
Abstract
The oral route is one of the most preferred routes of administration because of its convenience and safety. Nanostructured lipid carriers (NLCs) are the second-generation nanosize solid lipid nanocarriers that are composed of solid lipids, liquid lipids, and surfactants. The lipid matrix of NLCs has an imperfect structure which allows more drug loading. Other advantages offered by NLCs include biocompatibility, biodegradability, and high encapsulation efficiency. They are considered potential nanocarriers in oral drug delivery and have particle size in the range of 50–300 nm. NLCs have shown improved oral bioavailability of lipophilic drugs. They also bypass first-pass metabolism and inhibit the P-glycoprotein (P-gp) efflux mechanism of drugs. This review mainly highlights the role of NLCs in the oral delivery of drugs and different barriers that have to be overcome to achieve drug delivery by oral route.
Graphical abstract

Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
The oral route is a widely used and accepted route of administration. It offers many advantages such as self-administration of dosage forms, better patient compliance, painless, and economical route compared to other routes of administration. Despite these benefits, the oral route is still challenging because of low aqueous solubility and low penetration of drug across the gastrointestinal (GI) membrane. A drug that is administered orally must survive in the harsh environment of the GI tract and should be absorbed [1, 2]. Researchers are working on a nanotechnology-based delivery system to facilitate the oral administration of the poorly soluble drugs. Targeted and controlled release of drugs can be achieved using nanocarrier-based oral drug delivery system (DDS). In addition, nanocarriers that are given orally can improve the pharmacodynamic and pharmacokinetic performance of many drugs [3]. Thus, smart DDS should be formulated to overcome challenges associated with oral drug delivery [4].
Nanostructured lipid carriers
Different lipid-based DDS like liposomes, nanoemulsions, and micelles have been developed for oral drug delivery of drugs. Lipidic DDS have shown improved water solubility and oral absorption over conventional ones. But the conventional lipid-based DDS such as liposomes, nanoemulsions, and micelles get degraded in the GIT by the intestinal enzymes and show instability problems during their storage [5,6,7]. Muller et al. (1991) developed solid lipid nanoparticles (SLNs) using biodegradable and biocompatible solid lipid Dynasan 112 and solved stability issue and other limitations such as low drug loading and initial burst release of drugs that are associated with conventional lipidic DDS [8]. But on storage, these SLNs showed drug leakage problem because of their transformation to more ordered structure [9]. To avoid drawbacks associated with SLNs, modified lipidic nanocarriers, i.e., NLCs, came into existence in early 2000 and are considered second-generation SLNs [10]. The addition of liquid lipids into the solid matrix of SLNs results in the formation of imperfections in the solid matrix and is thus responsible for more drug payload by maintaining the physical stability of nanocarriers [3]. The newly formed nanoparticles containing imperfect matrices are termed NLCs [11,12,13]. The NLCs are formulated by heating and cooling crystallization of solid and liquid lipids mixture. NLCs offer many advantages over SLNs such as unstructured matrix that helps to avoid drug leakage and ensures high drug entrapment (Fig. 1) [14]. Both hydrophilic and lipophilic drugs can be incorporated into NLCs. Solid and liquid lipids used in the formulation of NLCs are biodegradable and show low toxicity profile [15].

Schematic representation of structure of SLNs and NLCs
NLCs can give controlled and site-specific drug delivery [5], and they ensure the accumulation of drug at the site of action because of enhancement in drug solubility [16]. NLCs show high sustainability because of the presence of biocompatible and physiological lipids [17]. The incorporation of liquid lipids into NLCs creates many imperfections in the core matrix which aids in higher drug entrapment [18,19,20].
Advantages of nanostructured lipid carriers
-
Good physical and chemical stability [21]
-
Low toxicity profile
-
Biodegradable and biocompatible
-
Organic solvents can be avoided for the formulation [10, 15].
-
Improved drug release pattern [22].
-
High drug payload [23]
-
Show good dispersibility in an aqueous medium [24].
Types of NLCs
There are three types of NLCs (Table 1, Fig. 2) which include the following:

Structural representation of imperfect, amorphous, and multiple types of NLCs
Composition of NLCs
The solid matrix of NLCs consists of solid and liquid lipids. The core ingredients of NLCs are solid lipids, liquid lipids, and surfactants. When these lipids and surfactant are blended, they form unstructured solid matrix. Solid and liquid lipids are generally used in a ratio of 70:30 to 99.9:0.1. The concentration of surfactants used in the formulation of NLCs is 1–5% [14].
Lipids
Lipids play an important role in the formulation of NLCs as they provide desired physical and chemical properties and stability. The selection of lipids is based on many factors such as they should be biocompatible and biodegradable and able to produce particles in the nano range [28]. Both the lipids used in the formulation of NLCs should be spatially incompatible, and also, there should not be the dissolution of solid lipids into liquid lipids. There should not be phase separation at a temperature lower than the melting point of lipids [16, 29, 30]. Selected lipids should not give toxic residue during the formulation of NLCs, and thus, lipids should have low toxicity profile [31].
Stearic acid and its derivatives are widely used as solid lipids. Stearic acid is a natural fat found in both animals and plants. It is also safe and biocompatible; therefore, it can be used in drug delivery [32, 33]. Sometimes, hard fats and waxes can also be used in the formulation as a part of the solid matrix. Waxes are isolated from animals and plants, while hard fats are hydrogenated products of unsaturated oils. Solid lipids are used in combination to decrease crystallinity of the lipid matrix which thereby increases drug payload [34, 35]. Other solid lipids used in the formulation of NLCs include palmitic acid, lauric acid, behenic acid, oleic acid, and myristic acid [36, 37].
Digestible natural oils are used as liquid lipids in the formulation of NLCs. Soyabean oil, sunflower oil, and cottonseed oil are the other natural edible oils used in the formulation of NLCs. Other than these liquid lipids, oleic acid and medium chain triglycerides can also be used as liquid lipids [38].
Surfactants
Surfactants possess hydrophilic head and hydrophobic tail. These agents decrease the interfacial tension between the oil and water phase. Surfactants are selected on the basis of HLB value, administration route of nanocarriers, and the surface modification of nanocarriers. Their ability to stop lipid degradation is also one of the criteria for the selection of surfactants [39, 40].
The most frequently used hydrophilic surfactants are tween and poloxamer 188. The lipophilic surfactants used in the formulation of NLCs are lecithins and span 80. The use of surfactants in combination is more helpful to avoid the agglomeration of particles. Surfactants are used in combination to prevent the aggregation of particles. The circulation of drugs can be improved by surface modification of nanocarriers with polyethylene glycol. Surface modification prevents drug uptake by reticuloendothelial system [41,42,43]. Examples of lipids and surfactants used for the formulation of NLCs are shown in Table 2.
Method of preparation of NLCs
Various formulation methods can be employed for the production of NLCs. Some of them include high-pressure homogenization, microemulsion technique, solvent injection method, solvent emulsification evaporation method, and double emulsion technique. High-pressure homogenization and microemulsion techniques are widely used methods for the formulation of NLCs (Table 3).
Characterization of NLCs
Particle size and particle morphology
The stability of NLCs can be influenced by particle size and particle size distribution. Smaller particles show more stability and less aggregation during storage. Particle size is also related to surface area and can further affect their solubility and drug release rate. The size of NLCs normally ranges from 10 to 1000 nm. NLCs for higher cellular uptake and site-specific drug delivery should range from 50 to 300 nm. For sustained drug release, NLCs should have a size above 300 nm [49].
Particle size determination of NLCs is done using photon correlation spectroscopy (PCS) and laser diffraction. PCS is the most widely used technique because of its suitability for the measurement. Both techniques can also be employed for the measurement of particle size distribution which is represented in terms of the polydispersity index (PI). If the PI value is equal to or near zero, then the distribution is monodispersed, while the distribution is polydispersed when the PI value is close to 1 [7, 31, 50]. Harshitha et al. (2019) formulated NLCs of paclitaxel using melt emulsification technique for the treatment of the liver cancer and characterized it for particle size and PDI (Fig. 3). The NLCs showed particle size and PDI of 153.82 ± 5.58 nm and 0.221 ± 0.026 [51].

Particle size of PTX-NLCs
Determination of structure and surface morphology of NLCs is done by transmission electron microscopy (TEM) and scanning electron microscopy (SEM). SEM involves scanning of the surface by using focused beam of electrons, thereby producing images, while TEM creates images when beam of electrons is transmitted through the sample. TEM gives an idea of particle size and the structure of the lipid matrix [52,53,54]. Atomic force microscopy (AFM) is another widely used technique for the determination of surface morphology. AFM gives three-dimensional images, while SEM and TEM give two-dimensional images [55]. AFM uses nanometer resolution to get detailed information about the structure and the topography of NLCs [56]. Zhu et al. (2020) prepared NLCs of nintedanib (BIBF) with a goal to enhance its oral absorption. BIBF NLCs were formulated using the melt-emulsification method and characterized for their morphology. NLCs demonstrated spherical morphology (Fig. 4) and acceptable entrapment efficiency [57].

TEM images of optimized BIBF-NLCs
Zeta potential
Zeta potential (ZP) is the electrical potential of particles that is related to the movement of particles in a liquid. The ZP is defined as the potential difference between the dispersion medium and the stationary layer of liquid attached to the particle. ZP depends on particles and conditions like nature ions, ionic strength, and pH [58]. It indicates the charge attained by the particles. The ZP gives an idea about the stability of NLCs during storage. High ZP prevents particle aggregation because of the electric repulsion between the particles and stabilizes NLCs. Low ZP results in the aggregation of particles as the attraction between the particles is greater than the repulsion. A ZP of more than +30 mV or less than −30 mV is required for the stabilization of NLCs. Doppler electrophoresis is utilized for the determination of the ZP of NLCs. When an electric field is applied across the sample, charged particles move with a velocity proportional to the magnitude of the ZP towards the opposite charge electrode. This magnitude of velocity is measured by Laser Doppler velocimetry [59]. Zhang et al. (2019) developed NLCs of tilmicosin (TMS) using high shear method combined with ultrasonic techniques and characterized them for ZP. TMS-NLCs showed ZP of −30.04 ± 1.36 mV (Fig. 5), indicating its good stability [60].

Zeta potential of TMS-NLCs
Polydispersity index
Measurement of the polydispersity index (PDI) is necessary as the colloidal particles are polydispersed. Determination of PDI is done by photon correlation spectroscopy [61]. Particle size distribution and stability of NLCs can be predicted using PDI. PDI values in the 0–0.5 range show a narrow particle size distribution, and the system is considered to be monodispersed [16], while PDI values greater than 0.5 indicate polydispersity. A PDI value of less than 0.3 is considered an optimum value [62, 63].
Degree of crystallinity
The encapsulation efficiency and rate of drug release from NLCs are influenced by the structure of the crystal lattice and its lipid components [64]. More imperfections in the crystal lattice ensure more drug encapsulation [65]. Increased lipid packing density and thermodynamic stability are observed with decrease in the drug incorporation rates in the following order: supercooled melt < α-modification < β-modification < β′-modification [66]. Determination of crystallinity is done by X-ray diffraction (XRD) and differential scanning calorimetry (DSC). In the case of DSC, physical and chemical changes within a sample are measured in terms of heat lost or gained as a function of temperature. DSC provides information about the nature of lipids as well as their melting and crystallization behavior [67, 68].
Wide angle X-ray diffraction (WXRD) can be used for the identification of some crystalline compounds. WXRD can identify arrangements of acyl chains in lipids. A change in peak intensity is measured by WXRD which is an indication of the formation of NLCs [64]. The ratio of bulk enthalpies can be determined by the degree of crystallinity of lipids from NLCs [69].
In XRD, monochromatic X-ray beam is diffracted at the angle determined by the spacing of the planes in the crystal type of arrangement of atoms which is further recorded as pattern by detector. For each type of crystalline compound, the intensity and position of the diffraction are unique. XRD can identify the arrangement of lipid molecules, the structure of lipids, and phase behavior [70, 71]. Pyo et al. (2020) enhanced the solubility and bioavailability of fenofibrate by formulating it as fenofibrate-loaded NLCs (FFB-NLCs) and coating it with a biodegradable polymer (chitosan) to allow controlled drug release. The FFB-NLCs were characterized for their crystallinity, and the diffractogram of pure fenofibrate demonstrated sharp characteristic peaks at diffraction angles of 11.8°, 14.3°, 16.1°, 16.6°, and 22.2°. These peaks showed crystallinity of fenofibrate and were not observed in the FFB-NLC formulations (Fig. 6) [31]. These results indicated that CF-NLCs were fabricated well by coating method [44].

X-ray diffractogram of FFB-NLCs
Encapsulation efficiency (EE)
Drug entrapment efficiency is defined as quantity of drug integrated into particles compared to total amount of drug available in the dispersion. Drug encapsulation efficiency is also called drug entrapment efficiency. The determination of encapsulation efficiency is done by the combination of separation and analytical techniques. Separation techniques may include ultrafiltration, dialysis, and centrifugation. The encapsulation efficiency of NLCs is generally more than 70% [72]. Encapsulation efficiency can affect drug release characteristics [73]. The lipophilic drugs are either uniformly distributed in the lipid matrix or embedded in the core, while hydrophilic drugs are embedded in interfacial and aqueous phases. Drug loading capacity depends on solubility of drugs in lipid phase [74].
The specified quantity of dispersion of NLCs is centrifuged, and concentration of the drug in the supernatant is measured. The free drug will be present in the supernatant. The concentration of entrapped drugs is calculated by subtracting the concentration of free drugs from the initial concentration of drugs present in the dispersion. Entrapment efficiency increases as the solubility of the drug in the lipid blend increases which will decrease drug expulsion from NLCs [75, 76]. Shevalkar et al. (2019) fabricated NLCs of ezetimibe to improve its bioavailability on oral administration by microemulsion method and characterized them for entrapment efficiency. The encapsulation efficiency of ezetimibe NLCs was found to be 34.60 ± 2.63%. This result demonstrated that ~35% of ezetimibe was entrapped in the NLC, while ~65% might be associated with the surfactant in micellar form [77].
Drug release
The sustained release of active ingredients from NLCs can lead to a prolonged half-life of drug. The release of active ingredients from NLCs depends on liquid lipid concentration, temperature, and surfactant concentration [64]. In the dialysis method, NLCs containing the drug is kept in a dialysis bag which is then immersed in a buffer solution at 37 °C. At a specific time interval, samples are removed and replaced with same volume of fresh dissolution medium. Another method that can be employed for drug release studies is the Franz diffusion cell method. In this method, the donor compartment contains drug-loaded NLCs, while acceptor compartment contains fresh buffer solution. These two compartments are separated by a cellulose membrane. At specific time intervals, samples are withdrawn from acceptor compartment and evaluated for the amount of drug released [78]. Drug release studies can provide insight into NLC performance in vivo [79]. Thapa et al. (2018) improved bioavailability of telmisartan by formulating telmisartan NLCs and characterized them for drug release study. The in vitro drug release of telmisartan NLCs in the simulated gastric fluid (acidic buffer, pH 1.2) and simulated intestinal fluid (phosphate buffer, pH 6.8) was found to be significantly improved compared to drug suspension and marketed formulation both due to increased solubilisation and entrapment efficiency (Fig. 7) [80].

In vitro drug release study of telmisartan NLCs, marketed formulation, and pure drug suspension in pH 6.8 phosphate buffer
Surface tension measurement
An increment in surfactant concentration reduces interfacial tension because of the emulsification process. Measurement of surface tension is done by Wilhelmy plate method. Surface tension can also be determined by measuring the contact angle [6]. Another instrument used for the measurement of surface tension is the Kibron instrument which is an easy to use torsion balance instrument with high precision. It is based on the use of the maximum pull force technique and ultrasensitive microbalance with the sensor. This instrument gives more accurate results compared to the platinum Wilhelmy plate and Du Nouy ring [81].
Oral drug delivery of drugs through NLCs
The oral route is highly preferred route of administration because of its safety, patient compliance, and ability to self-administer. Though it is the most suitable route of administration, many barriers present in the GIT make it challenging route of administration [82, 83]. The presence of goblet cells, enterocytes, and Peyer’s patches with M cells can make the intestinal epithelium a good platform for the absorption of drugs. The oral route is helpful in the case of diseases requiring frequent administration for a prolonged time. The oral route has advantages like painless route, self-administration, high patient compliance, and economic compared to other routes of drug delivery [84].
For effective drug delivery by the oral route, this route should overcome challenges like low aqueous solubility, harsh gastric environment, and stability of drug in the digestive enzymes [85]. Nanotechnology offers advantages in the field of oral drug delivery as it allows drug delivery intracellularly and transcellularly. It helps to deliver low aqueous soluble drugs and targets drugs at particular sites in the gastrointestinal tract. Nanotechnology enables drug transcytosis across tight intestinal barriers [86]. To overcome these challenges, nanocarriers like nanostructured lipid carriers can be a potential drug delivery system. This nanocarrier system offers many advantages such as improved oral uptake of the drug, enhanced oral bioavailability, improved drug stability, increased intracellular permeation, prevention of drug from P-glycoprotein efflux, and enzymatic degradation. In this system, drug is incorporated into lipid matrix, and controlled or sustained drug release can be achieved with the prolonged gastric residence of the drug. In addition, it prevents biochemical and enzymatic degradation of the drug in the GIT [87, 88].
Challenges associated with oral drug delivery and NLCs role in overcoming these barriers
Almost more than 60% of drugs that come into the market are available and are administered via the oral route. The drug absorption in GIT is based on physiological and anatomical barriers such as chemical and enzymatic barriers, permeability-related barriers like intestinal epithelium, and mucus layer. Oral delivery of many drugs is majorly affected by factors such as poor hydrophilicity and intrinsic dissolution rate.
Different drug delivery approaches were proposed to overcome these barriers and to enhance oral absorption of many drugs. The approaches to overcome these barriers include use of permeation enhancers, modification of drugs and GI transit time, prevention of drug degradation by first-pass metabolism, utilization of enzyme inhibitors, and development of new DDS such as NLCs. Luminal and proteolytic drug degradation of drugs can be prevented by designing targeted NLCs based DDS that can release drug at site of GIT where proteolytic activity is less which thereby enhances drug absorption and bioavailability [89].
The wide pH range of the gastrointestinal tract is one of the concerns for the chemical and physical stability of nanocarriers. In the stomach, more acidic conditions are found around pH 1–2.5, whereas in the colon pH conditions are found in the range of 7–8. Many nanocarriers possess ionizable groups on their surface, so pH near to their isoelectric point will lower or remove their surface charge. GI fluids contain different concentrations of electrolytes and salts which can show a negative surface charge on the nanocarriers. This causes destabilization that may result in their agglomeration in GI fluids. Gastric enzymes such as gelatinase or pepsin can also affect the stability of nanocarriers. Enzyme concentration is found to be higher in the duodenum due to the presence of biliary and pancreatic secretions that contain amylase, peptidase, and lipases [90]. The presence of lipases can help to digest lipid-based nanocarriers, leading to aggregation, solubilization, and precipitation of nanocarriers [91].
Mucus forms a barrier for the penetration of many drugs from the lumen to epithelium due to its viscoelastic and hydrogel-like structures. The two mucus layers that are the outer loosely adherent layer and the inner firm adherent layer restrict the penetration of various drugs. Goblet cells secrete mucus every 24–48 h which prevents the adhesion of harmful bacteria and compounds. Mucus has a gel-like structure that helps to entrap foreign materials. It mainly consists of mucin and glycoproteins [92]. Other constituents of mucus include carbohydrates, proteins, lipids, salts, nucleic acids, and antibodies [85]. Hence, mucus creates and provides safe and nutrient-rich surroundings for bacterial colonization and antibacterial drugs.
One of the critical biochemical barriers that affect the oral bioavailability of many drugs is pH and enzymatic degradation. The enzymes present in GIT and acidic pH of stomach degrade almost 90–94% of drugs because of deamination, hydrolysis, or oxidation [93]. In addition, digestive enzymes like pepsin are one of the challenges for oral drug delivery along with extremely acidic conditions of the stomach. The hydrophobic part of many drugs can be hydrolyzed by lipases present in the stomach. Digestive enzymes like chymotrypsin, carboxypeptidases, trypsins, and elastases which are present in high concentrations in the small intestine are also responsible for the digestion of many drugs [92]. Thus, lipid nanocarrier-based oral drug delivery is a difficult task as nanocarriers have to overcome extreme GI conditions and other physical and chemical barriers. Other barriers include GI fluids which have variable pH values and contain that may affect the nanocarrier stability. Gut microflora present in the mucus layer limits intestinal residence time or entry of nanocarriers into the intestinal epithelium leading to the efflux of drugs. This efflux of drugs results in low absorption of nanocarriers [90, 94]. If a proper study is conducted on these GI barriers and their interactions with nanocarriers, then these challenges can be converted into opportunities and will also help to design nanocarrier-based drug delivery systems that will facilitate oral drug delivery by enhancing oral absorption and bioavailability of poorly soluble drugs.
These challenges of the oral drug delivery can be effectively overcome by NLCs. For the absorption of any drug molecule by any mechanism, the required prerequisite is that the drug should be present in an aqueous solution. This fact is dependent on the drug’s aqueous solubility and rate of dissolution. Because of their increased specific surface area, lipid-based nanodrug delivery systems such as NLCs can enhance bioavailability. In addition, they increase the amount of nutrients in systemic circulation via systemic and lymphatic transport. Therefore, NLCs are safe and promising nanocarrier drug delivery system for lipophilic and poorly soluble molecules in an aqueous solution. The two main steps that decide the oral absorption of drug molecules are the dissolution rate and penetration rate of drug molecules across the cell membrane [95].
The formulation has altered properties when the lipids along with surfactants and cosurfactants are formulated as NLCs [96]. Alteration in the lipid’s physical state, interactions between molecules, and energy associated with lipids in the matrix and aqueous surfactant surrounding resulted in a change in the properties of NLCs. Drug-loaded NLCs can enhance bioavailability due to their nano-range particle size which in turn provide high surface area and energy required for the interaction between lipids-surfactant drug. Therefore, it can be concluded that small particle size can improve the bioavailability of drugs as well as the surface properties of NLCs significantly [97, 98]. Shah et al. formulated oral NLCs of raloxifene for enhancement of oral bioavailability. The pharmacokinetic study indicated increment bioavailability of raloxifene NLCs compared to drug suspension [107].
The oral pharmacokinetics can be assessed by different in vitro assays as extensive preclinical in vivo studies are time-consuming and costly. In addition, the in vivo animal pharmacokinetic data obtained from the studies is not reliable to predict oral bioavailability in humans [108, 109]. The oral bioavailability of many drugs can be predicted using economical and more efficient in vitro assays. Different parameters such as dissolution, permeability, solubility, and absorption can be assessed by in vitro assays. Different mechanistic approaches such as steady-state models, dynamic models, and quasi-equilibrium models are used to predict oral absorption. The dynamic models comprise of compartmental model and dispersion model. The steady-state models can only predict the extent of absorption, but not the rate of the absorption of the drug. The dynamic models are used for the prediction of the rate as well as the extent of oral drug absorption. The dynamic models can predict both the rate and extent of oral drug absorption [110,111,112,113]. In addition, dynamic models can assess plasma concentration-time drug profiles.
The pH-triggered release mechanisms are commonly utilized in oral delivery among many strategies for overcoming barriers. The pH-responsive NLCs for oral drug delivery have been shown to improve drug stability in the stomach and provide controlled release in the intestines [114]. In the acidic medium, pH-responsive NLCs undergo physical or chemical changes like dissociation or degradation, efficiently releasing the loaded drugs to achieve spatiotemporal control of targeted treatment [115,116,117]. The pH responsiveness of polymeric materials can be achieved by protonation of ionizable groups or acid hydrolysis of chemical bonds and conformation or chemical modifications [118]. The responsiveness of pH-sensitive nanosystem may be attributed to hydrophobic contacts, hydrogen bonds, stacking, or ionic bonds in the nano-core at the molecular level [119]. pH-sensitive NLC systems frequently consist of a surface polymer with acid-sensitive linkages that dissociate in response to ambient pH, resulting in targeted therapeutic release at the target disease tissues. pH-sensitive NLCs containing a targeting moiety or ligand can attach to their target cell, causing internalization. pH-responsive NLCs can dissociate and deliver drugs when coming into contact with an acidic intracellular environment. Thus, pH-sensitive NLCs can be used to selectively deliver drugs to target cells rather than nontarget cells [120]. Using pH-responsive materials such as PEG-PTMBPE, PEG-PH-PLLA, or PIA-PEG-FA-PHI maintains structural stability of oral delivery system to release the drug in a relatively short time when entering the intestine. This decreases release of drug in gastric secretion and prevents drug deposition on intestinal epithelium [121]. Kim et al. (2018) formulated RIPL peptide (IPLVVPLRRRRRRRRC)-conjugated NLCs for the selective drug delivery to hepsin (Hpn)-expressing cancer cells. pH-sensitive cleavable PEG (cPEG) was grafted onto RIPL-NLCs (cPEG-RIPL-NLCs) to attain steric stabilization. From the results, it was observed that PEG detachment from the cPEG-RIPL-NLCs was pH sensitive and time dependent. At 2-h incubation, cPEG-RIPL-NLCs and PEG-RIPL-NLCs exhibited comparable cellular uptake at pH 7.4, whereas cPEG-RIPL-NLC uptake was increased over twofold at pH 6.5 [122].
Mechanism of NLCs disposition
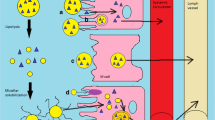
Many mechanisms were suggested for the disposition of NLCs. NLCs dispose of themselves inside the body by selective uptake through payer’s patches. As they pass through the GIT, NLCs undergo lipid digestion. Firstly, triglycerides present in NLCs are converted to monoglycerides and free fatty acids by duodenal pancreatic enzymes. The drug is released due to the breakdown of triglycerides and is then transported passively or actively by enterocytes or through the chylomicron-mediated pathway they enter into lacteals [123]. Further bile salts present in the intestinal chime interact with the drug and broken fragments of triglycerides. This interaction leads to the formation of micelles that cross the aqueous mucin layer and reach intestinal absorptive cells. Drugs, monoglycerides, and fatty acids come from these micelles. Further interaction between cholesterol, phospholipids, and lipid constituents results in the encapsulation of drugs in chylomicrons. Then this chylomicron-encapsulated drug undergoes exocytosis. Encapsulation of drugs with chylomicrons avoids first-pass metabolism by allowing the drug to enter through lacteals (Fig. 8) [10]. In the course of passage through the digestive system, NLCs can bypass the digestion process and enter paracellularly into portal blood, thereby circumventing enzymatic degradation. NLCs can also enter lymphatic circulation with the help of M cells [52]. Hu et al. formulated lipid nanoparticles to find out the absorption mechanism of these nanoparticles in vivo using fluorescent probes, but no evidence was found regarding the nanoparticles absorption through the intestinal membrane [123]. However, it was observed that surface modification of nanoparticles facilitates cellular uptake of nanoparticles and overcomes mucosal barriers [124, 125].

Mechanism of disposition of NLCs
Role of NLCs in oral delivery of drugs
Poorly soluble drugs
Poorly soluble drugs with low bioavailability can be given by the oral route effectively by formulating them as NLCs. The NLCs are considered potential oral drug carriers because of their high dispersivity which shows a high specific surface area for the intestinal lipase enzymatic attack. Other advantages offered by NLCs include enhanced drug absorption and bioavailability, improved drug entrapment, increased inclusion of drugs, high concentration of particles, and improved patient compliance [126]. NLCs possess an inherent GI solubilization property because of their transformation from crystalline form to amorphous form and ability to form mixed micelles in GI lumen (Fig. 9). The solubility of drugs also enhanced due to their property to combine with bile salts in GIT. In addition, NLCs use selective lymphatic transport for drugs showing extensive first-pass metabolism [127]. Therefore, NLCs have been potentially employed to enhance the solubility of many hydrophobic drugs. Bacalin, a poorly soluble drug when formulated as NLC, demonstrated increased oral bioavailability compared to its suspension because of enhanced solubility [102]. A poorly soluble drug repaglinide when formulated as NLCs has shown better antidiabetic property compared to its conventional dosage form because of its better solubility and oral absorption [128]. Fenofibrate-loaded NLCs showed a fourfold increase in area under curve in rats when administered orally as well as a good solubility profile [105].

Mechanism of solubility enhancement by NLCs
Restricted capacity of the process of biliary secretion emulsification
Gut enzymes degrade the lipids resulting in the formation of surface-active monoglycerides and diglycerides on the surface of NLCs. These molecules then separate and form micelles. During the separation and micelle formation process, the dissolved drug in the lipids is solubilized in the micelles. The formation of mixed micelles takes place when the previously formed micelles interact with surface-active bile salts like sodium cholate. Subsequently, along with the absorption of lipid degraded product, drug is absorbed together [129]. Simvastatin-loaded NLCs showed enhanced oral bioavailability due to the presence of solid and liquid lipids that are similar to fatty foods. This fatty food induced bile secretion and the formation of mixed micelles by the interaction of drug-loaded NLCs with bile salts which facilitates entry of NLCs into lymphatic vessels. This further prevents the first-pass metabolism of drugs and promotes drug absorption [22].
Efflux transporters such as P-glycoprotein
P-gp belongs to the ATP-binding cassette family, and its molecular weight is 170 kDa. It involves the transport of lipophilic, cationic, or neutral and high-molecular-weight compounds with no structural similarity [130]. P-gp is primarily found on the apical side of endothelial cells of the brain and epithelial cells of the liver, intestine, pancreas, and kidney [131, 132]. Lipids present in NLCs can modify the P-gp efflux process and thereby improve the pharmacokinetics of drugs to a large extent [133,134,135,136]. However, the mechanism involved in the inhibition of P-gp activity by these lipids is still unknown, but different theories propose that these lipids inhibit P-gp activity by altering the integrity of the cell membrane; interfering with the hydrolysis of ATP; blocking the binding sites allosterically, competitively, and non-competitively; and generating an ineffective cycle of ATP hydrolysis [137,138,139]. Saquinavir is a potential drug that shows P-gp efflux mechanism. When it is formulated as NLCs, the permeability of the drug is enhanced by 3.5-fold because of the prevention of P-gp efflux mechanism [140].
Extensive hepatic metabolism
NLCs can act as potential vehicles which are capable of safeguarding drugs from degradation because of hepatic metabolism during their transport across GIT. In the GIT, NLCs form mixed micelles by combining them with bile salts. Then they are selectively taken up in the lymphatic circulation, thereby bypassing the liver [22]. In addition, luminal solubilization of lipid-digested products is facilitated by mixed micelles and is also responsible for developing the concentration gradient required for absorption. This potential of NLCs to prevent drugs from hepatic metabolism further enhances the bioavailability of drugs and reduces dosing frequency [23, 141]. The drug vinpocetine undergoes extensive first-pass metabolism which is the main cause of its low oral absorption. NLCs loaded with vinpocetine promote the lymphatic uptake of drugs by preventing their hepatic degradation, thereby enhancing drug absorption [23].
Absorption of drugs through NLCs
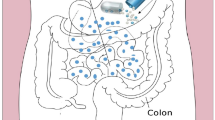
Different mechanisms have been proposed for improving the absorption of drugs. NLCs can adhere to the epithelium and thereby reduce fasted and fed state variability. Lipids enhance the absorption of drugs in a stepwise manner. Lipids are degraded by enzymes present in the gut resulting in the formation of mixed micelles together with bile salts. Within the micelle, the drug is entrapped and absorbed. The intestine takes up NLCs in a specific form and then transfers them to different organs of the lymphatic system in the body (Fig. 10) [23, 142]. The mechanism of this uptake can be uptake by M cells in the gut and intercellular or paracellular uptake [143]. In addition, this mechanism prevents hepatic metabolism by directing drugs to lymphatic absorption, thereby enhancing the bioavailability of drugs showing extensive hepatic metabolism [41]. Yu et al. developed sirolimus lipid-based nanostructured lipid carriers for improved oral bioavailability. It was observed that oral bioavailability of sirolimus NLCs in beagle dogs was 1.81-folds that of the commercial sirolimus tablets [144].

Mechanism of absorption of drugs through NLCs
Bioavailability enhancement
NLCs have great potential for improving the bioavailability of many drugs. Different mechanisms have been put forward to attain increased oral bioavailability of various drugs by NLCs. To a large extent, NLCs can modify P-gp-mediated efflux mechanism and thereby improve the pharmacokinetics of drugs [145]. After oral administration, the hydrophobic drugs are absorbed and diffuse across enterocytes of the intestine. There they combine with lipoproteins of enterocytes, leading to the secretion of the chylomicron-associated drug from the enterocyte to the lymphatic circulation rather than portal circulation which prevents first-pass metabolism by the liver [146]. Hence, the transport via the lymphatic route significantly improves oral bioavailability of drugs that are extensively metabolized by the liver [147, 148]. Kaithwas et al. developed NLCs of olmesartan with improved oral bioavailability. Compared to free drug, NLCs showed enhanced in vitro cellular when incubated with Caco-2 cells. AUCtotal and Cmax of OLM-NLC were observed to be significantly higher as compared to the free drug [149]. Nanocrystals can also improve solubility and bioavailability of water insoluble drugs by decreasing particle size of drug and thereby increasing the particle surface area and dissolution. But NLCs are more preferred over the nanocrystals as invisibility of nanocrystals avoid their identification by phagocytic system and eliminate them quickly [150]. Recent NLC formulation studies carried out to enhance oral bioavailability of lipophilic drugs have been shown in Table 4.
Toxicity aspects of NLCs
Normally, lipidic nanocarriers decrease risk of acute and chronic toxicity because they are well absorbed in the body and are composed of physiological compounds that lead to metabolic pathways [155, 156] and lower the toxicity profile of surfactants [157]. NLCs formulated with lipids and surfactants did not show cytotoxicity up to a 2.5% concentration of lipid [158]. However, toxicity of NLCs has not yet been reported. NLCs are formulated by using biodegradable and biocompatible lipids, therefore considered safe nanocarriers. In addition, they are well tolerated in in vivo and in vitro cytotoxicity studies. Compared to emulsions, NLCs contain low amount of surfactants which increases the safety profile of NLCs. Rahman et al. carried out oral toxicity studies of Zerumbone-loaded NLCs on mice [159]. No signs of toxicity in the liver, kidney, and lungs were observed after histopathological study [160].
Clinical trial approaches
Though NLCs show good ability to deliver drugs by oral route, still more preclinical and clinical studies are required. Hence, clinical trials need to be conducted with proper ethical guidelines. This might be because of insufficient research on the safety of NLCs as drug carriers. Lovastatin is used for the treatment of hypercholesterolemia. Lovastatin-loaded NLCs showed improved stability and clinical efficacy [161]. Therefore, NLCs should be further investigated for pharmacokinetic, pharmacodynamic, and toxicity studies. In addition, to ensure their safety, applications of NLCs should also be clinically investigated. Recent patents on NLCs formulations are shown in Table 5.
Conclusion
Oral drug delivery has come a long way from simple formulation to a novel nanotechnology-based formulation. Drug delivery by oral route offers many advantages like economic, self-administration, better patient compliance, and a painless route of administration. With a better understanding of gastrointestinal barriers, we can target many drugs by oral route in case of chronic diseases that require long treatment durations. Therefore, nanotechnology-based NLCs have been evolved to achieve oral drug delivery and to minimize drawbacks associated with SLNs. The main objective behind designing NLCs is to develop physically and chemically stable drug nanocarriers that will have potential marketability. NLCs have proven their potential to be given by oral route as they improve the bioavailability of many drugs and bypass first-pass metabolism. Compared to SLNs, NLCs show high drug payload capacity, less drug leakage, and more stability. Lipids used in the formulation of NLCs are biocompatible and biodegradable; therefore, they show a low toxicity profile. In addition, the drug efflux mechanism of NLCs helps in enhancing oral drug absorption by increasing intestinal transit time. Thus, NLCs can offer a promising future for oral delivery of lipophilic, poorly soluble drugs.
Current and future prospects
Many studies were carried out on NLCs since past few years. The improved knowledge about the mechanism for the transport of NLCs by oral route lead to increased development of the NLCs. NLCs are one of the better carriers for the drug delivery because of the presence of biocompatible and biodegradable lipids. In future, NLCs can be the efficient oral DDS for the poorly water-soluble drugs. Therefore, preclinical and clinical studies need to be conducted to target the oral drug delivery.
References
Hunter AC, Elsom J, Wibroe PP, Moghimi SM (2012) Polymeric particulate technologies for oral drug delivery and targeting: a pathophysiological perspective. Nanomedicine 1:S15–S20
Cheng Y, Xu Z, Ma M, Xu T (2008) Dendrimers as drug carriers: applications in different routes of drug administration. J Pharm Sci 97:123–143
Sao R, Vaish R, Sinha N (2015) Multifunctional drug delivery systems using inorganic nanomaterials: a review. J Nanosci Nanotechnol 15:1960–1972
Enayati-Fard R, Akbari J, Saeedi M, Morteza-Semnani K, Morad H, Nokhodchi A (2019) Preparation and characterization of atenolol microparticles developed by emulsification and solvent evaporation. Lat Am J Pharm 38:1342–1349
Subramanian S, Singireddy A, Krishnamoorthy K (2012) Nanosponges: a novel class of drug delivery system – review. J Pharm Pharm Sci 15:103–111
Fang CL, Al-Suwayeh SA, Fang JY (2013) Nanostructured lipid carriers (NLCs) for drug delivery and targeting. Recent Pat Nanotechnol 7:41–55
Mukherjee S, Ray S, Thakur RS (2009) Solid lipid nanoparticles: a modern formulation approach in drug delivery system. Indian J Pharm Sci 71:349–358
Silva AC, Santos D, Ferreira D, Lopes CM (2012) Lipid-based nanocarriers as an alternative for oral delivery of poorly water-soluble drugs: peroral and mucosal routes. Curr Med Chem 19:4495–4510
Souto EB, Doktorovova S (2009) Chapter 6 – Solid lipid nanoparticle formulations pharmacokinetic and biopharmaceutical aspects in drug delivery. Methods Enzymol 464:105–129
Das S, A. Chaudhury A (2011) Recent advances in lipid nanoparticle formulations with solid matrix for oral drug delivery. AAPS Pharm Sci Tech 12: 62–76
Müller RH, Radtke M, Wissing SA (2002) Nanostructured lipid matrices for improved microencapsulation of drugs. Int J Pharm 242:121–128
Muchow M, Maincent P, Muller RH (2008) Lipid nanoparticles with a solid matrix (SLN, NLC, LDC) for oral drug delivery. Drug Dev Ind Pharm 34:1394–1405
Shidhaye SS, Vaidya R, Sutar S, Patwardhan A, Kadam VJ (2008) Solid lipid nanoparticles and nanostructured lipid carriers-innovative generations of solid lipid carrier. Curr Drug Deliv 5:324–331
Pardeike J, Hommoss A, Muller RH (2009) Lipid nanoparticles (SLN, NLC) in cosmetic and pharmaceutical dermal products. Int J Pharm 366:170–184
Liu CH, Wu CT (2010) Optimization of nanostructured lipid carriers for lutein delivery. Colloid Surf A Physicochem Eng Asp 353:149–156
Muller R, Radtke M, Wissing S (2002) Solid lipid nanoparticles (SLN) and nanostructured lipid carriers (NLC) in cosmetic and dermatological preparations. Adv Drug Deliv Rev 54:S131–S155
Luo Q, Zhao J, Zhang X, Pan W (2011) Nanostructured lipid carrier (NLC) coated with chitosan oligosaccharides and its potential use in ocular drug delivery system. Int J Pharm 403:185–191
Davda J, Labhasetwar V (2002) Characterization of nanoparticle uptake by endothelial cells. Int J Pharm 233:51–59
Gupta AK, Gupta M, Yarwood SJ, Curtis AS (2004) Effect of cellular uptake of gelatin nanoparticles on adhesion, morphology and cytoskeleton organisation of human fibroblasts. J Control Release 95:197–207
Zhang XG, Miao J, Dai YQ, Du YZ, Yuan H, Hu FQ (2008) Reversal activity of nanostructured lipid carriers loading cytotoxic drug in multi-drug resistant cancer cells. Int J Pharm 361:239–244
Li Q, Cai T, Huang Y, Xia X, Cole S, Cai Y (2017) A review of the structure, preparation, and application of NLCs, PNPs, and PLNs. Nanomaterials 7:1–25
Tiwari R, K Pathak K (2011) Nanostructured lipid carrier versus solid lipid nanoparticles of simvastatin: comparative analysis of characteristics, pharmacokinetics and tissue uptake. Int J Pharm 415:232–243
Zhuang CY, Li N, Wang M, Zhang XN, Pan WS, Peng JJ (2010) Preparation and characterization of vinpocetine loaded nanostructured lipid carriers (NLC) for improved oral bioavailability. Int J Pharm 394:179–185
Araújo J, Gonzalez E, Egea MA, Garcia ML, Souto EB (2009) Nanomedicines for ocular NSAIDS: safety on drug delivery. Nanomedicine 5:394–401
Joshi MD, Müller RH (2009) Lipid nanoparticles for parenteral delivery of actives. Eur J Pharm Biopharm 71:161–172
Salvi VR, Pawar P (2019) Nanostructured lipid carriers (NLC) system: a novel drug targeting carrier. J Drug Deliv Sci Technol 51:255–267
Selvamuthukumar S, Velmurugan R (2012) Nanostructured lipid carriers: a potential drug carrier for cancer chemotherapy. Lipids Health Dis 159:1–8
Mitri K, Shegokar R, Gohla S, Anselmi C, Müller RH (2011) Lipid nanocarriers for dermal delivery of lutein: preparation, characterization, stability and performance. Int J Pharm 414:267–275
Doktorovová S, Araújo J, Garcia ML, Rakovský E, Souto EB (2010) Formulating fluticasone propionate in novel PEG-containing nanostructured lipid carriers (PEG-NLC). Colloids Surf B Biointerfaces 75:538–542
Radtke M, Müller RH (2001) Nanostructured lipid drug carriers. New Drugs 2:48–52
Tamjidi F, Shahedi M, Varshosaz J, Nasirpour A (2013) Nanostructured lipid carriers (NLC): a potential delivery system for bioactive food molecules. Innov Food Sci Emerg Technol 19:29–43
Gonzalez-Mira E, Egea MA, Garcia ML, Souto EB (2010) Design and ocular tolerance of flurbiprofen loaded ultrasound engineered NLC. Colloids Surf B Biointerfaces 81:412–421
Kelidari HR, Moazeni M, Babaei R, Saeedi M, Akbari J, Parkoohi PI (2017) Improved yeast delivery of fluconazole with a nanostructured lipid carrier system. Biomed Pharmacother 89:83–88
Kudarha R, Dhas NL, Pandey A, Belgamwar VS, Ige PP (2015) Box-Behnken study design for optimization of bicalutamide loaded nanostructured lipid carrier: stability assessment. Pharm Dev Technol 20:608–618
Garcia-Orue I, Gainza G, Girbau C, Alonso R, Aguirre JJ, Pedraz JL (2016) LL37 loaded nanostructured lipid carriers (NLC): a new strategy for the topical treatment of chronic wounds. Eur J Pharm Biopharm 108:310–316
Ferreira M, Chaves LL, Lima SA, Reis S (2015) Optimization of nanostructured lipid carriers loaded with methotrexate: a tool for inflammatory and cancer therapy. Int J Pharm 492:65–72
Monteiro LM, Lobenberg R, Cotrim PC, Barros de Araujo GL, Bou-Chacra N (2017) Buparvaquone nanostructured lipid carrier: development of an affordable delivery system for the treatment of leishmaniases. Biomed Res Int:1–11
Gramdorf S, Hermann S, Hentschel A, Schrader K (2008) Crystallized miniemulsions: influence of operating parameters during high-pressure homogenization on size and shape of particles. Colloids Surf A Physicochem Eng Asp 331:108–113
Attama A, Anthony A, Mumuni A, Momoh A, Philip F (2012) Lipid nanoparticulate drug delivery systems: a revolution in dosage form design and development. In: Sezer D (ed) Recent advances in novel drug carrier systems. IntechOpen, pp 107–140
Kaur S, Nautyal U, Singh R, Singh S, Devi A (2015) Nanostructure lipid carrier (NLC): the new generation of lipid nanoparticles. Asian Pac J Health Sci 2:76–93
Manjunath K, Venkateswarlu V (2005) Pharmacokinetics, tissue distribution and bioavailability of clozapine solid lipid nanoparticles after intravenous and intraduodenal administration. J Control Release 107:215–228
Sivaramakrishnan R, Nakamura C, Mehnert W, Korting HC, Kramer KD, Schäfer-Korting M (2004) Glucocorticoid entrapment into lipid carriers – characterisation by parelectric spectroscopy and influence on dermal uptake. J Control Release 97:493–502
Puri A, Loomis K, Smith B, Lee JH, Yavlovich A, Heldman E (2009) Lipid-based nanoparticles as pharmaceutical drug carriers: from concepts to clinic. Crit Rev Ther Drug Carrier Syst 26:523–580
Pyo Y-C, Tran P, Kim D-H, Park J-S (2020) Chitosan-coated nanostructured lipid carriers of fenofibrate with enhanced oral bioavailability and efficacy. Colloids Surf B Biointerfaces 196:1–9
Yunjing Z, Xue L, Cong L, Yihan K, Xing T, Yu Z (2020) Nanostructured lipid carriers as oral delivery systems for improving oral bioavailability of nintedanib by promoting intestinal absorption. Int J Pharm 586:1–11
Chahinez H, David A, Kamalinder KS (2020) Impact of liquid lipid on development and stability of trimyristin nanostructured lipid carriers for oral delivery of resveratrol. J Mol Liq 316:1–57
Rania KE, Sa D, Ebtessam AE, Gamal EM, Mona FA (2020) Chitosan coated nanostructured lipid carriers for enhanced in vivo efficacy of albendazole against Trichinella spiralis. Carbohydr Polym 232:115826
Archu S, Yub N, Bharti M, Kanchan K (2019) Nanostructured lipid carriers for oral bioavailability enhancement of exemestane: formulation design, in vitro, ex vivo, and in vivo studies. J Pharm Sci 108:3382–3395
Üner M (2015) Characterization and imaging of solid lipid nanoparticles and nanostructured lipid carriers. In: Aliofkhazraei M (ed) Handbook of nanoparticles. Springer, New York, pp 117–141
Mehnert W, Mäder K (2012) Solid lipid nanoparticles: productión, characterization and applications. Adv Drug Deliv Rev 64:83–101
Harshita BMA, Rizwanullah M, Beg S, Pottoo FH, Siddiqui S et al (2019) Paclitaxel-loaded nanolipidic carriers with improved oral bioavailability and anticancer activity against human liver carcinoma. AAPS PharmSciTech 20:87–104
Rizwanullah M, Amin S, Ahmed J (2017) Improved pharmacokinetics and antihyperlipidemic efficacy of rosuvastatin loaded nanostructured lipid carriers. J Drug Target 25:58–78
Gaba B, Fazil M, Khan S, Ali A, Baboota S, Ali J (2015) Nanostructured lipid carrier system for topical delivery of terbinafine hydrochloride. Bull Fac Pharm Cairo Univ 53:147–159
Lin WJ, Duh YS (2016) Nanostructured lipid carriers for transdermal delivery of acid labile lansoprazole. Eur J Pharm Biopharm 108:297–303
Shahgaldian P, DaSilva E, Coleman AW, Rather B, Zaworotko MJ (2003) Para-acyl-calix-arene based solid lipid nanoparticles (SLNs): a detailed study of preparation and stability parameters. Int J Pharm 253:23–38
Patel M, Souto EB, Singh KK (2013) Advances in brain drug targeting and delivery: limitations and challenges of solid lipid nanoparticles. Expert Opin Drug Deliv 10:889–905
Zhu Y, Liang X, Lu C, Kong Y, Tang X, Zhang Y et al (2020) Nanostructured lipid carriers as oral delivery systems for improving oral bioavailability of nintedanib by promoting intestinal absorption. Int J Pharm 586:119569
Xu R (2008) Progress in nanoparticles characterization: sizing and zeta potential measurement. Particuology 6:112–115
Harisa GI, Alomrani AH, Badran MM (2017) Simvastatin-loaded nanostructured lipid carriers attenuate the atherogenic risk of erythrocytes in hyperlipidemic rats. Eur J Pharm Sci 96:62–71
Qian Z, Haifeng Y, Benazir S, Xinyu L, Lin P, Xiuge G (2020) Nanostructured lipid carriers with exceptional gastrointestinal stability and inhibition of p-gp efflux for improved oral delivery of tilmicosin. Colloids Surf B Biointerfaces 187:110649
Xie S, Pan B, Wang M, Zhu L, Wang F, Dong Z (2010) Formulation, characterization and pharmacokinetics of praziquantel-loaded hydrogenated castor oil solid lipid nanoparticles. Nanomedicine 5:693–701
Anton N, Benoit JP, Saulnier P (2008) Design and production of nanoparticles formulated from nano-emulsion templates-a review. J Control Release 128:185–199
Zhang J, Fan Y, Smith E (2009) Experimental design for the optimization of lipid nanoparticles. J Pharm Sci 98:1813–1819
Hu FQ, Jiang SP, Du YZ, Yuan H, Ye YQ, Zeng S (2006) Preparation and characteristics of monostearin nanostructured lipid carriers. Int J Pharm 314:83–89
Han F, Li S, Yin R, Liu H, Xu L (2008) Effect of surfactants on the formation and characterization of a new type of colloidal drug delivery system: nanostructured lipid carriers. Colloids Surf A Physicochem Eng Asp 315:210–216
Müller RH, Mäder K, Gohla S (2000) Solid lipid nanoparticles (SLN) for controlled drug delivery - a review of the state of the art. Eur J Pharm Biopharm 50:161–177
Gönüllü U, Üner M, Yener G, Karaman EF, Aydoğmuş Z (2015) Formulation and characterization of solid lipid nanoparticles, nanostructured lipid carriers and nanoemulsion of lornoxicam for transdermal delivery. Acta Pharm 65:1–13
Shah R, Eldridge D, Palombo E, Harding I (2015) Lipid nanoparticles: production, characterization and stability. Springer, London
Castelli F, Puglia C, Sarpietro MG, Rizza L, Bonina F (2005) Characterization of indomethacin-loaded lipid nanoparticles by differential scanning calorimetry. Int J Pharm 304:231–238
Bunjes H, Steiniger F, Richter W (2007) Visualizing the structure of triglyceride nanoparticles in different crystal modifications. Langmuir 23:4005–4011
de Mendoza AEH, Rayo M, Mollinedo F, Blanco-Prieto MJ (2008) Lipid nanoparticles for alkyl lysophospholipid edel-fosine encapsulation: development and in vitro characterization. Eur J Pharm Biopharm 68:207–213
Crucho CIC, Barros MT (2017) Polymeric nanoparticles: a study on the preparation variables and characterization methods. Mater Sci Eng C 80:771–784
Joshi M, Patravale V (2008) Nanostructured lipid carrier based gel of celecoxib. Int J Pharm 346:124–132
Muller R, Mader K, Lippacher A, Jenning V (2000) Solid-liquid (semi-solid) liquid particles and method of producing highly concentrated lipid particle dispersions. German Patent Application, 199.
Fang G, Tang B, Chao Y, Xu H, Gou J, Zhang Y (2015) Cysteine functionalized nanostructured lipid carriers for oral delivery of docetaxel: a permeability and pharmacokinetic study. Mol Pharm 12:2384–2395
Elmowafy M, Shalaby K, Badran MM, Ali HM, Abdel-Bakky MS, Ibrahim HM (2018) Multifunctional carbamazepine loaded nanostructured lipid carrier (NLC) formulation. Int J Pharm 550:359–371
Shevalkar G, Vavia P (2019) Solidified nanostructured lipid carrier (S-NLC) for enhancing the oral bioavailability of ezetimibe. J Drug Deliv Sci Technol 53:101211
Fang YP, Lin YK, Su YH, Fang JY (2011) Tryptanthrin-loaded nanoparticles for delivery into cultured human breast cancer cells, Mcf7: the effects of solid lipid/liquid lipid ratios in the inner core. Chem Pharm Bull 59:266–271
Jenning V, Thunemann AF, Gohla SH (2000) Characterization of a novel solid lipid nanoparticle carrier system based on binary mixtures of liquid and solid lipids. Int J Pharm 199:167–177
Thapa C, Ahad A, Aqil M, Imam SS, Sultana Y (2018) Formulation and optimization of nanostructured lipid carriers to enhance oral bioavailability of telmisartan using Box Behnken design. J Drug Deliv Sci Technol 44:431–439
Schäfer-Korting M (2010) Drug delivery. In: Handbook of experimental pharmacology. Springer, Berlin
Shah MK, Khatri P, Vora N, Patel NK, Jain S, Lin S (2019) Lipid nanocarriers: preparation, characterization and absorption mechanism and applications to improve oral bioavailability of poorly water-soluble drugs. In: Grumezescu AM (ed) Biomedical applications of nanoparticles. William Andrew Publisher, Norwich, pp 117–147
Boyd BJ, Bergström CAS, Vinarov Z, Kuentz M, Brouwers J, Augustijns P (2019) Successful oral delivery of poorly water-soluble drugs both depends on the intraluminal behavior of drugs and of appropriate advanced drug delivery systems. Eur J Pharm Sci 137:1–27
Knipe JM, Chen F, Peppas NA (2015) Enzymatic biodegradation of hydrogels for protein delivery targeted to the small intestine. Biomacromolecules 16:962–972
Ensign LM, Cone R, Hanes J (2012) Oral drug delivery with polymeric nanoparticles: the gastrointestinal mucus barriers. Adv Drug Deliv Rev 64:557–570
Farokhzad OC, Langer R (2009) Impact of nanotechnology on drug delivery. ACS Nano 3:16–20
Bernkop-Schnurch A (2013) Nanocarrier systems for oral drug delivery: do we really need them? Eur J Pharm Sci 49:272–277
Park S, Garcia C, Shin G, Kim J (2017) Development of nanostructured lipid carriers for the encapsulation and controlled release of vitamin D3. Food Chem 225:213–219
Singh R, Lillard JW (2008) Past, present, and future technologies for oral delivery of herapeutic proteins. J Pharm Sci 97:2497–2423
Homayun B, Lin X, Choi HJ (2019) Challenges and recent progress in oral drug delivery systems for biopharmaceuticals. Pharmaceutics 11:1–29
Zhang R, McClements DJ (2018) Characterization of gastrointestinal fate of nanoemulsions. In: Jafari SM, McClements DJ (eds) Nanoemulsions. Elsevier Inc, USA, pp 577–612
Brown TD, Whitehead KA, Mitragotri S (2020) Materials for oral delivery of proteins and peptides. Nat Rev Mater 5:127–148
Moreno MR, Qi P, Wong YS, Xiong MG, Zhang Y, Venkatraman S (2019) Polymeric nanomaterials: methods of preparation and characterization. In: Mohapatra S, Ranjan S, Dasgupta N, Mishra R, Thomas S (eds) Nanocarriers for drug delivery. Elsevier, USA, pp 557–553
Kiptoo P, Calcagno AM, Siahaan TJ (2016) Physiological, biochemical, and chemical barriers to oral drug delivery. In: Wang B, Hu L (eds) Drug delivery. John Wiley & Sons, New York, pp 19–34
Poovi G, Damodharan N (2018) Lipid nanoparticles: a challenging approach for oral delivery of BCS class-II drugs. Fut. J Pharm Sci 4:191–205
Bummer PM (2004) Physical chemical considerations of lipid based oral drug delivery solid lipid NPs. Critical Rev Thera Drug Carrier Sys 21:1–19
Andrysek T (2003) Impact of physical properties of formulations on bioavailability of active substance: current and novel drugs with cyclosporine. Mol Immunol 39:1061–1065
Andrysek T (2006) Excipients and their role in absorption: influencing bioavailability of cyclosporine by triglycerides and polyglycerol esters. Biomed Pap Med Fac Univ Palacky Olomouc Czech Repub 150:227–233
Tan JS, Roberts CJ, Billa N (2018) Mucoadhesive chitosan-coated nanostructured lipid carriers for oral delivery of amphotericin B. Pharmaceut Dev Technol 24:504–512
Tian C, Asghar S, Wu Y, Amerigos DK, Chen Z, Zhang M (2017) N-acetyl-L-cysteine functionalized nanostructured lipid carriers for improving oral bioavailability of curcumin: preparation, in vitro and in vivo evaluations. Drug Deliv 24:1605–1616
Sun B, Luo C, Li L, Wang M, Du Y, Di D (2016) Core-matched encapsulation of an oleate prodrug into nanostructured lipid carriers with high drug loading capability to facilitate the oral delivery of docetaxel. Colloids Surf B Biointerfaces 143:47–55
Luan J, Zheng F, Yang X, Yu A, Zhai G (2015) Nanostructured lipid carriers for oral delivery of baicalin: in vitro and in vivo evaluation. Colloids Surf A Physicochem Eng Asp 466:154–159
Mandpe L, Pokharkar V (2015) Quality by design approach to understand the process of optimization of iloperidone nanostructured lipid carriers for oral bioavailability enhancement. Pharm Dev Technol 20:320–329
Abdelwahab SI, Sheikh BY, Taha ME, How C, Abdullah R, Yagoub U (2013) Thymoquinone loaded nanostructured lipid carriers: preparation, gastroprotection, in vitro toxicity, and pharmacokinetic properties after extravascular administration. Int J of Nanomedicine 8:2163–2172
Tran TH, Ramasamy T, Truong DH, Choi H, Yong C, Kim J (2014) Preparation and characterization of fenofibrate-loaded nanostructured lipid carriers for oral bioavailability enhancement. AAPS Pharm Sci Tech 15:1509–1515
Neupane YR, Srivastava M, Ahmad N, Kumar N (2014) Lipid based nanocarrier system for the potential oral delivery of decitabine: formulation design, characterization, ex vivo, and in vivo assessment. Int J Pharm 477:601–612
Shah NV, Seth AK, Balaraman R, Aundhia CJ, Maheshwari RA, Parmar GR (2016) Nanostructured lipid carriers for oral bioavailability enhancement of raloxifene: design and in vivo study. J Adv Res 7:423–434
Chu X, Bleasby K, Evers R (2013) Species differences in drug transporters and implications for translating preclinical findings to humans. Expert Opin Drug Metab Toxicol 9:237–252
Chanteux H, Staelens L, Mancel V, Gerin B, Boucaut D, Prakash C (2015) Cross-species differences in the preclinical pharmacokinetics of CT7758, an α4β1/α4β7 integrin antagonist. Drug Metab Dispos Biol Fate Chem 43:1381–1391
Ni PF, Ho NFH, Fox JF (1980) Theoretical model studies of intestinal drug absorption v: nonsteady-state fluid flow and absorption. Int J Pharm 5:33–47
Goodacre BC, Murray PJ (1981) A mathematical model of drug absorption. J Clin Hosp Pharm 6:117–133
Dressman JB, Fleisher D, Amidon GL (1984) Physicochemical model for dose-dependent drug absorption. J Pharm Sci 73:4–9
Dressman JB, Fleisher D (1986) Mixing-tank model for predicting dissolution rate control or oral absorption. J Pharm Sci 75:109–116
Han M, Fang QL, Zhan HW (2009) In vitro and in vivo evaluation of a novel capsule for colon-specific drug delivery. J Pharm Sci 98:2626–2635
Mura S, Nicolas J, Couvreur P (2013) Stimuli-responsive nanocarriers for drug delivery. Nat Mater 12:991–1003
Lu Y, Sun W, Gu Z (2014) Stimuli-responsive nanomaterials for therapeutic protein delivery. J Control Release 194:1–19
Apostolovic B, Danial M, Klok HA (2010) Coiled coils: attractive protein folding motifs for the fabrication of self-assembled, responsive and bioactive materials. Chem Soc Rev 39:3541–3575
Hoffman AS (2013) Stimuli-responsive polymers: biomedical applications and challenges for clinical translation. Adv Drug Deliv Rev 65:10–16
Li Y (2016) Non-covalent interactions in controlling pH-responsive behaviors of self-assembled nanosystems. Polym Chem 7:5949–5956
Zhang X, Zhao M, Cao N (2020) Construction of a tumor microenvironment pH-responsive cleavable PEGylated hyaluronic acid nano-drug delivery system for colorectal cancer treatment. Biomater Sci 8:1885–1896
Hossieni-Aghdam SJ, Behrouz FN, Zhila ZA, Solmaz MJ (2020) Facile fabrication and characterization of a novel oral pH-sensitive drug delivery system based on CMC hydrogel and HNT-AT nanohybrid. Int J Biol Macomol 207:2436–2449
Kim CH, Sa CK, Goh MS, Lee ES, Kang TH (2018) pH-sensitive PEGylation of RIPL peptide-conjugated nanostructured lipid carriers: design and in vitro evaluation. Int J Nanomed 13:6661–6675
Hu X, Fan W, Yu Z (2016) Evidence does not support absorption of intact solid lipid nanoparticles via oral delivery. Nanoscale 8:7024–7035
Zhou X, Zhang X, Ye Y (2015) Nanostructured lipid carriers used for oral delivery of oridonin: an effect of ligand modification on absorption. Int J Pharm 479:391–398
Beloqui A, Solinis MA, Des Rieux A (2014) Dextranprotamine coated nanostructured lipid carriers as mucuspenetrating nanoparticles for lipophilic drugs. Int J Pharm 468:105–111
Jaiswal P, Gidwani B, Vyas A (2016) Nanostructured lipid carriers and their current application in targeted drug delivery. Artif Cells Nanomed Biotechnol 44:27–40
Han HK, Lee BJ, Lee HK (2011) Enhanced dissolution and bioavailability of biochanin a via the preparation of solid dispersion: in vitro and in vivo evaluation. Int J Pharm 415:89–94
Date AA, Vador N, Jagtap A (2011) Lipid nanocarriers (GeluPearl) containing amphiphilic lipid gelucire 50/13 as a novel stabilizer: fabrication, characterization and evaluation for oral drug delivery. Nanotechnology 22:102–110
Khoo SM, Shackleford DM, Porter CJ, Edwards GA, Charman WN (2003) Intestinal lymphatic transport of halofantrine occurs after oral administration of a unit-dose lipid-based formulation to fasted dogs. Pharm Res 20:1460–1465
Linnets K, Ejsing TB (2008) A review on the impact of p-glycoprotein on the penetration of drugs into the brain. Focus on psychotropic drugs. Eur Neuropsychopharmacol 18:157–169
Lin JH, Yamazaki M (2003) Role of P-glycoprotein in pharmacokinetics: clinical implications. Clin Pharmacokinet 42:59–98
Fromm MF (2003) Importance of P-glycoprotein for drug disposition in humans. Eur J Clin Invest 33:6–9
Ahuja N, Katare OP, Singh B (2007) Studies on dissolution enhancement and mathematical modeling of drug release of a poorly water-soluble drug using water-soluble carriers. Eur J Pharm Biopharm 65:26–38
Batrakova EV, Li S, Li Y, Alakhov VY, Kabanov AV (2004) Effect of pluronic P85 on ATPase activity of drug efflux transporters. Pharm Res 21:2226–2233
Bardelmeijer HA, Ouwehand M, Beijnen JH, Schellens JH (2004) Efficacy of novel P-glycoprotein inhibitors to increase the oral uptake of paclitaxel in mice. Invest New Drugs 22:219–229
Chen MC, Wang JL, Tzen JT (2005) Elevating bioavailability of cyclosporine a via encapsulation in artificial oil bodies stabilized by caleosin. Biotechnol Prog 21:1297–1301
Rege BD, Kao JP, Polli JE (2002) Effects of nonionic surfactants on membrane transporters in caco-2 cell monolayers. Eur J Pharm Sci 16:237–246
Romsicki Y, Sharom FJ (1999) The membrane lipid environment modulates drug interactions with the P-glycoprotein multidrug transporter. Biochemistry 38:6887–6896
Woodcock DM, Linsenmeyer ME, Chojnowski G, Kriegler AB, Nink V, Webster LK (1992) Reversal of multidrug resistance by surfactants. Br J Cancer 66:62–68
Beloqui A, Solinís MÁ, Gascón AR, del Pozo-Rodríguez A, des Rieux A, Préat V (2013) Mechanism of transport of saquinavir loaded nanostructured lipid carriers across the intestinal barrier. J Control Release 166:115–123
Shete H, Chatterjee S, De A (2013) Long chain lipid based tamoxifen NLC. Part II: Pharmacokinetic, biodistribution and in vitro anticancer efficacy studies. Int J Pharm 454:584–592
Parhi R, Suresh P (2010) Production of solid lipid nanoparticles-drug loading and release mechanism. J Chem Pharm Res 2:211–227
Kreuter J (1991) Peroral administration of nanoparticles. Adv Drug Deliv Rev 7:71–86
Qin Y, Xiongwei H, Yuhua M, Yunchang X, Yi L, Jianping Q, Li X (2016) Lipids-based nanostructured lipid carriers (NLCs) for improved oral bioavailability of sirolimus. Drug Deliv:1–7
Batrakova EV, Li S, Miller DW, Kabanov AV (1999) Pluronic P85 increases permeability of a broad spectrum of drugs in polarized BBMEC and caco-2 cell monolayers. Pharm Res 16:1366–1372
Trevaskis NL, Charman WN, Porter CJ (2008) Lipid-based delivery systems and intestinal lymphatic drug transport: a mechanistic update. Adv Drug Deliv Rev 60:702–716
Cense HA, Eijck CH, Tilanus HW (2006) New insights in the lymphatic spread of oesophageal cancer and its implications for the extent of surgical resection. Best Pract Res Clin Gastroenterol 20:893–906
Arya M, Bott SR, Shergill IS, Ahmed HU, Williamson M, Patel HR (2006) The metastatic cascade in prostate cancer. Surg Oncol 15:117–128
Vikram K, Chander PD, Varun K, Sanyog J (2017) Nanostructured lipid carriers of olmesartan medoxomil with enhanced oral bioavailability. Colloids Surf B Biointerfaces 154:10–20
Bujnakova Z, Dutkova E, Balaz M, Turianicova E, Balaz P (2017) Mechanochemistry of chitosan-coated zinc sulfide (ZnS) nanocrystals for bio-imaging applications. Nanoscale Res Lett 12:328–337
Natarajan J, Prashant KH, Radhakrishnan A, Jubie S, Arigo A, Sathianarayanan S (2017) Enhanced oral bioavailability of an antipsychotic drug through nanostructured lipid carriers. Int J Biol Macromol 110:269–275
Thi M, Dong H, Young G, Minki J, Minwoo J, Ha-Eun K (2022) Enhancement of S (+)-zaltoprofen oral bioavailability using nanostructured lipid carrier system. Arch Pharm Res 1:1–11
Marzia C, Lavinia M, Francesca M, Natascia M, Paola M, Carla G (2018) Design, characterization and in vivo evaluation of nanostructured lipid carriers (NLC) as a new drug delivery system for hydrochlorothiazide oral administration in pediatric therapy. Drug Deliv 25:1910–1921
Moraes S, Marinho A, Lima S, Granja A, Araújo JP, Reis S (2020) Targeted nanostructured lipid carriers for doxorubicin oral delivery. Int J Pharm 592:120029
Wissing SA, Kayser O, Muller RH (2004) Solid lipid NPs for parenteral drug delivery. Adv Drug Deliv Rev 56:1257–1272
Rawat M, Singh D, Saraf S (2004) Nanocarriers: promising vehicle for bioactive drugs. Biol Pharm Bull 29:1790–1808
Mehnert W, Mader K (2001) Solid lipid NPs: production, characterization and applications. Adv Drug Deliv Rev 47:165–196
Muller RH, Souto EB, Goppert T (2005) Nanotechnologies for the life sciences biological and pharmaceutical nanomaterials. Wiley, United states
Rahman H, Sulaiman R, Abdullah O, Hemn H, Chartrand MS, Namvar F (2014) Acute toxicity study of zerumbone-loaded nanostructured lipid carrier on BALB/c mice model. Biomed Res Int 2014:563930
Zhou L, Chen Y, Zhang Z, He J, Meng D, Qingqing W (2012) Preparation of tripterine nanostructured lipid carriers and their absorption in rat intestine. Int J Pharm Sci 67:304–310
Chen CC, Tsai TH, Huang ZR, Fang JY (2010) Effects of lipophilic emulsifiers on the oral administration of lovastatin from nanostructured lipid carriers: physicochemical characterization and pharmacokinetics. Eur J Pharm Biopharm 74:474–482
Catarina EL, Seabra CD, Claudia DO, Maria DE (2019) Nanostructurated lipid carriers, methods and uses thereof. EP3474864A1
Fox CB, Khandhar AP, Van HN, Erasmus JG, Lin SS (2020) Nanostructured lipid carriers and stable emulsions and uses thereof. EP3638207.
Xiao Y, Yu A, Wu Y,Wang Z, Yu F, Jin X (2016) A kind of curcumin nano-lipid carrier that the N-acetylL-cysteine for oral administration is modified. CN106176677.
Tiange C, Yu C, Fengde T (2017) Psoralen-doxorubicin-loaded composite nanostructured lipid carrier preparation and preparation method thereof. CN104367549A.
Ying L, Nianping F, Zhiqiang C, Rong T, Shan W (2010) Nanostructured lipid carrier, preparation method and application thereof. CN101890170.
Acknowledgements
The authors thank the management and principal for their support of our work on NLCs.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
The authors declare that they have no conflict of interest.
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Springer Nature or its licensor (e.g. a society or other partner) holds exclusive rights to this article under a publishing agreement with the author(s) or other rightsholder(s); author self-archiving of the accepted manuscript version of this article is solely governed by the terms of such publishing agreement and applicable law.
About this article

Cite this article
Jiwankar, M., Sabale, V. Potential of nanostructured lipid carriers in oral delivery of the poorly soluble drugs. J Nanopart Res 25, 188 (2023). https://doi.org/10.1007/s11051-023-05840-0
Received:
Accepted:
Published:
DOI: https://doi.org/10.1007/s11051-023-05840-0