
Abstract
Iron based nanoparticles have shown promise in biomedical applications by possessing diagnostic and therapeutic capabilities. Currently, superparamagnetic iron oxide nanoparticles (SPIONs) are the standard magnetic materials used for magnetic based imaging and hyperthermia therapeutics due to their good magnetic properties, biocompatibility, and stability. However, further improvement of iron-based nanoparticles for such applications can be achieved by creating nanoparticles that possess a pure iron core. Therefore, this study investigates a one-pot synthesis of superparamagnetic iron core–shell nanoparticles using a thermal decomposition of iron pentacarbonyl. Reaction time and surfactant quantities were modified to investigate possible size, shape, and dispersion variations. Characterizations included transmission electron microscopy, x-ray diffraction, Mössbauer spectroscopy, vibrating sample magnetometry, zeta potential, and cytotoxicity studies. From this method, core–shell, monodispersed nanoparticles averaging 14 nm in size were produced and displayed good colloidal stability and high magnetic saturation up to 130 emu/g. Iron nanoparticles were coated with 1,2- distearoyl-sn-glycero-3-phosphoethanolamine-N-[methoxy- (polyethylene glycol)-2000] (ammonium salt) (DSPE-mPEG) polymer to increase biocompatibility and showed low cytotoxicity. The nanoparticles reported display high potential for magnetic imaging and magnetic hyperthermia applications.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Magnetic nanoparticles (MNPs) have been extensively studied for various biomedical applications as theranostic agents, drug delivery systems, and biosensors [1,2,3]. Among such biomedical applications, uses of MNPs for magnetic imaging applications including magnetic resonance imaging (MRI) and magnetic particle imaging (MPI) are of particular interest. MRI has become a powerful tool to accurately diagnose and treat diseases. MRI offers excellent depth penetration and spatial resolution, making it one of the most commonly used diagnostic imaging tools [4,5,6]. Gadolinium (Gd) based contrast agents are among the most commonly used MRI agents. However, in 2006, Gd based contrast agents were shown to cause nephrogenic systemic fibrosis in patients experiencing chronic kidney disease [3, 7, 8]. Therefore, superparamagnetic iron oxide nanoparticles (SPIONs) have been increasingly used as a safer alternative for T2 contrast agents [9].
Compared to MRI, MPI is a newer imaging modality that was proposed in the early 2000s by Gleich and Weizenecker [10, 11]. The signal for MPI is derived from the electronic magnetization of SPIONs, which allows the signal to be 22 million times stronger than MRI’s nuclear magnetization of water in a 7 Tesla field [12]. The tracers create a positive contrast in the image, similar to positron emission tomography (PET) and single-photon emission computed tomography (SPECT) [11, 13]. MPI offers increased depth penetration, no radiation, no background signal from tissues, and allows for real-time quantitative analysis of cells [14, 15]. Currently, SPIONs between 20–30 nm in size, coated with a polymer, have displayed good MPI results [16, 17]. In addition, current FDA approved iron oxide nanoparticles such as Ferucarbotran (Resovist ®, Bayer Healthcare) and Feridex ® (Fermuoxides, Berlex Laboratories) used for MRI applications have been used for MPI applications [12]. However, the magnetic saturation values of SPIONs used for MPI and MRI applications only reaches to around 70 emu/g [18]. In order to obtain higher signal intensity and spatial resolution in MPI, and good contrast in MRI, the MNPs used should possess higher magnetic saturation values, while continuing to be superparamagnetic [19].Utilizing pure iron nanoparticles (FeNPs) for magnetic imaging applications could assist in achieving improved sensitivity for MRI applications and resolution for MPI applications when compared to SPIONs currently used. Indeed, pure iron possesses higher magnetic saturation per unit volume compared to iron oxide [20]. Therefore, studies in creating metallic iron nanoparticles for MRI and MPI applications are needed.
Synthesis of monodisperse iron nanoparticles (FeNPs) have been previously reported, with thermal decomposition of various metallic precursors being the most favored. Effectively, when compared to other synthetic processes, thermal decomposition of organometallic precursors often yields monodispersed, spherical particles with superparamagnetic properties. Thermal decomposition using organo-metallic precursors such as iron bis(trimethylsilyl)amides (Fe[N(SiMe3)2]2) [21], iron oleates (C18H33FeO2−) [22], iron (III) acetylacetonate [Fe (acac)3] [23], and Iron cupferronates [Fe(III)(Cup)3] Cup = N-nitrosophenylhydroxylamine) [24] are among those studied. However, decomposition of iron pentacarbonyl [Fe(CO)5] is one of the most common pathways [25,26,27,28,29]. Therefore, in the present study, an investigation in the synthesis and characterization of iron nanoparticles using this particular method was performed to obtain iron nanoparticles which may find application in magnetic resonance imaging and magnetic particle imaging applications. Variations in the reaction time and initial surfactant amounts were studied to determine if such parameter variations affected the particle size, shape, and distribution.
Materials and methods
Chemicals
Iron (0) pentacarbonyl [Fe(CO)5 > 99.99%], hexadecylamine (HDA, 98%), oleic acid (90%), 1-octadecene (90%), diethylether (anhydrous, ≥ 99.0%), pentane (anhydrous, ≥ 99%), tetrahydrofuran (THF, ≥ 99.9%), hexanes, oleylamine (70%), chloroform, 1,2- distearoyl-sn-glycero-3-phosphoethanolamine-N-[methoxy- (polyethylene glycol)-2000] (ammonium salt) (DSPE-mPEG), and aqueous hydrochloric acid (HCl, 37%) were purchased from Sigma Aldrich. Anhydrous ethanol was purchased from Decon Labs.
Synthesis of HDA.HCl
Synthesis of hexadecylamine-hydrochloride (HDA.HCl) was performed a day before FeNP synthesis by referencing a procedure described by Dash et al., [27]. First, 0.488 g of HDA was dissolved in 100 mL of pentane and stirred with a magnetic stir bar. A mixture of 250 μl of HCl in 30 mL diethylether was slowly poured into the HDA-pentane solution and stirred for 1 min 30 s. The resulting white precipitate was gravity filtered through Whatman filter paper and washed three times with 20 mL of tetrahydrofuran by centrifugation at 9000 RPM in a Beckman Coulter C0650 fixed-angle rotor for 5 min. The resulting white product was dried in a vacuum oven overnight at 30 °C.
Synthesis of FeNPs
Initially, 0.2779 g of HDA.HCl was dissolved in 0.084 mL of oleylamine, 0.207 mL oleic acid, and 20 mL of 1-octadecene in a 100 mL four-neck round-bottom flask. The solution was stirred with a magnetic stir bar and vacuumed and purged with Argon gas 3 times under a Schlenk line. The solution was heated to 120 °C and held for 2 h. The temperature was then raised to 180 °C at a 2 °C/ min rate and 0.7 mL of Fe(CO)5 was directly injected into the solution. The solution turned black upon injection and a yellowish-green gas formed inside the flask. The solution was stirred for 90 min at 180 °C and then cooled to room temperature. A permanent magnet was placed at the bottom of the flask and the liquid solution was removed after decantation, leaving the collected nanoparticles on the magnetic stir bar. Hexane was used to collect particles from the magnetic stir bar. The particles were washed with 60 mL of 200 proof absolute ethanol by centrifugation three times at 8000 rpm for 10 min. Half of the total yield was dispersed in 5 mL of chloroform and kept in a refrigerator for subsequent PEG coating; the other half was dried into powder and stored in an Argon atmosphere glovebox for further characterization.
From the original procedure, quantities of oleylamine and oleic acid (OAm/OA) were varied to determine if the overall amount of OAm and OA to iron pentacarbonyl affected the size or shape of the particles. Varying amounts of OAm/OA tested included 1.5x, 2x, and 3 × the original starting amounts and were compared to the original 1 × amount. The reaction time and temperature after Fe(CO)5 injection was kept consistent with a 90 min reaction time at 180 °C.
In addition, various reaction times of the initial synthesis were performed to investigate possible effects on particle size and crystallinity. The initial synthesis of HDA∙HCl and FeNPs was the same except for the reaction time after the Fe(CO)5 was injected. These reaction times were 30 min, 60 min, 80 min, 90 min, and 24 h.
DSPE-mPEG coating
Iron nanoparticles were coated with 1,2- distearoyl-sn-glycero-3-phosphoethanolamine-N-[methoxy- (polyethylene glycol)-2000] (ammonium salt) (DSPE-mPEG). This Y-shaped PEG polymer was used to increase biocompatibility and colloidal stability [27, 30, 31]. 2 mL of chloroform dispersed nanoparticles were mixed with 0.4 mL of DSPE-mPEG and 9.6 mL of chloroform in a 100 mL round bottom flask. The solution was evaporated with a rotary evaporator for 1 h in a 60 °C water bath under vacuum. 4 mL of ultrapure water was added to the flask after evaporation and sonicated for 1 min to evenly disperse particles in water. The solution was pipetted into Vivaspin Turbo 15 filter tubes (Sartorius) and centrifuged at 5000 rpm for 1 h to remove excess DPSE-mPEG from the solution. Coated particles in the filter tube were pipetted into a storage vial and 1 mL of ultrapure water was added to redisperse the particles into an aqueous medium. Particles were stored in a refrigerator for further cytotoxicity testing.
Characterizations
Transmission electron microscopy, x-ray diffraction, and Mössbauer spectroscopy were performed on all samples. However, due to a lack of difference in the varied reaction time series results when compared to the OAm/OA series, only the samples from the OAm/OA series were further characterized with vibrating sample magnetometry, zeta potential measurements, and cytotoxicity studies.
Transmission Electron Microscopy (TEM)
TEM was performed at the Vanderbilt Institute of Nanoscience and Engineering (VINSE), Vanderbilt University. An FEI Tecnai Osiris TEM system was operated between 80 kV – 200 kV. Diameters were determined using ImageJ processing software. Around 200 nanoparticles were measured for each sample to determine average nanoparticles sizes and standard deviation values.
X-ray Diffraction (XRD)
Powder XRD measurements were performed using a Rigaku SmartLab3kW X-ray Diffractometer with a Cu Kα X-ray source in a 2θ range of 20° to 90°.
Mössbauer spectroscopy
Powdered samples were loaded and sealed into Mössbauer sample cups and characterized at 293 K and 6 K. Mössbauer spectroscopy experiments were performed with a 57Co source, a Janis SHI-850–5 cryogen-free cryostat, and Lakeshore 325 temperature controller. Hyperfine interaction parameters were analyzed using a least-squares fitting model with Mössbauer GenFit Software (R. S. Preston and D. E. Brown).
Vibrating Sample Magnetometry (VSM)
VSM measurements were performed at the Institute for Advanced Materials and Manufacturing (IAMM) at the University of Tennessee Knoxville. Measurements were performed in a ± 1000 mT field at 300 K. OriginLabs was used to graph and fit M-H curves and determine magnetic saturation values.
Zeta potential
Zeta potential measurements were performed using a Particulate Systems NanoPlus Zeta/Nano Particle Analyzer on PEG coated samples to determine the surface charges. Analysis was performed using the NanoPlus software program.
Inductively Coupled Plasma- Optical Emission Spectroscopy (ICP-OES)
An Agilent 5110 inductively coupled plasma optical emission spectrometer at the Civil and Environmental Engineering building at Vanderbilt University was used for ICP-OES measurements. DSPE-mPEG coated samples were tested prior to biocompatibility tests to calculate proper Fe dose concentrations. Preparation of samples followed SW-846 Test Method 6010. 500 μL of coated FeNPs dispersed in water was mixed with 2 mL of 1 M nitric acid and 2 mL of 37% HCl in a 25 mL volumetric flask. The solution was sonicated for 5 min before heating in an 80 °C water bath for 1 h to allow particles to fully dissolve. Whatman filter paper with a pore size of 0.45 μm was used to gravity filter the solution into a 200 mL volumetric flask. The initial volumetric flasks were rinsed with ultrapure water and poured onto the filter for the solution to drip into the 200 mL volumetric flask. The final solution was brought to the appropriate 200 mL volume with ultrapure water to obtain a 50 mg/L iron concentration. 10 mL of the final solution was used for final ICP-OES measurements.
Cytotoxicity
Cytotoxicity studies were performed at VINSE using 4T1 murine breast cancer cell lines. 4T1 cells were seeded into black-walled, clear-bottom 96-well plates with cell media 24 h prior to FeNP introduction. 3 samples of coated FeNPs were introduced to the cells that included the 1 × OAm/OA, 2 × OAm/OA and 3 × OAm/OA samples at 4 varying doses. Serial dilutions were performed with cell media to create 4 doses that included 100, 50, 25, and 12.5 μg/mL. Cells incubated for 24 h after FeNP doses were introduced, and a Celltiter-Glo assay (Promega) was performed to determine cell viability.
Results and discussion
Transmission electron microscopy (TEM) images of samples from the varied OAm/OA series are shown in Fig. 1 and average particle size, iron core, and iron oxide shell sizes are summarized in Table 1. An average of 200 nanoparticles were measured to determine means and standard deviation (SD) values. A thin iron oxide layer is present as a lighter shell surrounding the darker iron core, similar to previous reports [32,33,34]. Elemental mapping to confirm the iron core- iron oxide shell structure is shown in Figure S1. TEM images of the varied reaction time series are shown in Fig. 2 with a summary of the iron core and iron oxide shell size for each sample shown in Table 2. The reaction times of 30 min, 60 min, 80 min, and 90 min displayed similar overall particle sizes as well as iron core and iron oxide shell ratios. Standard deviations of averaged results for all samples were comparable to previous reports [33, 35,36,37]. In addition, all particles appeared to be spherical and monodispersed.

TEM images of samples from varied OAm/OA series with mean diameters ± SD displayed at the top left of images

TEM images of samples from the varying reaction time series. The nanoparticle mean diameters ± SD are displayed at the top left of each TEM image
From TEM results, increasing the OAm/OA surfactant amounts had little effect in size and shape of the nanoparticles. Although no drastic size and structure changes appeared in the TEM images from the various OAm/OA samples, it was found that after synthesis, increasing the amount of surfactant made the particles more difficult to wash. Longer washing procedures were needed in order to properly wash all solvents and surfactants from the particles before further coating steps could be performed. However, longer washing is not optimal for the particles as the longer they are exposed to ambient conditions, the higher the likelihood that oxidation could occur. In addition, little variation in size and shape occurred when the reaction time varied from 30 to 90 min. Alterations in the shape of the particles began to occur in the 24 h reaction time sample and particles began to form irregular shapes and break apart.
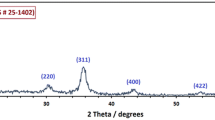
XRD spectra of the 1x, 1.5x, 2x, and 3 × OAm/OA samples are shown in Fig. 3a. Little difference in XRD spectra of the samples was found as all spectra displayed characteristic α-Fe at 45, 65, and 82 degrees (DB # 1,100,108). XRD spectra of the varying reaction time series is shown in Fig. 3b. Little difference in XRD spectra of the 30 min, 60 min, 80 min, and 90 min was found as each spectrum displays characteristic α-Fe peaks. However, the 24-h spectrum indicated peaks characteristic of ⍺-Fe and small peaks attributed to either Fe3O4 or γ-Fe2O3 at 35, 37, 43, and 63 degrees (DB # 2,108,027). Similar iron oxide peaks were also present in the 30 min sample at lower intensity. Due to Fe3O4 and γ-Fe2O3 possessing the same cubic structure and nearly identical lattice parameters, distinguishing between the two from XRD analysis alone is difficult [34, 38]. However, from further Mössbauer studies, the iron oxide present is determined to be γ-Fe2O3.

A) XRD stacked plot of varying OAm/OA surfactant series b) XRD stacked plot of varying reaction time series
XRD analysis agreed with TEM analysis for both series. All of the OAm/OA samples displayed similar XRD spectra, with distinct α-Fe peaks that can be attributed to the iron core of the nanoparticles. No oxide peaks were present, which could be due to the iron oxide shell possessing a small crystalline domain size. Therefore, significant peak broadening and low signal intensity was observed, making it difficult to discern the iron oxide peaks [39, 40]. In addition, distinct α-Fe peaks were obtained for all samples in the varying reaction time series. However, small iron oxide peaks were present in the 24 h and 30 min samples. Lower intensity of the iron oxide peaks for the 30 min sample were observed compared to the 24 h sample. Due to the longer reaction time of 24 h, crystallinity of the iron oxide shell may increase compared to the 30-, 60-, 80-, and 90-min samples as longer reaction times are known to increase the crystalline structure [41]. In addition, the oxide layer could become slightly thicker to where XRD could begin to detect some presence of iron oxide.
Mössbauer spectroscopy studies were performed on all samples since it possesses higher sensitivity than XRD for distinguishing different types of iron components in the samples [42, 43]. Mössbauer studies were performed at 293 K and 6 K on all samples. Stacked Mössbauer spectra at 293 K and 6 K of all OAm/OA samples are shown in Fig. 4. Values from hyperfine fitting parameters for the 293 K and 6 K spectra are displayed in Tables 3 and 4 respectively. For the 293 K spectra, all sample spectra consisted of 2 components, a narrow sextet, and a broad doublet pattern. Component 1 for all OAm/OA samples consisted of a narrow sextet pattern with a hyperfine field between 31.5 – 34.8 T, isomer shift of 0.00 mm/s, and quadrupole splitting 0.00 mm/s, which is attributed to α-Fe. The second component includes a broad doublet pattern with a hyperfine field of 0.00 T, isomer shift between 0.28 – 0.36 mm/s, and quadrupole splitting between 0.98 – 1.09 mm/s, which is indicative of γ-Fe2O3.

Stacked 57Fe Mössbauer spectra of varying OAm/OA series at 293 K and 6 K
Low temperature measurements were performed on all of the samples to better determine the iron oxide component and magnetic properties. For the 6 K measurements for the OAm/OA series, all spectra for the samples were similar, consisting of 2 components, a narrow and broad sextet. Component 1 was comprised of a narrow sextet with a hyperfine field of 34.3 T, isomer shift of 0.12 mm/s and quadrupole splitting of 0.00 mm/s, attributed to α-Fe. Component 2 consisted of a broad sextet with a hyperfine field between 47.5 – 48.0 T, isomer shift between 0.45 – 0.52 mm/s, and quadrupole splitting of 0.00 mm/s, which is indicative of γ-Fe2O3. From low temperature measurements, the γ-Fe2O3 component displays superparamagnetic properties due to the relaxation from a doublet pattern in 293 K spectra, to a sextet at 6 K.
The stacked Mössbauer results of the varying reaction time series measured at 293 K and 6 K are shown in Fig. 5. Values from hyperfine fitting parameters for 293 K and 6 K are displayed in Tables 5 and 6 respectively. From Mössbauer measurements at 293 K, little variation in iron and iron oxide composition was displayed among all time increments All spectra consisted of 2 components that includes a narrow sextet and a doublet pattern. Component 1 possessed a narrow sextet pattern with a hyperfine field around 33.7 T, isomer shift of 0.00 mm/s, and quadrupole splitting 0.00 mm/s, characteristic of α-Fe. Component 2 consists of a broad doublet pattern with a hyperfine field of 0.00 T, isomer shift between 0.28—0.47 mm/s, and quadrupole splitting around 0.5–1.19 mm/s, which is indicative of γ-Fe2O3.

Stacked 57Fe Mössbauer spectra of varying reaction time series at 293 K and 6 K
Low temperature Mössbauer measurements at 6 K for all samples from the varied reaction time series confirmed the magnetic components of the spectrum. All spectra were similar, consisting of 2 sextet components. Component 1 displayed a narrow sextet with a hyperfine field of 34.3 T, isomer shift of 0.12 mm/s and quadrupole splitting 0.00 mm/s, indicative of α-Fe. The second component contained a broad sextet with a hyperfine field between 47.7- 49.6 T, isomer shift between 0.45 – 0.48 mm/s, and quadrupole splitting between 0.00 – 0.04 mm/s, which is associated with γ-Fe2O3.
From the varied OAm/OA surfactant series, Mössbauer spectroscopy results aligned with XRD and TEM data. Increasing the initial OAm/OA surfactant amounts caused an increase in overall iron oxide percentage and less α-Fe content. Although no apparent XRD peaks for iron oxide are present for samples 1x, 1.5x, 2x, and 3 × OAm/OA, increases in γ-Fe2O3 content are apparent in the Mössbauer spectra. Mössbauer studies were also consistent with TEM and XRD results for the varying reaction time series. For the reaction time series, the particles consisted of 2 components, an α-Fe and iron oxide component. After further Mössbauer investigation, the iron oxide present was γ-Fe2O3.
From low temperature Mössbauer studies, the γ-Fe2O3 oxide shell component displayed superparamagnetic characteristics for all samples in the time series and varying OAm/OA surfactant series. The doublet component associated with γ-Fe2O3 in the 293 K spectrum relaxed into a sextet pattern at 6 K, which is characteristic of superparamagnetic behavior [44].
Vibrating sample magnetometry (VSM) was performed to obtain magnetic saturation and coercivity. Since little difference from the samples in the varying time series were found when compared to the OAm/OA series, VSM, Zeta potential and cytotoxicity measurements were only performed on samples from the varying OAm/OA surfactant series. The M-H curves of the 1x, 1.5x, 2x, and 3 × OAm/OA samples at 300 K are shown in Fig. 6. From the M-H curves, all samples displayed superparamagnetic characteristics due to the little to no hysteresis present [45]. In addition, all samples possessed high magnetic saturation between 100 – 130 emu/g, with the exception of 3 × OAm/OA that had slightly lower magnetic saturation around 60 emu/g.

M-H curves of samples from the varied OAm/OA series at 300 K
From magnetization measurements, the initial 1 × OAm/OA sample possessed the highest magnetic saturation around 130 emu/g. As the amount of OAm/OA increased for samples, the magnetic saturation value decreased. This is most likely due to a higher presence of iron oxide than α-Fe in the samples, which was found from Mössbauer measurements. As previously mentioned, higher amounts of OAm/OA caused more difficulty in washing the particles, which caused longer exposure to ambient conditions. Therefore, higher chances of oxidation could have occurred as the washing time increased as OAm/OA amounts increased. Due to the larger percent of iron oxide present in samples with higher amounts of OAm/OA used, a decrease in overall magnetic saturation was observed.
The 1x, 2x, and 3x, OAm/OA samples were chosen to be coated with DSPE-mPEG. Further TEM and XRD characterization of the coated samples are shown in Figures S2 and S3 respectively to confirm the iron core–shell structure and composition was maintained after polymer coatings. Zeta potential measurements are shown in Table 7. The 1x, 2x, and 3 × OAm/OA coated nanoparticle samples were prepared to a 100 μg/ mL concentration in water. Zeta potential for samples was found to be between -21 to -25 mV, suggesting good colloidal stability; these are typical values for surfaces coated with PEG polymers to provide effective electrostatic stabilization [46].
For cytotoxicity studies, the 1 × OAm/OA, 2 × OAm/OA and 3 × OAm/OA coated samples were introduced to cells at 4 varying doses. Serial dilutions were performed with cell media to create 4 doses that included 100, 50, 25, and 12.5 μg/mL. 4T1 cells were seeded into 96-well plates 24 h before FeNP introduction. The cells incubated for 24 h after FeNPs were introduced, and a Celltiter-Glo assay (Promega) was performed to determine cell viability. Each sample dose was carried out in triplicate and averaged to determine mean values and standard deviations.
Results of the biocompatibility tests are shown in Fig. 7. Values are shown as means ± standard deviation (SD) from triplicate measurements. Statistical significance was analyzed using a 2-way ANOVA followed by a Tukey test for pairwise comparisons to examine the effect of sample and dose on cell viability. Calculations were performed using OriginLab software.

Cell viability results of coated 1x, 2 × and 3 × OAm/OA samples at 100, 50, 25, and 12.5 μg/mL doses. All results are an average of three measurements and given as mean ± SD. Statistical significance was assumed for p-values < 0.05: *p < 0.02, **p < 0.0001
There was significant difference between the 2 × and 3 × OAm/OA samples when compared to the 1 × OAm across all doses with a p-value < 0.0001. Little variation in cell viability was found among the varying doses for each sample, with the exception of the 3 × OAm/OA sample. The 3 × OAm/OA sample displayed a significant decrease in cell viability for the 50, 25, and 12.5 ug/ml doses when compared to the 100 ug/ml dose with a p value < 0.001. However, the highest dose of 100 μg/mL yielded around 95% cell viability for the 1 × OAm/OA sample, 88% from 2 × OAm/OA sample, and 93% from the 3 × OAm/OA sample. From these results, it is concluded that these particles have low cytotoxicity. Due to the low cytotoxicity of the particles, further in vitro and in vivo tests would be viable in the future.
Conclusion
Monodispersed FeNP’s were synthesized by a thermal decomposition of Fe(CO)5. Two series were performed that included variations in the reaction time and starting OAm/OA surfactant amounts. The results show that very little change occurred when reaction time was varied, but a 24 h reaction time began to produce more irregularly shaped particles and an increased presence of iron oxide. Increasing the OAm/OA surfactant ratio produced an increase in iron oxide content of particles as well. Therefore, from these studies, optimal FeNPs parameters were determined to be 1 × OAm/OA surfactant amounts with a 90-min reaction time after injection of Fe(CO)5. Particle size averaged around 14 nm that consisted of distinct core–shell structures. Samples displayed good magnetic saturation up to 130 emu/g, and possessed superparamagnetic characteristics. From cytotoxicity studies, DPSE-mPEG coated FeNPs displayed excellent cell viability. From this study, we believe these nanoparticles would assist in providing enhanced performance for magnetic imaging applications and magnetic hyperthermia applications.
Data Availability
The data used to support the findings of this study are included within the article.
References
Vangijzegem T, Stanicki D, Laurent S (2019) Magnetic iron oxide nanoparticles for drug delivery: applications and characteristics. Expert Opin Drug Deliv 16(1):69–78. https://doi.org/10.1080/17425247.2019.1554647
Dadfar SM et al (2019) Iron Oxide Nanoparticles: Diagnostic, Therapeutic and Theranostic Applications. Adv Drug Deliv Rev 138. https://doi.org/10.1016/j.addr.2019.01.005
Yeo D, Wiraja C, Chuah YJ, Gao Y, Xu C (2015) A Nanoparticle-Based Sensor Platform for Cell Tracking and Status/Function Assessment. Nature. https://doi.org/10.1038/srep14768
Ahrens ET, Bulte JW (2013) Tracking immune cells in vivo using magnetic resonance imaging. Nat Rev Immunol 13(10):755–763. https://doi.org/10.1038/nri3531
Bulte JW (2009) In vivo MRI cell tracking: clinical studies. AJR Am J Roentgenol 193(2):314–325. https://doi.org/10.2214/AJR.09.3107
Zeng JF et al (2014) “Anchoring Group Effects of Surface Ligands on Magnetic Properties of Fe304 Nanoparticles: Towards High Performance MRI Contrast Agents,” (in English). Adv Mater 26(17):2694–2698. https://doi.org/10.1002/adma.201304744
Wahsner J, Gale EM, Rodriguez-Rodriguez A, Caravan P (2019) Chemistry of MRI Contrast Agents: Current Challenges and New Frontiers. Chem Rev 119(2):957–1057. https://doi.org/10.1021/acs.chemrev.8b00363
Grobner T (2006) Gadolinium–a specific trigger for the development of nephrogenic fibrosing dermopathy and nephrogenic systemic fibrosis? Nephrol Dial Transplant 21(4):1104–1108. https://doi.org/10.1093/ndt/gfk062
Marckmann P et al (2006) “Nephrogenic systemic fibrosis: Suspected causative role of gadodiamide used for contrast-enhanced magnetic resonance imaging,” (in English). J Am Soc Nephrol 17(9):2359–2362. https://doi.org/10.1681/Asn.2006060601
Gleich B, Weizenecker R (2005) “Tomographic imaging using the nonlinear response of magnetic particles,” (in English). Nature 435(7046):1214–1217. https://doi.org/10.1038/nature03808
Goodwill PW et al (2012) X-space MPI: magnetic nanoparticles for safe medical imaging. Adv Mater 24(28):3870–3877. https://doi.org/10.1002/adma.201200221
Chandrasekharan P et al (2018) A perspective on a rapid and radiation-free tracer imaging modality, magnetic particle imaging, with promise for clinical translation. Br J Radiol 91(1091):20180326. https://doi.org/10.1259/bjr.20180326
Wu LC et al (2019) A Review of Magnetic Particle Imaging and Perspectives on Neuroimaging. AJNR Am J Neuroradiol 40(2):206–212. https://doi.org/10.3174/ajnr.A5896
Yu EY et al (2017) Magnetic Particle Imaging: A Novel in Vivo Imaging Platform for Cancer Detection. Nano Lett 17(3):1648–1654. https://doi.org/10.1021/acs.nanolett.6b04865
Zheng B et al (2016) “Quantitative Magnetic Particle Imaging Monitors the Transplantation, Biodistribution, and Clearance of Stem Cells In Vivo,” (in English). Theranostics 6(3):291–301. https://doi.org/10.7150/thno.13728
Ludwig F et al (2014) Self-consistent magnetic properties of magnetite tracers optimized for magnetic particle imaging measured by ac susceptometry, magnetorelaxometry and magnetic particle spectroscopy. J Magn Magn Mater 360:169–173. https://doi.org/10.1016/j.jmmm.2014.02.020
Bauer LM, Situ SF, Griswold MA, Samia ACS (2016) “High-performance iron oxide nanoparticles for magnetic particle imaging - guided hyperthermia (hMPI),” (in English). Nanoscale 8(24):12162–12169. https://doi.org/10.1039/c6nr01877g
Arami H et al (2017) “Tomographic magnetic particle imaging of cancer targeted nanoparticles,” (in English). Nanoscale 9(47):18723–18730. https://doi.org/10.1039/c7nr05502a
Gloag L et al (2020) Zero valent iron core-iron oxide shell nanoparticles as small magnetic particle imaging tracers. Chem Commun (Camb) 56(24):3504–3507. https://doi.org/10.1039/c9cc08972a
Hadjipanayis CG, Bonder MJ, Balakrishnan S, Wang X, Mao H, Hadjipanayis GC (2008) Metallic Iron Nanoparticles for MRI Contrast Enhancement and Local Hyperthermia. Small 4(11):1925–1929. https://doi.org/10.1002/smll.200800261
Dumestre F, Chaudret B, Amiens C, Renaud P, Fejes P (2004) Superlattices of iron nanocubes synthesized from Fe[N(SiMe3)2]2. Science 303(5659):821–823. https://doi.org/10.1126/science.1092641
Hao R, Xing R, Xu Z, Hou Y, Gao S, Sun S (2010) Synthesis, functionalization, and biomedical applications of multifunctional magnetic nanoparticles. Adv Mater 22(25):2729–2742. https://doi.org/10.1002/adma.201000260
Park J et al (2004) Ultra-large-scale syntheses of monodisperse nanocrystals. Nat Mater 3(12):891–895. https://doi.org/10.1038/nmat1251
Majidi S, Sehrig FZ, Farkhani SM, Goloujeh MS, Akbarzadeh A (2016) Current methods for synthesis of magnetic nanoparticles. Artif Cells Nanomed Biotechnol 44(2):722–734. https://doi.org/10.3109/21691401.2014.982802
Peng S, Wang C, Xie J, Sun S (2006) Synthesis and Stabilization of Monodisperse Fe Nanoparticles. J Am Chem Soc 128. https://doi.org/10.1021/ja063969h
Shao HP, Lee H, Huang YQ, Ko IY, Kim C (2005) “Control of iron nanoparticles size and shape by thermal decomposition method,” (in English). IEEE Trans Magn 41(10):3388–3390. https://doi.org/10.1109/Tmag.2005.855206
Dash A et al (2021) “Colloidally Stable Monodisperse Fe Nanoparticles as T-2 Contrast Agents for High-Field Clinical and Preclinical Magnetic Resonance Imaging,” (in English). Acs Appl Nano Mater 4(2):1235–1242. https://doi.org/10.1021/acsanm.0c02848
Mehta JP et al. (2018) Advances in the Synthesis and Long-Term Protection of Zero-Valent Iron Nanoparticles," (in English). Part Part Syst Char 35(8). https://doi.org/10.1002/ppsc.201800120
Farrell D, Ding Y, Majetich SA, Sanchez-Hanke C, Kao CC (2004) “Structural ordering effects in Fe nanoparticle two- and three-dimensional arrays,” (in English). J Appl Phys 95(11):6636–6638. https://doi.org/10.1063/1.1688644
Xiao SL et al (2019) “Synthesis Of PEG-Coated, Ultrasmall, Manganese-Doped Iron Oxide Nanoparticles With High Relaxivity For T-1/T-2 Dual-Contrast Magnetic Resonance Imaging,” (in English). Int J Nanomed 14:8499–8507. https://doi.org/10.2147/Ijn.S219749
Tong S, Hou SJ, Zheng ZL, Zhou J, Bao G (2010) “Coating Optimization of Superparamagnetic Iron Oxide Nanoparticles for High T-2 Relaxivity,” (in English). Nano Lett 10(11):4607–4613. https://doi.org/10.1021/nl102623x
Wang CM, Baer DR, Amonette JE, Engelhard MH, Antony J, Qiang Y (2009) “Morphology and Electronic Structure of the Oxide Shell on the Surface of Iron Nanoparticles,” (in English). J Am Chem Soc 131(25):8824–8832. https://doi.org/10.1021/ja900353f
Mahin J, Torrente-Murciano L (2020) Continuous synthesis of monodisperse iron@iron oxide core@shell nanoparticles," (in English). Chem Eng J 396. https://doi.org/10.1016/j.cej.2020.125299
Nemati Z, Alonso J, Khurshid H, Phan MH, Srikanth H (2016) “Core/shell iron/iron oxide nanoparticles: are they promising for magnetic hyperthermia?,” (in English). Rsc Adv 6(45):38697–38702. https://doi.org/10.1039/c6ra05064f
Singh G et al (2014) “Tunability in Crystallinity and Magnetic Properties of Core-Shell Fe Nanoparticles,” (in English). Part Part Syst Char 31(10):1054–1059. https://doi.org/10.1002/ppsc.201400032
Yoon TJ, Lee H, Shao HL, Weissleder R (2011) “Highly Magnetic Core-Shell Nanoparticles with a Unique Magnetization Mechanism,” (in English). Angew Chem Int Edit 50(20):4663–4666. https://doi.org/10.1002/anie.201100101
Farrell D, Majetich SA, Wilcoxon JP (2003) “Preparation and characterization of monodisperse Fe nanoparticles,” (in English). J Phys Chem B 107(40):11022–11030. https://doi.org/10.1021/jp0351831
Kim W et al (2012) A new method for the identification and quantification of magnetite-maghemite mixture using conventional X-ray diffraction technique. Talanta 94:348–352. https://doi.org/10.1016/j.talanta.2012.03.001
Holder CF, Schaak RE (2019) “Tutorial on Powder X-ray Diffraction for Characterizing Nanoscale Materials,” (in English). ACS Nano 13(7):7359–7365. https://doi.org/10.1021/acsnano.9b05157
Famiani S, LaGrow AP, Besenhard MO, Maenosono S, Thanh NTK (2018) “Synthesis of Fine-Tuning Highly Magnetic Fe@FexOy Nanoparticles through Continuous Injection and a Study of Magnetic Hyperthermia,” (in English). Chem Mater 30(24):8897–8904. https://doi.org/10.1021/acs.chemmater.8b04056
Karade VC et al (2018) “Effect of reaction time on structural and magnetic properties of green-synthesized magnetic nanoparticles,” (in English). J Phys Chem Solids 120:161–166. https://doi.org/10.1016/j.jpcs.2018.04.040
Holmes-Hampton GP, Tong WH, Rouault TA (2014) Biochemical and biophysical methods for studying mitochondrial iron metabolism. Methods Enzymol 547:275–307. https://doi.org/10.1016/B978-0-12-801415-8.00015-1
Ajinkya N, Yu X, Kaithal P, Luo H, Somani P, Ramakrishna S (2020) Magnetic Iron Oxide Nanoparticle (IONP) Synthesis to Applications: Present and Future. Materials (Basel) 13(20). https://doi.org/10.3390/ma13204644
Lyubutin IS, Starchikov SS, Lin CR, Gervits NE, Korotkov NY, Bukreeva TV (2015) "Structural and Magnetic Properties of Iron Oxide Nanoparticles in Shells of Hollow Microcapsules Designed for Biomedical Applications," (in English). Croat Chem Acta 88(4). https://doi.org/10.5562/cca2739
Mamiya H, Jeyadevan B (2014) Magnetic Hysteresis Loop in a Superparamagnetic State," (in English). IEEE Trans Magn 50(1). https://doi.org/10.1109/Tmag.2013.2274072
Gonçalves LC, Seabra AB, Pelegrino MT, de Araujo DR, Barnardes JS, Haddad PS (2017) Superparamagnetic Iron Oxide Nanoparticles Dispersed in Pluronic F127 Hydrogel: Potential Uses in Topical Applications. R Soc Chem 7. https://doi.org/10.1039/C6RA28633J
Acknowledgements
We thank Vanderbilt University for the use of their facilities. We thank Shrusti Patel (Vanderbilt University) for cytotoxicity analyses.
Funding
Funding was provided by the Center of Laser Applications at the University of Tennessee Space Institute.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflicts of interest
The authors declare no conflict of interest.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Below is the link to the electronic supplementary material.
Rights and permissions
Springer Nature or its licensor (e.g. a society or other partner) holds exclusive rights to this article under a publishing agreement with the author(s) or other rightsholder(s); author self-archiving of the accepted manuscript version of this article is solely governed by the terms of such publishing agreement and applicable law.
About this article

Cite this article
Williams, A.G., Liu, L., Johnson, C.E. et al. One-pot synthesis of iron core—iron oxide shell nanoparticles with potential for magnetic imaging. J Nanopart Res 25, 201 (2023). https://doi.org/10.1007/s11051-023-05851-x
Received:
Accepted:
Published:
DOI: https://doi.org/10.1007/s11051-023-05851-x