SBA-15, MCM-41, and MCM-48. Grafting methods use different silylated linkers such as amines, thiols, sulfonic acid, alkyl halide, and pyridines to covalently attach the catalyst to the surface of pre-prepared mesoporous silica. This creates strong binding but grafting yields can be low compared to condensation methods. Co-condensation allows the incorporation of the silylated catalyst during the synthesis of the silica, which can improve the catalyst loading, but could affect the nanostructure order and porosity of the obtained material. In this method, the mono-bi- or multi-silylated catalyst will be co-condensed with a silica source (e.g., TEOS), with ≤ 15% of the catalyst. Therefore, mono-silylated catalyst will appear onto the surface, while bi- and multi-silylated catalyst will be incorporated into the silica wall, while periodic mesoporous organosilicas (PMOs) involve the condensation of bi-or multi-silylated organic precursors in the presence of a template without any other silica source. In this method, catalysts will be incorporated into silica pore walls with maximum loading. Coordination method is the simplest and the most used pathway to immobilize metalloSalen and metalloSalphen complexes into mesoporous silica. In this method, the catalyst is immobilized onto the silica surface via coordination bond between a heteroatom of a ligand pre-grafted onto the silica surface and the metal ion center. This method usually does not affect the silica texture properties and nanostructure order; however, the catalyst loading is limited to the heteroatom density. Compared to unsupported metalloSalen and metalloSalphen catalysts, the silica-supported systems consistently achieve higher conversions and product selectivity in industrial processes such as the oxidations and hydrogenation of important hydrocarbons, including styrene, methylstyrene, cyclohexene, and cyclohexane. Recoverability and reusability advantages make them prime candidates for sustainable and industrial scale-up utilization.
Data availability
No datasets were generated or analysed during the current study.
References
Schiff H (1869) Untersuchungen über Salicinderivate. Justus Liebigs Ann Chem 150:193–200. https://doi.org/10.1002/jlac.18691500206
Pfeiffer P, Breith E, Lübbe E, Tsumaki T (1933) Tricyclische orthokondensierte Nebenvalenzringe. Justus Liebigs Ann Chem 503:84–130. https://doi.org/10.1002/jlac.19335030106
Pfeiffer P, Breith E, Lubbe E, Tsumaki T (1932) Thcylische Orthokondesierte Nebenvalenzringe. Annalen Der. Chemie 492:81–127
Sani U, Na’ibi HU, Dailami SA (2018) In vitro antimicrobial and antioxidant studies on N-(2- hydroxylbenzylidene) pyridine -2-amine and its M(II) complexes. Nig J Bas App Sci 25:81. https://doi.org/10.4314/njbas.v25i1.11
More MS, Joshi PG, Mishra YK, Khanna PK (2019) Metal complexes driven from Schiff bases and semicarbazones for biomedical and allied applications: a review. Mater Today Chem 14:100195. https://doi.org/10.1016/j.mtchem.2019.100195
Yaseen AA, Al-Tikrity ETB, Al-Mashhadan MH et al (2021) An overview: using different approaches to synthesis new Schiff bases materials. JUAPS 15:3–59. https://doi.org/10.37652/juaps.2022.172453
Novoa-Ramírez CS, Silva-Becerril A, Olivera-Venturo FL et al (2020) N/N bridge type and substituent effects on chemical and crystallographic properties of Schiff-base (salen/salphen) Niii complexes. Crystals 10:616. https://doi.org/10.3390/cryst10070616
Räisänen MT, Nieger M, Slawin AMZ et al (2011) Two- and three-dimensional packing diagrams of M(salophen) complexes. CrystEngComm 13:4701. https://doi.org/10.1039/c0ce00926a
Ambroziak K, Szypa M (2007) A synthesis of unsymmetrical chiral salen ligands derived from 2-hydroxynaphthaldehyde and substituted salicylaldehydes. Tetrahedron Lett 48:3331–3335. https://doi.org/10.1016/j.tetlet.2007.03.082
Atwood DA, Harvey MJ (2001) Group 13 compounds incorporating salen ligands. Chem Rev 101:37–52. https://doi.org/10.1021/cr990008v
Ding B, Solomon MB, Leong CF, D’Alessandro DM (2021) Redox-active ligands: recent advances towards their incorporation into coordination polymers and metal-organic frameworks. Coord Chem Rev 439:213891. https://doi.org/10.1016/j.ccr.2021.213891
Chiari B, Cinti A, Crispu O, et al (2001) Binuclear Co(ii)Co(ii), Co(ii)Co(iii) and Co(iii)Co(iii) complexes of “short” salen homologues derived from the condensation of salicylaldehyde and methanediamine or phenylmethanediamines. Synthesis, structures and magnetism. J Chem Soc, Dalton Trans 3611–3616. https://doi.org/10.1039/b105594c.Electronic supplementary information (ESI) available: Appendix, derivation of the expression for the average magnetic susceptibility of a Co(ii)Co(ii) dimer. See http://www.rsc.org/suppdata/dt/b1/b105594c/
Chiari B, Cinti A, Crispu O, et al (2002) New pentanuclear mixed valence Co( II )–Co( III ) complexes of “short” salen homologues. J Chem Soc, Dalton Trans 4672–4677. https://doi.org/10.1039/B206221F
Pasini A, Demartin F, Piovesana O, et al (2000) Novel copper(II) complexes of “short” salen homologues. Structure and magnetic properties of the tetranuclear complex [Cu2(L2)2]2 [H2L2 = phenyl-N,N ′-bis(salicylidene)methanediamine] †. J Chem Soc, Dalton Trans 3467–3472. https://doi.org/10.1039/b003825n
Rigamonti L, Zardi P, Carlino S et al (2020) Selective formation, reactivity, redox and magnetic properties of MnIII and FeIII dinuclear complexes with shortened salen-type Schiff base ligands. IJMS 21:7882. https://doi.org/10.3390/ijms21217882
Clarke RM, Herasymchuk K, Storr T (2017) Electronic structure elucidation in oxidized metal–salen complexes. Coord Chem Rev 352:67–82. https://doi.org/10.1016/j.ccr.2017.08.019
Erxleben A (2018) Transition metal salen complexes in bioinorganic and medicinal chemistry. Inorg Chim Acta 472:40–57. https://doi.org/10.1016/j.ica.2017.06.060
Hobday MD, Smith TD (1973) N, N’-Ethylenebis(salicylideneiminato) transition metal ion chelates. Coord Chem Rev 9:311–337. https://doi.org/10.1016/S0010-8545(00)82081-010.1016/S0010-8545(00)82081-0
Nabeshima T, Yamamura M (2013) Cooperative formation and functions of multimetal supramolecular systems. Pure Appl Chem 85:763–776. https://doi.org/10.1351/PAC-CON-12-08-02
Weber B, Jäger E (2009) Structure and magnetic properties of iron(II/III) complexes with N 2 O 2 2– coordinating Schiff base like ligands. Eur J Inorg Chem 2009:465–477. https://doi.org/10.1002/ejic.200800891
Bennani YL, Hanessian S (1997) trans -1,2-Diaminocyclohexane derivatives as chiral reagents, scaffolds, and ligands for catalysis: applications in asymmetric synthesis and molecular recognition. Chem Rev 97:3161–3196. https://doi.org/10.1021/cr9407577
Kleij AW (2009) Nonsymmetrical salen ligands and their complexes: synthesis and applications. Eur J Inorg Chem 2009:193–205. https://doi.org/10.1002/ejic.200800936
Huber A, Müller L, Elias H et al (2005) Cobalt( II ) complexes with substituted salen-type ligands and their dioxygen affinity in N, N -dimethylformamide at various temperatures. Eur J Inorg Chem 2005:1459–1467. https://doi.org/10.1002/ejic.200400888
Mazzoni R, Roncaglia F, Rigamonti L (2021) When the metal makes the difference: template syntheses of tridentate and tetradentate salen-type Schiff base ligands and related complexes. Crystals 11:483. https://doi.org/10.3390/cryst11050483
Akine S, Sunaga S, Taniguchi T et al (2007) Core/shell oligometallic template synthesis of macrocyclic hexaoxime. Inorg Chem 46:2959–2961. https://doi.org/10.1021/ic062327s
Pessoa JC, Correia I (2019) Salan vs. salen metal complexes in catalysis and medicinal applications: virtues and pitfalls. Coord Chem Rev 388:227–247. https://doi.org/10.1016/j.ccr.2019.02.035
Specklin D, Fliedel C, Hild F et al (2017) Mononuclear salen-gallium complexes for iso-selective ring-opening polymerization (ROP) of rac-lactide. Dalton Trans 46:12824–12834. https://doi.org/10.1039/C7DT02730C
Baleizão C, Garcia H (2006) Chiral salen complexes: an overview to recoverable and reusable homogeneous and heterogeneous catalysts. Chem Rev 106:3987–4043. https://doi.org/10.1021/cr050973n
Matsumoto K, Saito B, Katsuki T (2007) Asymmetric catalysis of metal complexes with non-planar ONNO ligands: salen, salalen and salan. Chem Commun 3619. https://doi.org/10.1039/b701431g
Libíková H, Pogády J, Wiedermann V, Breier S (1975) Search for herpetic antibodies in the cerebrospinal fluid in senile dementia and mental retardation. Acta Virol 19:493–495
Nelson SG, Peelen TJ, Wan Z (1999) Mechanistic alternatives in Lewis acid-catalyzed acyl halide aldehyde cyclocondensations. Tetrahedron Lett 40:6541–6543. https://doi.org/10.1016/S0040-4039(99)01309-X
Osamura Y, Terada K, Kobayashi Y et al (1999) A molecular orbital study of the mechanism of chlorination reaction of benzene catalyzed by Lewis acid. J Mol Struct (Thoechem) 461–462:399–416. https://doi.org/10.1016/S0166-1280(98)00452-7
Santelli M, Pons J-M (1996) Lewis acids and selectivity in organic synthesis. CRC Press, Boca Raton https://doi.org/10.5860/choice.34-0936
Yoon TP, Dong VM, MacMillan DWC (1999) Development of a new Lewis acid-catalyzed Claisen rearrangement. J Am Chem Soc 121:9726–9727. https://doi.org/10.1021/ja9919884
Dalton CT, Ryan KM, Wall VM et al (1998) Recent progress towards the understanding of metal–salen catalysed asymmetric alkene epoxidation. Top Catal 5:75–91. https://doi.org/10.1023/A:1019102003494
Venkataramanan NS, Kuppuraj G, Rajagopal S (2005) Metal–salen complexes as efficient catalysts for the oxygenation of heteroatom containing organic compounds—synthetic and mechanistic aspects. Coord Chem Rev 249:1249–1268. https://doi.org/10.1016/j.ccr.2005.01.023
Lidskog A, Li Y, Wärnmark K (2020) Asymmetric ring-opening of epoxides catalyzed by metal–salen complexes. Catalysts 10:705. https://doi.org/10.3390/catal10060705
Kokubo C, Katsuki T (1996) Highly enantioselective catalytic oxidation of alkyl aryl sulfides using Mn-salen catalyst. Tetrahedron 52:13895–13900. https://doi.org/10.1016/0040-4020(96)00851-4
Ready JM, Jacobsen EN (2001) Highly active oligomeric (salen)Co catalysts for asymmetric epoxide ring-opening reactions. J Am Chem Soc 123:2687–2688. https://doi.org/10.1021/ja005867b
Shimazaki Y (2013) Oxidation Chemistry of Metal(II) Salen-type complexes. In: Khalid M (ed) Electrochemistry. InTech https://doi.org/10.5772/48372
Cozzi PG (2004) Metal-Salen Schiff base complexes in catalysis: practical aspects. Chem Soc Rev 33:410–421. https://doi.org/10.1039/B307853C
Wezenberg SJ, Kleij AW (2008) Material applications for salen frameworks. Angew Chem Int Ed 47:2354–2364. https://doi.org/10.1002/anie.200702468
Jacobsen EN, Zhang W, Muci AR et al (1991) Highly enantioselective epoxidation catalysts derived from 1,2-diaminocyclohexane. J Am Chem Soc 113:7063–7064. https://doi.org/10.1021/ja00018a068
Räisänen MT, Korpi H, Sundberg MR et al (2013) Synthesis and characterization of binuclear Co(II) complexes with bis(salen-type) ligands. Inorg Chim Acta 394:203–209. https://doi.org/10.1016/j.ica.2012.08.007
Matsunaga S, Shibasaki M (2014) Recent advances in cooperative bimetallic asymmetric catalysis: dinuclear Schiff base complexes. Chem Commun 50:1044–1057. https://doi.org/10.1039/C3CC47587E
Kobayashi S, Sugiura M, Kitagawa H, Lam WW-L (2002) Rare-earth metal triflates in organic synthesis. Chem Rev 102:2227–2302. https://doi.org/10.1021/cr010289i
Finelli A, Abram S-L, Hérault N et al (2020) Bimetallic salen-based compounds and their potential applications. Cryst Growth Des 20:4945–4958. https://doi.org/10.1021/acs.cgd.9b01610
Leoni L, Dalla Cort A (2018) The supramolecular attitude of metal–salophen and metal–salen complexes. Inorganics 6:42. https://doi.org/10.3390/inorganics6020042
Whiteoak CJ, Salassa G, Kleij AW (2012) Recent advances with π-conjugated salen systems. Chem Soc Rev 41:622–631. https://doi.org/10.1039/C1CS15170C
Oliveri IP, Di Bella S (2023) Lewis Acidic Zinc(II) Complexes of tetradentate ligands as building blocks for responsive assembled supramolecular structures. Chemistry 5:119–137. https://doi.org/10.3390/chemistry5010010
Asatkar AK, Senanayak SP, Bedi A et al (2014) Zn( ii ) and Cu( ii ) complexes of a new thiophene-based salphen-type ligand: solution-processable high-performance field-effect transistor materials. Chem Commun 50:7036–7039. https://doi.org/10.1039/C4CC01360C
Escudero-Adán EC, Belmonte MM, Martin E et al (2011) A short desymmetrization protocol for the coordination environment in bis-salphen scaffolds. J Org Chem 76:5404–5412. https://doi.org/10.1021/jo2008065
Pietrangelo A, Sih BC, Boden BN et al (2008) Nonlinear optical properties of Schiff-base-containing conductive polymer films electro-deposited in microgravity. Adv Mater 20:2280–2284. https://doi.org/10.1002/adma.200702582
Akine S, Taniguchi T, Nabeshima T (2001) Synthesis and crystal structure of a novel triangular macrocyclic molecule, tris(H 2 saloph), † †H2saloph=N, N′-disalicylidene-o-phenylenediamine. and its water complex. Tetrahedron Lett 42:8861–8864. https://doi.org/10.1016/S0040-4039(01)01943-8
Frischmann PD, Jiang J, Hui JK-H et al (2008) Reversible−irreversible approach to Schiff base macrocycles: access to isomeric macrocycles with multiple salphen pockets. Org Lett 10:1255–1258. https://doi.org/10.1021/ol8001317
Jiang J, MacLachlan MJ (2010) Unsymmetrical triangular Schiff base macrocycles with cone conformations. Org Lett 12:1020–1023. https://doi.org/10.1021/ol100028s
Hui JK-H, MacLachlan MJ (2006) [6 + 6] Schiff-base macrocycles with 12 imines: giant analogues of cyclohexane. Chem Commun 2480. https://doi.org/10.1039/b603985e
Gerloch M, Lewis J, Mabbs FE, Richards A (1968) The preparation and magnetic properties of some Schiff base–iron( III ) halide complexes. J Chem Soc A, 112–116. https://doi.org/10.1039/J19680000112
Castilla AM, Curreli S, Escudero-Adán EC et al (2009) Modular synthesis of heterobimetallic salen structures using metal templation. Org Lett 11:5218–5221. https://doi.org/10.1021/ol902149p
Wezenberg SJ, Anselmo D, Escudero-Adán EC et al (2010) Dimetallic activation of dihydrogen phosphate by Zn(salphen) chromophores. Eur J Inorg Chem 2010:4611–4616. https://doi.org/10.1002/ejic.201000455
Decortes A, Kleij AW (2011) Ambient fixation of carbon dioxide using a Zn II salphen catalyst. ChemCatChem 3:831–834. https://doi.org/10.1002/cctc.201100031
Kresge CT, Leonowicz ME, Roth WJ et al (1992) Ordered mesoporous molecular sieves synthesized by a liquid-crystal template mechanism. Nature 359:710–712. https://doi.org/10.1038/359710a0
Slowing II, Vivero-Escoto JL, Trewyn BG, Lin VS-Y (2010) Mesoporous silica nanoparticles: structural design and applications. J Mater Chem 20:7924. https://doi.org/10.1039/c0jm00554a
ALOthman ZA (2012) A Review: Fundamental Aspects of Silicate Mesoporous Materials. Materials (Basel) 5:2874–2902. https://doi.org/10.3390/ma5122874
Beck JS, Vartuli JC, Roth WJ et al (1992) A new family of mesoporous molecular sieves prepared with liquid crystal templates. J Am Chem Soc 114:10834–10843. https://doi.org/10.1021/ja00053a020
Yu C, Yu Y, Zhao D (2000) Highly ordered large caged cubic mesoporous silica structures templated by triblock PEO–PBO–PEO copolymer. Chem Commun 575–576. https://doi.org/10.1039/b000603n
Bagshaw SA, Prouzet E, Pinnavaia TJ (1995) Templating of mesoporous molecular sieves by nonionic polyethylene oxide surfactants. Science 269:1242–1244. https://doi.org/10.1126/science.269.5228.1242
Che S, Garcia-Bennett AE, Yokoi T et al (2003) A novel anionic surfactant templating route for synthesizing mesoporous silica with unique structure. Nature Mater 2:801–805. https://doi.org/10.1038/nmat1022
Chew T-L, Ahmad AL, Bhatia S (2010) Ordered mesoporous silica (OMS) as an adsorbent and membrane for separation of carbon dioxide (CO2). Adv Coll Interface Sci 153:43–57. https://doi.org/10.1016/j.cis.2009.12.001
Nnamdi Chibuike Iheaturu (2014) Synthesis, preparation and characterization of nanoporous core-shell-clay epoxy composites.https://doi.org/10.13140/RG.2.2.23461.91361
Yamaguchi T, Yoshida K, Ito K et al (2011) Thermal behavior, structure, and dynamics of low temperature water confined in mesoporous materials MCM-41. Bunseki Kagaku 60:115–130. https://doi.org/10.2116/bunsekikagaku.60.115
Janus R, Wądrzyk M, Lewandowski M et al (2020) Understanding porous structure of SBA-15 upon pseudomorphic transformation into MCM-41: Non-direct investigation by carbon replication. J Ind Eng Chem 92:131–144. https://doi.org/10.1016/j.jiec.2020.08.032
Andas J, Ekhbal SH, Ali TH (2021) MCM-41 modified heterogeneous catalysts from rice husk for selective oxidation of styrene into benzaldehyde. Environ Technol Innov 21:101308. https://doi.org/10.1016/j.eti.2020.101308
Bhattacharyya S, Lelong G, Saboungi M-L (2006) Recent progress in the synthesis and selected applications of MCM-41: a short review. J Exp Nanosci 1:375–395. https://doi.org/10.1080/17458080600812757
García-Martínez J (2015) Mesostructured zeolites: bridging the gap between zeolites and MCM-41. ChemViews. https://doi.org/10.1002/chemv.201500056
Yamada SA, Hung ST, Thompson WH, Fayer MD (2020) Effects of pore size on water dynamics in mesoporous silica. J Chem Phys 152:154704. https://doi.org/10.1063/1.5145326
Laskowski Ł, Laskowska M, Vila N et al (2019) Mesoporous silica-based materials for electronics-oriented applications. Molecules 24:2395. https://doi.org/10.3390/molecules24132395
Dhaneswara D, Sofyan N (2016) Effect of different pluronic P123 triblock copolymer surfactant concentrations on SBA-15 pore formation. IJTech 7:1009. https://doi.org/10.14716/ijtech.v7i6.3412
Da Silveira T, Awano CM, Donatti DA et al (2014) About the thermal stability and pore elimination in the ordered hexagonal mesoporous silica SBA-15. Microporous Mesoporous Mater 190:227–233. https://doi.org/10.1016/j.micromeso.2014.02.023
Fedyna M, Śliwa M, Jaroszewska K et al (2020) Procedure for the synthesis of AlSBA-15 with high aluminium content: characterization and catalytic activity. Microporous Mesoporous Mater 292:109701. https://doi.org/10.1016/j.micromeso.2019.109701
Mahato BN, Krithiga T (2022) Recent developments in metal-doped SBA-15 catalysts for heterogeneous catalysis and sustainable chemistry. Can J Chem 100:9–17. https://doi.org/10.1139/cjc-2021-0201
An K, Somorjai GA (2015) Nanocatalysis I: synthesis of metal and bimetallic nanoparticles and porous oxides and their catalytic reaction studies. Catal Lett 145:233–248. https://doi.org/10.1007/s10562-014-1399-x
Gkiliopoulos D, Tsamesidis I, Theocharidou A et al (2022) SBA-15 mesoporous silica as delivery vehicle for rhBMP-2 bone morphogenic protein for dental applications. Nanomaterials 12:822. https://doi.org/10.3390/nano12050822
Larki A, Saghanezhad SJ, Ghomi M (2021) Recent advances of functionalized SBA-15 in the separation/preconcentration of various analytes: a review. Microchem J 169:106601. https://doi.org/10.1016/j.microc.2021.106601
Shakeri M, Khatami Shal Z, Van Der Voort P (2021) An overview of the challenges and progress of synthesis, characterization and applications of plugged SBA-15 materials for heterogeneous catalysis. Materials 14:5082. https://doi.org/10.3390/ma14175082
Wang J, Wang G, Zhang Z et al (2021) Effects of mesoporous silica particle size and pore structure on the performance of polymer-mesoporous silica mixed matrix membranes. RSC Adv 11:36577–36586. https://doi.org/10.1039/D1RA05125C
Yates TJV, Thomas JM, Fernandez J-J et al (2006) Three-dimensional real-space crystallography of MCM-48 mesoporous silica revealed by scanning transmission electron tomography. Chem Phys Lett 418:540–543. https://doi.org/10.1016/j.cplett.2005.11.031
Kumar D, Schumacher K, Von Hohenesche CD et al (2001) MCM-41, MCM-48 and related mesoporous adsorbents: their synthesis and characterisation. Colloids Surf A: Physicochem Eng Asp 187–188:109–116. https://doi.org/10.1016/S0927-7757(01)00638-0
Taralkar US, Kasture MW, Joshi PN (2008) Influence of synthesis conditions on structural properties of MCM-48. J Phys Chem Solids 69:2075–2081. https://doi.org/10.1016/j.jpcs.2008.03.004
Jana S, Dutta B, Honda H, Koner S (2011) Mesoporous silica MCM-41 with rod-shaped morphology: synthesis and characterization. Appl Clay Sci 54:138–143. https://doi.org/10.1016/j.clay.2011.07.018
Brankovic M, Zarubica A, Andjelkovic T, Andjelkovic D (2017) Mesoporous silica (MCM-41): Synthesis/modification, characterization and removal of selected organic micro-pollutants from water. Adv techn 6:50–57. https://doi.org/10.5937/savteh1701050B
Pratiwi FW, Kuo CW, Wu S-H, et al (2018) The bioimaging applications of mesoporous silica nanoparticles. In: The Enzymes. Elsevier, pp 123–153 https://doi.org/10.1016/bs.enz.2018.07.006
Chaudhary V, Sharma S (2017) An overview of ordered mesoporous material SBA-15: synthesis, functionalization and application in oxidation reactions. J Porous Mater 24:741–749. https://doi.org/10.1007/s10934-016-0311-z
Kim T-W, Chung P-W, Lin VS-Y (2010) Facile synthesis of monodisperse spherical MCM-48 mesoporous silica nanoparticles with controlled particle size. Chem Mater 22:5093–5104. https://doi.org/10.1021/cm1017344
Pantazis CC, Trikalitis PN, Pomonis PJ, Hudson MJ (2003) A method of synthesis of silicious inorganic ordered materials (MCM-41–SBA-1) employing polyacrylic acid–CnTAB–TEOS nanoassemblies. Microporous Mesoporous Mater 66:37–51. https://doi.org/10.1016/j.micromeso.2003.08.017
Florensa M, Llenas M, Medina-Gutiérrez E et al (2022) Key parameters for the rational design, synthesis, and functionalization of biocompatible mesoporous silica nanoparticles. Pharmaceutics 14:2703. https://doi.org/10.3390/pharmaceutics14122703
Miceli M, Frontera P, Macario A, Malara A (2021) Recovery/reuse of heterogeneous supported spent catalysts. Catalysts 11:591. https://doi.org/10.3390/catal11050591
Ganesan V, Yoon S (2020) Direct heterogenization of salphen coordination complexes to porous organic polymers: catalysts for ring-expansion carbonylation of epoxides. Inorg Chem 59:2881–2889. https://doi.org/10.1021/acs.inorgchem.9b03247
Yilmaz F, Karaağaç Y, Kani̇ İ (2017) Heterogenization of homogeneous perfluorothiophene Rh(I) complex and examination of hydrogenation activity in scCO2 media. Anadolu University Journal of Science and Technology A-Applied Sciences and Engineering 1–1. https://doi.org/10.18038/aubtda.331159
Lantos J, Kumar N, Saha B (2024) A comprehensive review of fine chemical production using metal-modified and acidic microporous and mesoporous catalytic materials. Catalysts 14:317. https://doi.org/10.3390/catal14050317
Liu Y, Chen L, Yang L et al (2024) Porous framework materials for energy & environment relevant applications: a systematic review. Green Energy & Environment 9:217–310. https://doi.org/10.1016/j.gee.2022.12.010
Wang H, Liu X, Liu X et al (2023) Fluidisable mesoporous silica composites for thermochemical energy storage. Energy 275:127255. https://doi.org/10.1016/j.energy.2023.127255
Mohan A, Jaison A, Lee Y-C (2023) Emerging trends in mesoporous silica nanoparticle-based catalysts for CO 2 utilization reactions. Inorg Chem Front 10:3171–3194. https://doi.org/10.1039/D3QI00378G
D’Elia V, Kleij AW (2022) Surface science approach to the heterogeneous cycloaddition of CO2 to epoxides catalyzed by site-isolated metal complexes and single atoms: a review. Green Chemical Engineering 3:210–227. https://doi.org/10.1016/j.gce.2022.01.005
Luts T, Suprun W, Hofmann D et al (2007) Epoxidation of olefins catalyzed by novel Mn(III) and Mo(IV) Salen complexes immobilized on mesoporous silica gelPart I. Synthesis and characterization of homogeneous and immobilized Mn(III) and Mo(IV) Salen complexes. J Mol Catal A: Chem 261:16–23. https://doi.org/10.1016/j.molcata.2006.07.035
Carvalho PA, Comerford JW, Lamb KJ et al (2019) Influence of mesoporous silica properties on cyclic carbonate synthesis catalysed by supported aluminium(salen) complexes. Adv Synth Catal 361:345–354. https://doi.org/10.1002/adsc.201801229
Pal N, Bhaumik A (2013) Soft templating strategies for the synthesis of mesoporous materials: inorganic, organic–inorganic hybrid and purely organic solids. Adv Coll Interface Sci 189–190:21–41. https://doi.org/10.1016/j.cis.2012.12.002
O. Lintang H, Yuliati L (2019) Designed mesoporous materials toward multifunctional organic silica nanocomposites. In: Krishnappa M (ed) Mesoporous materials - properties and applications. IntechOpen .https://doi.org/10.5772/intechopen.84875
Hoffmann F, Fröba M (2011) Vitalising porous inorganic silica networks with organic functions—PMOs and related hybrid materials. Chem Soc Rev 40:608–620. https://doi.org/10.1039/C0CS00076K
Pande V, Kothawade S, Kuskar S, et al (2023) Fabrication of mesoporous silica nanoparticles and its applications in drug delivery. In: Ranjan Sahu D (ed) Nanotechnology and nanomaterials. IntechOpen. https://doi.org/10.5772/intechopen.112428
Karimi B, Ganji N, Pourshiani O, Thiel WR (2022) Periodic mesoporous organosilicas (PMOs): from synthesis strategies to applications. Prog Mater Sci 125:100896. https://doi.org/10.1016/j.pmatsci.2021.100896
Goscianska J, Olejnik A, Nowak I (2017) APTES-functionalized mesoporous silica as a vehicle for antipyrine – adsorption and release studies. Colloids Surf, A 533:187–196. https://doi.org/10.1016/j.colsurfa.2017.07.043
Erigoni A, Diaz U (2021) Porous silica-based organic-inorganic hybrid catalysts: a review. Catalysts 11:79. https://doi.org/10.3390/catal11010079
Wentz CM, Tsinas Z, Forster AL (2023) A synthetic methodology for preparing impregnated and grafted amine-based silica composites for carbon capture. JoVE 65845. https://doi.org/10.3791/65845
Vallet-Regí M, Schüth F, Lozano D et al (2022) Engineering mesoporous silica nanoparticles for drug delivery: where are we after two decades? Chem Soc Rev 51:5365–5451. https://doi.org/10.1039/D1CS00659B
Yu K, Gu Z, Ji R et al (2007) Effect of pore size on the performance of mesoporous material supported chiral Mn(III) salen complex for the epoxidation of unfunctionalized olefins. J Catal 252:312–320. https://doi.org/10.1016/j.jcat.2007.09.009
Ma L, Su F, Guo W et al (2013) Epoxidation of styrene catalyzed by mesoporous propylthiol group-functionalized silica supported manganese(III) salen complexes with different pore morphologies. Microporous Mesoporous Mater 169:16–24. https://doi.org/10.1016/j.micromeso.2012.10.013
Gill CS, Venkatasubbaiah K, Jones CW (2009) Asymmetric cyclopropanation with immobilized Ru(II)-salen-Py2. Synfacts 2009:1050–1050. https://doi.org/10.1055/s-0029-1217653
Zhao D, Zhao J, Zhao S, Wang W (2007) Preparation of MCM-41 supported heterogenized chiral salen Mn (III) complex and the catalytic activity in the asymmetric epoxidation. J Inorg Organomet Polym 17:653–659. https://doi.org/10.1007/s10904-007-9157-9
Bigi F, Moroni L, Maggi R, Sartori G (2002) Heterogeneous enantioselective epoxidation of olefins catalysed by unsymmetrical (salen)Mn(iii) complexes supported on amorphous or MCM-41 silica through a new triazine-based linker. Chem Commun 716–717. https://doi.org/10.1039/b110991j. Electronic supplementary information (ESI) available: synthesis of compounds 1, 3A, 3B, 4A, 4B and 1H NMR spectra. See http://www.rsc.org/suppdata/cc/b1/b110991j/
Kim G-J, Shin J-H (1999) The catalytic activity of new chiral salen complexes immobilized on MCM-41 by multi-step grafting in the asymmetric epoxidation. Tetrahedron Lett 40:6827–6830. https://doi.org/10.1016/S0040-4039(99)01407-0
Yu K, Lou L-L, Lai C et al (2006) Asymmetric epoxidation of unfunctionalized olefins catalyzed by Mn(III) salen complex immobilized on MCM-48. Catal Commun 7:1057–1060. https://doi.org/10.1016/j.catcom.2006.05.017
Ortiz-Bustos J, Martín A, Morales V et al (2017) Surface-functionalization of mesoporous SBA-15 silica materials for controlled release of methylprednisolone sodium hemisuccinate: influence of functionality type and strategies of incorporation. Microporous Mesoporous Mater 240:236–245. https://doi.org/10.1016/j.micromeso.2016.11.021
Ghaedi H, Zhao M (2022) Review on template removal techniques for synthesis of mesoporous silica materials. Energy Fuels 36:2424–2446. https://doi.org/10.1021/acs.energyfuels.1c04435
Hoffmann F, Cornelius M, Morell J, Fröba M (2006) Silica-based mesoporous organic–inorganic hybrid materials. Angew Chem Int Ed 45:3216–3251. https://doi.org/10.1002/anie.200503075
Barczak M (2018) Template removal from mesoporous silicas using different methods as a tool for adjusting their properties. New J Chem 42:4182–4191. https://doi.org/10.1039/C7NJ04642A
Barker RE, Guo L, Mota CJA et al (2022) General approach to silica-supported salens and salophens and their use as catalysts for the synthesis of cyclic carbonates from epoxides and carbon dioxide. J Org Chem 87:16410–16423. https://doi.org/10.1021/acs.joc.2c02104
Amarasekara AS, Oki AR, McNeal I, Uzoezie U (2007) One-pot synthesis of cobalt-salen catalyst immobilized in silica by sol–gel process and applications in selective oxidations of alkanes and alkenes. Catal Commun 8:1132–1136. https://doi.org/10.1016/j.catcom.2006.11.001
Amarasekara AS, McNeal I, Murillo J et al (2008) A simple one-pot synthesis of Jacobson-Katsuki type chiral Mn(III)–salen catalyst immobilized in silica by sol–gel process and applications in asymmetric epoxidation of alkenes. Catal Commun 9:2437–2440. https://doi.org/10.1016/j.catcom.2008.06.009
Birault A, Molina E, Toquer G et al (2018) Large-Pore Periodic Mesoporous Organosilicas as Advanced Bactericide Platforms. ACS Appl Bio Mater 1:1787–1792. https://doi.org/10.1021/acsabm.8b00474
Asefa T, MacLachlan MJ, Coombs N, Ozin GA (1999) Periodic mesoporous organosilicas with organic groups inside the channel walls. Nature 402:867–871. https://doi.org/10.1038/47229
Inagaki S, Guan S, Fukushima Y et al (1999) Novel mesoporous materials with a uniform distribution of organic groups and inorganic oxide in their frameworks. J Am Chem Soc 121:9611–9614. https://doi.org/10.1021/ja9916658
Melde BJ, Holland BT, Blanford CF, Stein A (1999) Mesoporous sieves with unified hybrid inorganic/organic frameworks. Chem Mater 11:3302–3308. https://doi.org/10.1021/cm9903935
Ha C-S, Park SS (2019) PMOs for catalytic applications. In: Periodic mesoporous organosilicas. Springer Singapore, Singapore, pp 125–187 https://doi.org/10.1007/978-981-13-2959-3_5
Liu B, Li H, Quan K et al (2023) Periodic mesoporous organosilica for chromatographic stationary phases: from synthesis strategies to applications. TrAC, Trends Anal Chem 158:116895. https://doi.org/10.1016/j.trac.2022.116895
Zhu L, Liu X, Chen T et al (2012) Functionalized periodic mesoporous organosilicas for selective adsorption of proteins. Appl Surf Sci 258:7126–7134. https://doi.org/10.1016/j.apsusc.2012.04.011
Sarkar S, Sharmah B, Kabir ME, et al (2023) Periodic mesoporous organosilica-based nanocomposite hydrogels for biomedical applications. In: Functional nanocomposite hydrogels. Elsevier, pp 199–213 https://doi.org/10.1016/B978-0-323-99638-9.00008-3
Poscher V, Salinas Y (2020) Trends in degradable mesoporous organosilica-based nanomaterials for controlling drug delivery: a mini review. Materials 13:3668. https://doi.org/10.3390/ma13173668
Wang W, Lofgreen JE, Ozin GA (2010) Why PMO? Towards functionality and utility of periodic mesoporous organosilicas. Small 6:2634–2642. https://doi.org/10.1002/smll.201000617
Fujita S, Inagaki S (2008) Self-organization of organosilica solids with molecular-scale and mesoscale periodicities. Chem Mater 20:891–908. https://doi.org/10.1021/cm702271v
Mizoshita N, Tani T, Inagaki S (2011) Syntheses, properties and applications of periodic mesoporous organosilicas prepared from bridged organosilane precursors. Chem Soc Rev 40:789–800. https://doi.org/10.1039/C0CS00010H
Park SS, Santha Moorthy M, Ha C-S (2014) Periodic mesoporous organosilicas for advanced applications. NPG Asia Mater 6:e96–e96. https://doi.org/10.1038/am.2014.13
Van Der Voort P, Esquivel D, De Canck E et al (2013) Periodic mesoporous organosilicas: from simple to complex bridges; a comprehensive overview of functions, morphologies and applications. Chem Soc Rev 42:3913–3955. https://doi.org/10.1039/C2CS35222B
Baleizão C (2004) Periodic mesoporous organosilica incorporating a catalytically active vanadyl Schiff base complex in the framework. J Catal 223:106–113. https://doi.org/10.1016/j.jcat.2004.01.016
Hartono SB, Hadisoewignyo L, Yang Y et al (2016) Amine functionalized cubic mesoporous silica nanoparticles as an oral delivery system for curcumin bioavailability enhancement. Nanotechnology 27:505605. https://doi.org/10.1088/0957-4484/27/50/505605
Suteewong T, Sai H, Bradbury M et al (2012) Synthesis and formation mechanism of aminated mesoporous silica nanoparticles. Chem Mater 24:3895–3905. https://doi.org/10.1021/cm301857e
Yang F, Gao S, Xiong C et al (2015) Coordination of manganese porphyrins on amino-functionalized MCM-41 for heterogeneous catalysis of naphthalene hydroxylation. Chin J Catal 36:1035–1041. https://doi.org/10.1016/S1872-2067(15)60836-1
Liu L, Hu J, He J et al (2016) Aminopropyl group-modified SBA-15 covalent attachment Mn(salen) complexes as catalysts for styrene epoxidation. Phosphorus Sulfur Silicon Relat Elem 191:1–6. https://doi.org/10.1080/10426507.2014.979983
Saikia L, Srinivas D, Ratnasamy P (2007) Comparative catalytic activity of Mn(salen) complexes grafted on SBA-15 functionalized with amine, thiol and sulfonic acid groups for selective aerial oxidation of limonene. Microporous Mesoporous Mater 104:225–235. https://doi.org/10.1016/j.micromeso.2007.02.026
Lou L-L, Du H, Yu K et al (2013) Immobilized chiral Mn(III) salen complexes on co-condensed imidazole-functionalized mesoporous materials: effective catalysts for asymmetric epoxidation of olefins. J Mol Catal A: Chem 377:85–91. https://doi.org/10.1016/j.molcata.2013.04.028
Mandal M, Nagaraju V, Sarma B et al (2015) Enantioselective epoxidation of styrene by manganese chiral Schiff base complexes immobilized on MCM-41. ChemPlusChem 80:749–761. https://doi.org/10.1002/cplu.201402446
Shi X, Fan B, Li H et al (2014) Preparation and catalytic properties of RuSalen-functionalized periodic mesoporous silicas. Microporous Mesoporous Mater 196:277–283. https://doi.org/10.1016/j.micromeso.2014.05.022
Rayati S, Khodaei E, Nafarieh P et al (2020) A manganese( iii ) Schiff base complex immobilized on silica-coated magnetic nanoparticles showing enhanced electrochemical catalytic performance toward sulfide and alkene oxidation. RSC Adv 10:17026–17036. https://doi.org/10.1039/D0RA02728F
Wu G, Wang X, Liu X et al (2014) Environmental benign oxidation of benzyl alcohol catalyzed by sulphonato-salphen–chromium(III) complexes immobilized on MCM-41. Catal Lett 144:364–371. https://doi.org/10.1007/s10562-013-1102-7
Wang R, Wang J, Zi H et al (2017) Catalytic transfer hydrogenation of ethyl levulinate to γ-valerolactone over zirconium (IV) Schiff base complexes on mesoporous silica with isopropanol as hydrogen source. Molecular Catalysis 441:168–178. https://doi.org/10.1016/j.mcat.2017.07.026
Funding
The authors extend their appreciation to the Deanship of Scientific Research at King Khalid University for funding this work through a large group Research Project under grant number RGP2/280/45.
Author information
Authors and Affiliations
Contributions
S.A Writing the original draft M.A Review & editing.
M.A: review and editing.
Corresponding author
Ethics declarations
Ethical approval
Not applicable.
Conflict of interests
The authors declare no competing interests.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Springer Nature or its licensor (e.g. a society or other partner) holds exclusive rights to this article under a publishing agreement with the author(s) or other rightsholder(s); author self-archiving of the accepted manuscript version of this article is solely governed by the terms of such publishing agreement and applicable law.
About this article

Cite this article
Alshehri, S., Abboud, M. Mesoporous silica–supported metalloSalen and metalloSalphen: synthesis and application in heterogeneous catalysis. J Nanopart Res 26, 177 (2024). https://doi.org/10.1007/s11051-024-06089-x
Received:
Accepted:
Published:
DOI: https://doi.org/10.1007/s11051-024-06089-x