
Abstract
A glass-based surface-enhanced Raman scattering (SERS) substrate using 90 nm gold nanourchin (GNU) was fabricated as a localized surface plasmon resonance (LSPR) sensor. Self-assembly of GNU monolayer is achieved by silanizing the glass surface with amino-terminated (3-aminopropyl) triethoxysilane (APTES) and covalently grafting with thiol-terminated mercaptoundecanoic (MUA). Six substrates are used in vertical direction for immobilization of GNU at two concentrations of 5 pM (S1, S2, S3) and 15 pM (S4, S5, S6) for three sets of time 1, 3, and 5 h in each case, respectively, and one substrate (S7) is immersed horizontally at 15 pM for 5 h. The substrates are assessed using reflective surface-enhanced Fourier transform near-infrared (SEFT-NIR) and SERS. The results show that S3 (5 pM, 3 h) ≈ 96%, S4 (15 pM, 1 h) ≈ 97%, and S7 ≈ 97.8% have the highest GNU coverage, respectively. However, higher GNU coverage does not necessarily produce the maximum SERS signal as in our case S2 > S3 > S1 and S4 > S5 > S6. Principal component analysis (PCA) results can be interpreted as showing significant variance between the groups with less variation between the trials in the SEFT-NIR data, while the PC plots of the Raman data show that the spectra of all groups look very similar. Both techniques showed S3 and S4, and S7 as optimized substrates. Scanning electron microscopy (SEM) and atomic force microscopy (AFM) indicated a dispersed distribution of GNU with peak heights of ≤ 10 nm. This work is fundamentally important for designing and modeling LSPR-based nanosensors.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
In recent years, nanotechnology has advanced significantly where various nanostructures can be engineered as nanoplatforms and play a great role in various applications such as nanomedicine and biomedical engineering including diagnosis [1, 2], imaging [3,4,5], photothermal therapy [6,7,8], pharmaceutical, and drug delivery [9,10,11]. It is well known that the conduction band electrons of plasmonic nanoparticles (PNPs) such as gold nanoparticles (AuNP) undergo a coherent collective oscillation when excited by an external electric field, which exhibits strong optical absorption and scattering due to localized surface plasmon resonance (LSPR) [12, 13]. The main advantages of optical properties include strong LSPR, surface-enhanced scattering, nonlinear optical properties, tunable resonance in the Vis–NIR, and time-dependent properties because of adjustable nanoparticle size and shape [14,15,16], biocompatibility because of their inert surface, nontoxicity, relatively easy synthesis, functionalization, and surface conjugation chemistry, absence of photobleaching, and very low oxidation [17]. The colloidal PNPs can be integrated into advanced materials, which in turn offers flexibility and functionality as an aspect of sensor engineering [18]. Raman scattering (RS) is an inelastic scattering of photons as a result of the interaction between the incident light and a molecule. However, the weak intensity of the Raman signal can be enhanced (105–1014) times by the adsorption of sample molecules onto a metal surface via surface-enhanced Raman scattering (SERS) spectroscopy. The main mechanisms of SERS are due to (a) enhancement of the incident electromagnetic field due to LSPR where the induced dipole caused by plasmon oscillation enhances the local electric field at the surface of the PNP, and thus, strong light absorption and scattering occur at the SPR frequency [15]. For scattering to occur, the surface must be roughened to produce a perpendicular component to the plasma; thus, the plasmon energy causes the RS to occur in the analyte molecule, and (b) is the chemical enhancement where the chemical bonding of the target analyte to the PNP results in charge transfer between the adsorbed molecule and the metal surface. SERS is one of the most ultrasensitive and non-destructive spectroscopic analytical techniques that can provide fingerprint information for individual molecules and is widely used in scientific research [19], environmental monitoring [20], and medicine [21, 22]. Therefore, if monolayer-targeted AuNPs of uniform size distribution are efficiently immobilized on a solid surface as a SERS substrate, the biosensor will produce strong and reliable label-free signals corresponding to a chemical-binding event through changes in the LSPR spectrum. In addition, the field enhancement critically depends on the NPs size, shape, orientation, and aggregation [23].
Despite extensive research and well-established literature on SERS biosensors, there are generally three acknowledged limitations considered for the applicability of SERS technology: (a) preparation of an ideal substrate consisting of highly ordered hotspots, (b) reproducible detection of single molecules in a liquid medium, and (c) quantitative analysis in highly diluted liquids. SERS active substrates can be colloidal, i.e., solution-based and solid-supported substrates where various types, shapes, and sizes of nanostructures including spherical nanoparticles, nanorods, nanostars, nanoshells, and many others can be utilized [24]. The solution-based systems are common because of easy preparation using wet chemistry, the sample damage can be minimized due to the Brownian motion of the dispersed colloids. However, this can make them less reproducible [25, 26]. On the other hand, solid-supported SERS substrates offer number of advantages over the solution type as the nanostructures are confined to or embedded in a surface, which facilitates easy incorporation with microfluidics using an ultra low volumes, thus allowing simple sample preparation such as surface functionalization and washing [27].
Interest in immobilization of AuNPs onto solid substrates has grown in recent years across various fields of research in chemistry, materials, photonics, and biomedical engineering because it advances sensors based on LSPR, SERS, and electrochemistry. The immobilization state on substrate is a crucial issue for optimization of such applications performance. Controlled immobilization of AuNPs at various solid substrates such as planar electrodes [28], glass slides [18, 29], quartz, and silicon wafers [30] can be achieved by physical (or direct) and chemical methods. The former includes techniques such as ion reduction [31], thermal evaporation [32], and electrodeposition [33], and the latter can be either non-covalent self-assembly via ionic nature including hydrogen bonding, hydrophobic forces, van der Waals forces, or electrostatic interactions or covalent self-assembly via bond formation to generate functional nanostructure [34, 35]. Self-assembled 3D AuNP can improve the SERS activity by increasing the number of hot spots or nanogaps between nanoparticles, which can effectively amplify the electric field and enhance the Raman signal.
Traditionally, non-covalent self-assembly of AuNPs on the solid surface is carried out by using the positive charge of the amine group of (3-aminopropyl) triethoxysilane (APTES), which is an aminosilane frequently used for surface functionalization. APTES has three hydrolysable ethoxy groups that attach to the metal nanoparticles during the silanization process, leaving the amine (NH2) from the aminopropyl groups pointing away from the surface for further functionalization as shown in Fig. 1. In the self-assembly process, NH2 can be folded to form hydrogen bonds with free silanol groups, which leads to multilayer deposition of APTES molecules [36]. The formation of a high-quality silane monolayer is essential to prevent the aggregation of AuNPs on APTES-functionalized glass. However, they are easily assembled or disassembled due to thermal fluctuation, pH, light, concentration, solvents, or ions, thus limiting their lifetime for applications [36]. The chemical and mechanical stability can be enhanced via covalent bonding. In our work, we have used combined APTES functionalization of the glass surface and thiol group of mercaptoundecanoic (MUA) where S–H head groups are used on AuNP due to the strong affinity of sulfur in these noble metals. The sulfur-AuNP interaction is covalent with a strength of ≈45 kCal/mol [37]. The result is a 3D non-covalent self-assembly of APTES and covalent bonding of thiol-AuNP (Fig. 1). There are different APTES molecule orientations that can be formed on the surface of the glass, which can determine the surface contact angle properties hence the strength of bonding. The first type is when ethoxy groups, i.e., OCH2CH3, from APTES molecules bond with hydroxylated glass leading to lower availability of free NH2 terminals. In the second type, hydrogen bonds can be formed between NH2 groups and –OH from the hydroxylated glass surface, leading the –NH2 terminal group toward the glass, and the third type is when APTES molecules are completely adsorbed on the –OH terminated glass surface where Si bond would be covalently bonded the oxygen from SiO2 surface seen as in Fig. 1, with –NH2 oriented away from the surface available to bind with MUA.

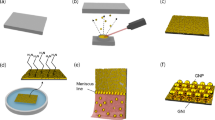
Fabrication of SERS substrate starting with a cleaned glass substrate, b amino-silanized with APTES, c covalently grafted with activated MUA, and d covalent binding of GNU to MUA via the strong thiol (SH) affinity to gold to make the final SERS substrate (e)
Among the various available plasmonic nanostructures, GNU as anisotropic 3-D nanocrystals have unique optical properties compared to smooth spherical gold nanoparticles of the same core diameter, where the spiky uneven surface causes a redshift in the SPR peak and a larger enhancement of the electromagnetic field at the GNU spike tips in smaller and more localized volume [38]. GNU are photostable, suitable for long-term tracking, multiplexing, and non-invasive imaging in living organisms. One advantage of using sharp branched nanostructures such as GNU in IR region is their SPR position in the near IR (680 nm for 100 nm diameter as in this case), thus the reduction of fluorescence background. In addition, the SERS enhancement factor increases when the excitation laser wavelength and the LSPR position, i.e., the peak wavelength, move to a longer regime. It is suggested that star or branched shape-based substrates exhibit the strongest Raman intensity, 7.4 times higher than gold spherical nanoparticles and 3.4 times higher than silver spherical nanoparticles [39, 40]. The suggested SERS substrates have a surface roughness defined by the diameter of the GNU and an average variable interparticle spacing. Therefore, it is expected that covalently immobilized GNU monolayers play a key role in enhanced SERS results [41]. Despite numerous amounts of research on SERS solid substrate, not much work is particularly reported about the effect of immersion direction and time of the glass substrate in colloidal solution on NPs distribution and SERS signals. It has been reported that MUA-functionalized APTES produced a better dispersion of gold nanoparticles than APTES functionalized surface alone [42]. To investigate this concept, we have covalently self-assembled a three-layer nanostructure of APTES, MUA, and GNU, fabricated on glass by immersing the flat substrates functionalized with a bifunctional cross-linker vertically and horizontally into GNU solution. The resultant morphology is modified by varying the immersion conditions. The aim of the work was to gain a better understanding and insight into the possible effect of the direction of substrate immersion within the solution for immobilization of GNU. However, knowing that the immobilization of nanoparticles is also a function of time and concentration, these factors were studied as well. The applications of SERS are numerous as mentioned above; however, by optimization of immobilization conditions it is expected to obtain a more sensitive SERS substrate for biological and chemical sensing, biological imaging, and targeted delivery where nanostructures with sufficient hotspots and high aspect ratio are requisite. Therefore, the increased surface area of the spiky core allows a greater degree of surface functionalization hence higher targeting species and therapeutic agents. Another example in the field of analytical chemistry where recently much attention has been focused on is the detection of a single molecule with high SERS intensity and enhancement factor. To achieve these goals, experiments were designed such that we could systematically investigate the optical properties of GNU immobilized on glass surface with the specific goal of optimizing the performance as a SERS substrate biosensor via the following steps: (a) covalently immobilize citrate-stabilized GNU on amino-silanized (APTES)-MUA glass slides in vertical and horizontal directions, (b) characterize the above substrates using reflective FT-NIR and SERS spectroscopy, and (c) study the effect of colloid concentration, immersion direction, and time of submersion of the glass on GNU distribution using SEM and AFM. The novelty of this work lies in the use of GNU for improved plasmonic sensitivity due to the branched tips, and optimization as well as statistical analysis of vertical and horizontal immersion methods.
Materials and methods
Glass microscope slides (Pearl, 7101) were used to make the substrates. DI water, nitric acid (HNO3, 69%), hydrochloric acid (HCl, ACS Reagent, 37%), sodium hydroxide (NaOH, BioXtra, anhydrous), ammonium hydroxide (NH4OH, 28%), hydrogen peroxide (H2O2, 30%), (3-aminopropyl) triethoxysilane (APTES; NH2 (CH2)3Si (OCH2CH3)3), N-(3-dimethylaminopropyl)-N′-ethyl carbodiimide hydrochloride (EDC), N-hydroxy succinimide (NHS), 11-mercaptoundecanoic acid (MUA), reagent alcohol, toluene, and 2-propanol were purchased from Sigma-Aldrich. Ethanol (70%) and ultrapure water were purchased from Thermo Fisher. Citric acid-stabilized gold nanourchins (GNU) of 90 nm diameter at an 8.92 pM concentration were purchased from Cytodiagnostics.
Functionalization of glass surface with APTES
The experimental details of glassware pre-treatment, substrate preparation, and cleaning are available in the Supporting Information. The glass forks were functionalized with APTES after cleaning. A 10% v/v APTES in toluene solution was prepared in a clean Coplin staining jar (BR472800, Sigma-Aldrich). In the biosafety cabinet, 0.6 mL of APTES was extracted and added to 5.4 mL of toluene in the staining jar. The jar was left on an orbital shaker (RK-51700–13, Cole-Parmer) for 1 min at 150 rpm. In the biosafety cabinet, the APTES solution was heated to 70 °C on a hotplate. After 10 min of heating, all 6 glass forks were immersed in the APTES solution (at least 5 mm deep). The staining jar was then capped, and the substrates were incubated for 3 h at 70 °C. After functionalization, the glass forks were rinsed in toluene, and a 50 mL Pyrex beaker was filled with toluene up to the 20 mL mark in which the substrates were immersed. It was then sonicated for 10 min at 80 kHz, 80% power, to remove excess APTES. The APTES-functionalized forks were then transferred to the slide rack and cured in an oven for 1.5 h at 90 °C. The slide rack of glass forks was then stored in a desiccator in the dark overnight until the next day for characterization.
Functionalization of glass + APTES with MUA
The glass forks were then further functionalized with MUA. A molar ratio of 1:1:1 of MUA:EDC:NHS was prepared in reagent alcohol (anhydrous ethanol). First, 0.49152 g of EDC, 0.29508 g of NHS, and 0.55988 g of MUA were weighed on separate weigh papers using an analytical balance (Sartorius, USA). The EDC, NHS, and MUA powders were transferred to a volumetric flask. The flask was then filled up to the 6 mL mark with anhydrous ethanol. The contents of the flask did not dissolve well with orbital shaking, so the MUA solution was sonicated at 37 kHz, 70% power, for 3 min to disperse. In the biosafety cabinet (Class II A2, Labgard, USA), the dispersed MUA solution was transferred to a clean Coplin staining jar (BR472800, Sigma-Aldrich), which was then heated to 25 °C on a hotplate stirrer (OF-04805–64, Cole-Parmer). The MUA was left to activate for 1.5 h while stirring with a magnetic stir bar. After the activation period, the 6 glass forks were immersed in the MUA solution (at least 5 mm deep) while stirring. The staining jar was then capped, and the substrates were incubated for 3 h at 25 °C. After functionalization, the glass forks were rinsed in anhydrous ethanol, then sonicated twice in anhydrous for 10 min at 80 kHz, 80% power, to remove excess MUA. The APTES-MUA-functionalized forks were then transferred to the slide rack (Uxcell) and dried in an oven with air for 1.5 h at 90 °C. The slide rack of glass forks was then stored in a desiccator in the dark overnight until the next day for characterization.
Immobilization of GNU on substrate
Vertical immersion
The first day of GNU immobilization was done at a low concentration (C1: 5 pM). The supplied GNU solution (8.92 pM) was left on the orbital shaker (RK-51700–13, Cole-Parmer) for 10 min at 150 rpm to disperse the GNU. To 8 microcentrifuge tubes (Biolynx), 1.05 mL of the supplied GNU was added. The tubes balanced each other in the microcentrifuge (OF-17710–11, Cole-Parmer) and were centrifuged at 2700 rpm for 35 min. For each tube, 1.025 mL of the supernatant was removed with a micropipette to not disturb the GNU in the remaining 25 µL. Then, in the biosafety cabinet (Class II A2, Labgard, USA), 1.025 mL of ultrapure water was added to each tube. The tubes were then shaken vigorously to disperse the GNU. From the microcentrifuge tubes, 2.8 mL of resuspended GNU was transferred to each of three clean Coplin staining jars (BR472800, Sigma-Aldrich). In the biosafety cabinet, 2.2 mL of ultrapure water was extracted and dispensed in each staining jar. The total volume of 5 mL of dispersed GNU in each staining jar was steadily shaken and dispersed. They were now each at a constant concentration of 5 pM, and the glass forks labeled as S1 [C1, t1 (1 h)], S2 [C1, t2 (3 h)], and S3 [C1, t3 (5 h)] were taken from the desiccator and were immersed alone in each one of the staining jars. The jars were then sonicated (Elmasonic P30H) at 80 kHz, 80% power, for 5 h. The staining jars were removed from the sonication bath at t1, t2, and t3 after sonication. Each time a jar was removed, the glass fork inside was rinsed under DI two times and left to air dry. The glass prongs were then snapped on the score line, yielding two separate substrates. These substrates were kept in clean sample containers and placed in the desiccator, in the dark. Each sample container that was used had been wiped and rinsed with 2-propanol, left to air dry, and lined with weigh paper beforehand. The second day of GNU immobilization was done the same as day one at a high concentration (C2: 15 pM) except in the biosafety cabinet 0.599 mL of ultrapure water was added to each tube. The tubes were then shaken vigorously by hand to disperse the GNU. From the microcentrifuge tubes, 4.992 mL of resuspended GNU was transferred to each of three clean Coplin staining jars. In the biosafety cabinet, 8 µL of ultrapure water was extracted and dispensed in each staining jar. The total volume of 5 mL of dispersed GNU in each staining jar was swirled around to disperse. They were now each at constant concentration, and the substrates were labeled as S4 [C2, t1 (1 h)], S5 [C2, t2 (3 h)], and S6 [C2, t3 (5 h)].
Horizontal immersion
The procedure was similar to the vertical direction with some minor changes. For each GNU tube, the supernatant was removed with a micropipette to leave 25 µL so that the GNU pellets remained undisturbed. Then, in the biosafety cabinet, ultrapure water was added to each GNU tube in a volume of 0.56 mL to tubes I and II and 0.565 mL to tube III. The GNU pellets were then redispersed in the tubes with a micropipette. From the microcentrifuge tubes, 1.685 mL of resuspended GNU was transferred to a clean Coplin staining jar. In the biosafety cabinet, 0.35 mL of ultrapure water was extracted and dispensed into the staining jar. The staining jar GNU solution was dispersed with the micropipette and was now at a GNU concentration of 15 pM (used for immobilization). The glass substrates were immersed in the staining jar, then the jar was sonicated at 80 kHz, 80% power for 5 h, and labeled as S7.
Characterization
The solutions at various stages were characterized using a UV–Vis spectrometer (Jenway 7205, Cole Parmer, Canada). Reflectance FTIR of the active area of each substrate was done using a 200 µm bifurcated reflectance probe (RP28, Thorlabs, USA) connected to the spectrometer (NanoQuest 2.5, Ocean Insight) with a spectral bandwidth of 4000–7400 cm−1 corresponding to 2.5–1.3 μm. The parameters were set to 5 s scan time and 8 nm resolution using the reflectance setting. The light source was an 8.8 mW output power tungsten halogen (HL-2000-HP, Ocean Insight) with a wavelength range between 360 and 2500 nm. A solid white foam surface was therefore used for background readings before characterizing the substrate active area. The SERS experiment was performed using a benchtop 637 nm laser, 10 mW tunable output power (S1FC637-ThorLabs-Canada) connected to 638 nm Raman probe (RIP-RPB-638-FC-APC-SMA-Ocean Insight-USA) with 9.5 mm diameter. It consists of a fiber bundle to direct excitation light from the laser to the sample and collect Raman scattered light. In the Ocean View software, the parameters were set to 5 s integration time, 3 scan average, and 5 boxcar width. The nonlinearity correction was applied, and the clean peaks option was selected. A dark background with the 637 nm laser shutter closed was taken. The spectral range is 300–3900 cm−1 with a working distance of 7.5 mm. Light at the laser wavelength (i.e., Rayleigh scattering) is rejected on the path to the spectrometer (HDX-Vis–NIR-Ocean Insight) with a spectral sensitivity between 150 and 3400 cm−1 (i.e., 66.67–2.94 μm) by a dichroic filter to avoid saturating the detector via the second arm of the cable.
Results and discussion
We fabricated covalently self-assembled SERS substrates based on GNU immobilized on functionalized glass via APTES-thiol bonding. The details of glassware pre-treatment, substrate preparation, and cleaning are available in the Supporting Information. Figure 1 represents schematically the fabricated SERS substrate where the cleaned glass substrate in Fig. 1a is amino-silanized by APTES (Fig. 1b) and then activated by MUA to create the covalent bonding due to strong affinity of S–H groups to the surface of GNU (Fig. 1c) and finally, it is immobilized by citrate-capped negatively charged GNU (Fig. 1c). The final SERS substrate is illustrated in Fig. 1e.
UV–Vis absorbance
The optical characterization of the GNU in solution is shown in Fig. 2a where the extinction spectra were measured between 300 and 800 nm with two distinct peaks at 337 and 646 nm corresponding to PBS and SPR of 90 nm GNU as supplied, respectively. The intensity of SPR at C1 is decreased by ≈44% and increased at C2 by ≈11% compared to the supplied sample. Based on the Lambert–Beer law, A = εLC where A is the absorbance, ε is the molar extinction coefficient, L is the optical path length, and \(C=\frac{{N}_{Total}}{{NVN}_{A}}\) is the concentration of the sample, \({N}_{Total}\) is the total number of gold atoms, N is the average number of gold atoms per GNU, V is the volume of the reaction solution in liter, and NA is the Avogadro’s number. Thus, it expected that by decreasing C, A also decreases, as seen in Fig. 2a. In addition, it has been suggested that spiky nanostructures have at least two co-linear branches implying they have higher aspect ratio than the others hence a tunable LSPR [43, 44]. Therefore, by changing the concentration of the solution, the branch length of the GNU could be tuned.

UV–Vis absorbance of a GNU as supplied, at C1 (5 pM), and C2 (15 pM), b APTES in toluene, and c the addition of MUA to the APTES solution. Note the hyperchromic shift and the increase in intensity
Figure 2b shows the results for APTES in toluene with peaks at 460, 495 510, 525, and 560 nm. When MUA was added to the APTES solution, the peaks remained in the same positions, but it showed a hyperchromic effect \(\left(\pi \to \pi *\right)\), i.e., the intensity was increased as seen in Fig. 2c.
Vertical immersion
Reflective FT-NIR and SEM
NIR spectroscopy is an analysis method that uses the NIR region of the electromagnetic spectrum (800–2500 nm). It measures the absorption of light from the sample within this range at different wavelengths and reflects or transmits the rest. The recorded NIR spectrum consists of overtones and combination vibrations of molecules that contain CH, NH, or OH groups. The commercial microscope slides are mainly made of borosilicate or soda lime, with chemical compositions of SiO2 (80%), B2O3 (13%), Na2O (4%), and Al2O3 (3%). FT-NIR of silicate glass provides useful information about structural network organization such as the connectivity of Si–O bonds. It is noteworthy that the different functionalized substrates, i.e., glass only, glass-APTES, and glass-MUA, reflect radiation differently. Figure 3a shows the dominant lines for glass such as 4000, 4500, 5338, 5475, and 6617 cm−1. The intensity of the lines is significantly reduced when the glass was functionalized by APTES in Fig. 3b indicating the change of dipole moment and the possibility of the transition of energy levels. Figure 3c shows when MUA is added to APTES where the profile and the intensity remains almost the same except a strong line at 4121 cm−1 is observed. It is noteworthy that the reflective index is closely related to the absorption and the amount of reflection is determined by the refractive index; thus, the reflection changes wherever there is an absorption band. Therefore, if the surface absorbs a wavelength of light, its relative intensity decreases.

Reflectance FT-NIR of a glass microscope slide alone, b glass silanized with APTES, and c glass silanized with APTES grafted with MUA
Figure 4 indicates the results of FT-NIR obtained for S1, S2, and S3 where Fig. 4a covers between 4000 and 4500 cm−1 with the highest number of peaks corresponding to S3, i.e., a greater number of vibrational freedoms of the molecules. Between 4500 and 5000 cm−1 in Fig. 4b, the intensity of the lines of S3 slightly reduced, which means the change of dipole moment and the possibility of the transition of energy levels is reduced but the number of lines of S2 is increased with two sharp peaks corresponding to S1. In the range of 5000–6000 cm−1 (Fig. 4c), S1 shows sharp peaks at 5313, 5450, 5822, and 5959 cm−1, S2 three lines at 5214, 5537, and 5723 cm−1 and S3 only one line at 5350 cm−1, respectively. However, between 6000 and 7000 shown in Fig. 4d, only S1 with two lines at 6245 and 6493 cm−1 and S2 with one line at 6369 cm−1 showed response, respectively. It appears that S3, with longer immersion time in the GNU responds stronger at longer wavelengths between 2.5 and 2 μm and very weakly at shorter wavelengths.

a–d Reflectance SEFT-NIR of S1, S2, and S3 at GNU concentration of 5 pM and immersion times of 1, 3, and 5 h, respectively. SEM images of S1, S2, and S3 at e 10 k and f 120 k magnification
Figure 4e indicates the SEM images of GNU deposited on S1, S2, and S3, respectively, at a low magnification of 10 k where the surface area covered by conjugated GNU increases by immersion time. Figure 4f shows part of the covered area at a higher magnification of 120 k where the surface density of immobilized GNU on glass increases linearly with solution immersion time at a constant concentration of 5 pM. It is suggested that APTES/MUA functionalization allows for a superior gold nanoparticle dispersion with a large fraction of isolated particles whereas, with an APTES functionalized surface, a higher number of large aggregates are formed on the surface [42]. The immobilization of GNU at the surface can also be related to the number of accessible free-NH2 terminal groups, i.e., lower coverage represents lower free-NH2 available and as in S1 and as the immersion time increases, more GNU is attached to the groups via electrostatic interaction as in S3. For all specimens, it was noticed that GNU was immobilized near the meniscus during the immersion in the GNU solution. This may be caused by the change in surface tension across the vertically immersed substrate, combined with the heating of the solution during prolonged sonication. The long sonication times heat up the solution over time, causing some evaporation of the solution, evidenced by condensed drops of GNU solution found on the caps of the staining jars during GNU immersion. To achieve a more even and uniform distribution, especially in the immersion conditions where the formation of multilayered GNU at the meniscus site was observed, one of the substrates was immersed horizontally at a lower solution volume [S7 (15 pM, 5 h)] to compare the effect and possible differences.
Similarly, when the experiment was repeated for the second set of the samples at a higher concentration of 15 pM, S4 exhibits the highest number of reflections particularly a very strong peak at 4083 cm−1 as seen in Fig. 5a. Figure 5b shows the reflections between 4500 and 5000 cm−1 with S4 having the highest number of distributions and highest intensity, S5 hardly shows any significant line except one at 4965 cm−1, and S6 at 4170, 4319, and 4829 cm−1, respectively. However, between 5000 and 6000 where most of the lines occur in Fig. 5c, S4 shows a few lines at 4928, 5884, 6307, and 6878 cm−1, S5 with lines of least intensity at 5164, 5363, and 5350 cm−1, and S6 with a higher number of lines than S5 at 4568, 4630, 4680, 4829, and 4928 cm−1, respectively. Figure 5d indicates the response between 6000 and 7000 cm−1 where S4 distinctly exhibits a higher number of lines including 6307, 6456, 6667, 6692, 6741, and 6878 cm−1, and more intense than S4 and S5. Figure 5e shows the SEM images for S4, S5, and S6 at a lower magnification of 10 k, respectively, and the corresponding SEMs at a higher magnification of 120 k is shown in Fig. 5f.

a–d Reflectance SEFT-NIR of S4, S5, and S6 at GNU concentration of 15 pM and immersion times of 1, 3, and 5 h, respectively. SEM images of S4, S5, and S6 at e 10 k and f 120 k magnification
As the immersion time increases, the degree of aggregation and clustering seems to increase. In contrast to the single GNU layer densely packed in a two-dimensional plane, the clusters not only exist at the top layer but also at the crossing areas due to the spatially overlapping of the dual-layer clusters. The substrates (S1–S6) respond differently due to variations in the free-end linker of the GNU-conjugated structure. Table 1 summarizes the lines observed in the groups and substrates, and the lines of groups that are repeated in each substrate are determined as common lines. The ratio of intensity is then calculated by dividing the intensity of a given common line in the substrate by the corresponding intensity in that group. Therefore, the percentage of the substrate covered by GNU is determined by subtracting the total average in each group from 100 as shown in Table 2.
Figure 6 shows the SEM images of (a) S5 and (b) S6 from a frontal view. It was noticed that more GNU are populated near the edge in S6 forming clusters than in S5.

SEM images of a S5 and b S6 from a frontal view at 15 k and 120 k, respectively
RS of glass + APTES + MUA
Figure 7a represents the RS results for glass, glass + APTES, and glass + APTES + MUA between 500 and 3500 cm−1 excited at 4 mW. The pristine ternary Na borosilicate glass with O–Si–O bending and stretching modes exhibits the main lines given in Table 3, and the typical hydroxyl (OH) and H2O vibrational modes of water and surface silanol groups are seen in Fig. 7b. The symmetric stretching mode \(\left({v}_{s}\right)\) of B–O of planar metaborate ring structure is detected near 621 cm−1. The lines between ≈780–840 cm−1 represent the silica bending modes \({\updelta }_{\mathrm{bbens}}\left(\mathrm{Si}-\mathrm{O}-\mathrm{Si}\right)\), and the symmetric stretching modes of the above-mentioned amorphous structure of silica tetrahedron are between 900 and 1250 cm−1. Bond vibrations of BO3 units in boroxol rings and the stretching bands of molecular oxygen (1558 cm−1) were also detected.

a, b Raman spectra of the three functionalization stages from 500 to 3500 cm−1 at 4 mW. c, d shows the effect of power at 2 and 4 mW on glass + APTES, and similarly, e, f corresponds to glass + APTES + MUA. Note that the intensity of each profile varies with wavenumber, and that it is smaller at lower power between 500 and 2000 cm−1 and higher at longer range (2000–3500 cm−.1)
When the glass is functionalized with APTES (Fig. 7a), the intensity of the spectrum profile is significantly increased, and some new lines are observed as highlighted in Table 3, indicating the detection of new functional groups at the surface. However, following the functionalization with MUA, the intensity of the overall profile decreases and red-shifted (†) with respect to APTES shown in Table 3. It is noticed that some of the lines for glass have no corresponding lines when APTES and MUA are added, which may indicate the absence of functionalization binding sites and that APTES and MUA are Raman inactive at that frequency range. For a vibrational transition to be Raman active, the molecule must undergo a change in polarizability, i.e., distorting electrons from their original position, during the vibration. Figure 7c shows the effect of laser power on the RS spectra for glass + APTES between 500 and 3500 cm−1 where after irradiating at 2 mW (a) the intensity of the lines between 500 and 1000 cm−1 decreases by ≈75%, indicating the lower transition probability hence less population of excited states; (b) the maximum intensity at 1369 cm−1 decreases by 87.5%; (c) the width of the profile becomes more spread out, i.e., more spectral uncertainties are involved; and (d) most of the lines are blue-shifted (*) as shown in Table 3. The expanded range between 2500 and 3500 cm−1 is shown in Fig. 7d where situation reverses and the lines at 2 mW showed higher intensity compared to 4 mW.
Similar results are observed when MUA is grafted to APTES as seen in Fig. 7e and f. The main points are observed at 2 mW (a) the intensity of the first peak at 621 cm−1 increases by ≈25%; (b) the width of the profile between 500 and 1000 cm−1 remains constant, i.e., the no changes occurs in the energy levels of atomic and molecular system; (c) the maximum intensity at 1541 cm−1 decreases by 75% corresponding to 1558 cm−1 indicating lower transition probability hence less population of excited states; and (d) the width of the profile between 1000 and 2000 cm−1 at 4 mW is reduced to 1200–2000 cm−1.
According to Rayleigh theory, when a beam of oscillating unpolarized light Ei interacts with particles such as atoms or molecules (e.g., APTES and MUA), it causes the negatively charged electrons to oscillate at the same frequency as the incident light (637 nm in our case). The field \({\overline{E} }_{{\mu }_{d}}\) associated with the oscillating electric dipole \({\mu }_{d}\) of the molecule interacts with the incident light and is scattered elastically with an isotropic intensity distribution since the oscillatory wavelets emitted by the oscillating charges are approximately in phase with one another [45]:
where \({R}_{mo}\) is the molecule radius, \({\varepsilon }_{mo}\) and \({\varepsilon }_{en}\) are the molecule and the medium dielectric constants, respectively, and \(\omega\) is the field angular frequency. The scattered light field \({E}_{s}\) will be proportional to \(\left(1/{c}^{2}\right)\left({d}^{2}{\mu }_{d}/{dt}^{2}\right)\) where c is the speed of light, and the second derivative of \({\mu }_{d}\) is the acceleration of the charge on the dipole moment. The intensity of the scattered light \({I}_{s}\) is directly proportional to the polarizability \({\alpha }_{p}\) of the molecule, which in turn depends on the molecular weight and on the particle size. Therefore, the higher the particle \({\alpha }_{p}\), the higher will be the magnitude of \({\mu }_{d}\) induced. In addition, the oscillating dipole reradiates light energy \(\left(Er\right)\) at the same frequency as the incident light. Since \({\overline{E} }_{{\mu }_{d}}\) induced in the molecule is proportional to its volume \({V}_{mo}\), the \({I}_{s}\) is proportional to \({V}_{mo}^{2}\) and the intensity of reradiated light \({I}_{r}={\left|{\overline{E} }_{{\mu }_{d}}\right|}^{2}\), thus proportional to \({V}_{mo}^{2}\). Therefore, the scattered intensity will be stronger at higher power. In Raman Stokes scattering, the emitted dipole radiation is red-shifted \(\left({\omega }_{0}-{\omega }_{R}\right)\), and in anti-Stokes, blue-shifted \(\left({\omega }_{0}+{\omega }_{R}\right)\) where \({\omega }_{0}\) and \({\omega }_{R}\) are the angular frequencies of the incident laser field and the corresponding normal mode, respectively. In the case of APTES, 537 cm−1 (1.8 μm) at 2 mW changes to 647 cm−1 (1.54 μm) at 4 mW, respectively, and in the case of MUA at 2 mW, 602 cm−1 (1.66 μm) and 685 cm−1 (1.46 μm) changes to 621 cm−1 (1.61 μm) and 780 cm−1 (1.28 μm) at 4 mW, respectively.
SERS of glass + APTES + MUA + GNU
At this stage, the functionalized substrates are immobilized by GNU where it is expected when the molecules adjacent to the metallic surface exhibit significantly enhanced Raman signals. Figure 8a shows the SERS spectra for S1, S2, and S3 between 500 and 3500 cm−1, respectively, where S2 distinctly shows a stronger response than S3 and S1. This can be caused by the clustering of GNU, which causes Mie scattering where the scattered light is shifted to a longer wavelength with corresponding lower energy. Considering that the intensity is inversely proportional to the interacting volume, thus producing decreased signal intensity. The overall response profile of S3 is red-shifted with respect to S2 due to an increase in the size of the GNU cluster. Figure 8b shows that the response of S3 significantly exceeded S1 between 2000 and 2500 cm−1 with the major peaks at 2062, 2228, 2290, 2333, and 2380, and S2 showed no response at all unlike in Fig. 8a. Similarly, in Fig. 8c, S3 showed the highest response between 2900 and 3500 cm−1 with the strongest line at 2989 cm−1 but S1 had more lines distributed between 2500 and 3500 cm−1 than S2 and S3 with S2 the least signal response.

a SERS spectra of vertically immersed S1, S2, and S3 from 500 to 3500 cm−1 at 4 mW, b expanded spectra between 2000 and 2500 cm−1, c between 2500 and 3500 cm−1, and d the schematic diagram of the LSPR detection based on SERS imaging. Note that the response of the substrates varies within different wavenumber ranges as S2 > S3 > S1 between 500 and 2000 cm−1 and S3 > S1 > S2 between 2000 and 3500 cm−.1
Therefore, the SERS signals of the substrates vary with wavenumber, i.e., the wavelength of the emitted radiation from the substrate, which represents the molecular rotational and vibrational energy of specific chemical bonds and chemical composition. In our case, S2, S3, and S1 indicated the strongest response between 20 and 5, 5 and 4.2, and 4.2 and 3.4 µm, respectively, and between 3.4 and 2.8 µm the response was in the order of S3 > S2 > S1. It is well known that the plasmon coupling between nanoparticles at high surface density leads to stronger hotspots [38, 46], and a red-shift of the spectrum defined as the ratio of center-to-center distance D to radius of the GNU r, i.e., \(D/r\). Therefore, chemisorbed GNUs on glass with lower \(D/r\) ratio should exhibit stronger resonance coupling, and thus enhancement of SERS [18]. As observed in the SEMs in Figs. 4 and 5, the randomly distributed air gaps between the individual GNU or even clusters can lead to the excitation of gap plasmon modes [47]. Larger GNU exhibited an LSPR peak at longer wavelengths because the tips support more extended longitudinal plasmon resonance [38].
Figure 9a indicates the whole SERS spectra between 500 and 3500 cm−1 for S4, S5, and S6, respectively. Within 500–2000 cm−1 in Fig. 9b, the SERS responses were in the order of S4 > S5 > S6. The response at 608 cm−1 was considerably diminished for S5 and S6 equally. Interestingly, the peaks 1403, 1558, 1740, and 1937 cm−1 were observed unchanged in all three substrates of S4, S5, and S6 with the intensity ratios of S4 ≈ 2 (S5) ≈ 2 (S6). Throughout the spectrum between 2000 and 2500 cm−1 in Fig. 9c, the substrates exhibited a similar profile. Between 2000 and 2100 cm−1, only S4 and S6 showed peaks at 2072 cm−1 with S4 > > S6. In the range of 2200–2300 cm−1, the response of S4 was greater than both S5 and S6 which were equal. The intensity of all lines increased significantly between 2300 and 2350 cm−1, in the order of S5 > S4 ≈ S6, and beyond that, i.e., between 2350 and 2500 cm−1, S6 > S4≈ S5. In Fig. 9d, most of the molecule vibrational lines are overlapped and the intensities are relatively enhanced between 2500 and 3500 cm−1 (i.e., 4–2.8 µm), particularly with the dominant line at 2989 cm−1 (i.e., 3.3 µm). It is noteworthy that a non-linear trend was observed for the first set, i.e., S2 > S3 > S1 and a linear behavior in the second set, i.e., S4 > S5 > S6, respectively, i.e., increasing the immersion time for GNU immobilization degrades the quality of the SERS signal despite the higher GNU deposition. Thus, the optimum results are produced by S2 (5 pM, 3 h) and S4 (15 pM, 1 h) between 500 and 2000 cm−1, and at the shorter range by S3 (5 pm, 5 h) and S6 (15 pm, 5 h), respectively. The overall results are summarized in Table 4.

SERS spectra of vertically immersed S4, S5, and S6 from 500 to 3500 cm−.1 at 4 mW. The response from each substrate varies in different wavelength regions, as noted S4 > S5 > S6 (a–c), and S6 dominates at longer wavelengths (d)
The effect of linearly increasing signal deterioration by increasing the immersion time in the second set can be explained first by considering that the SERS intensity of a molecule may be described as [48]:
where P is the laser power, N is the number of Stokes-active scatterers or number of molecules within the laser focal field, \({\sigma }_{SERS}\) is the scattering cross-section, and \({E}_{0}\) and \({E}_{loc}\) are the amplitudes of the incident and local, i.e., enhanced electric field, respectively, and \({I}_{0}={\left|{E}_{0}\right|}^{2}\) is the incident intensity. Equation (2) indicates that the SERS signal is proportional to the fourth power of the field enhancement factor \(\frac{\left|{E}_{loc}\right|}{\left|{E}_{0}\right|}\) due to LSPR in the GNU, which is highly effective in recording SERS spectra. Therefore, the field enhancement critically depends on the NPs size, shape, orientation, and aggregation. The strong relationship between the size of the metal nanoparticles and their SERS activity is due to the variation of the LSPR with NP size. Aggregation of the colloid produces a range of cluster sizes, each with a different resonance maximum, and is important factor in signal enhancement due to the presence of hot spots [49]. The effect of clustering is that the SERS intensity is at a maximum away from molecular resonance maximum. This is due to the greater enhancement obtained from aggregates even if fewer particles are involved. Therefore, a stronger enhancement effect will be produced when single NPs form aggregates of two multiple NPs due to coupling of the electromagnetic field. According to the surface selection rule, vibrational modes whose polarization tensor elements are perpendicular to the metal surface should be strongly enhanced in a SERS spectrum [50].
Horizontal immersion (S7)
Reflective SEFT-NIR
Compared to the vertical case, several new lines with high intensity appear with APTES on glass as seen in Fig. 10a including 4220, 4282, 4532, 4630, 4804, and 4866 cm−1. However, the intensity at 4518 cm−1 is reduced, and 4779 cm−1 in the vertical case was not observed in the horizontal. Interestingly, the intensity of the entire spectrum profile of Fig. 10a is increased compared to the vertical profile. In addition, the lines 5350, 6083, 6269, and 6841 in the vertical were red-shifted to corresponding values of 5400, 6332, 6269, and 6841, respectively. In Fig. 10b, when MUA is added, the lines between 4000 and 5500 cm−1 are blue-shifted but between 5500 and 7000 cm−1 are red-shifted. However, the intensity of the lines between 4000 and 5500 cm−1 is generally increased unlike in Fig. 10a but between 5500 and 7000 cm−1 is decreased. When GNU is conjugated in Fig. 10c, the intensity of the lines between 4000 and 4500 cm−1 decreases, and a very strong and dominant line is observed at 4891 cm−1. No response was observed between 5000 and 6500 cm−1. Two new lines were observed at 6692 and 6754 cm−1. Generally, the intensity of the lines is decreased compared to vertical, which can be due to the previously proposed argument that the higher the number of deposited GNU, the lower the glass space available. This is consistent with the observation that in the horizontal case, more GNU is immobilized on the surface under the same condition of 5 h and 15 pM as in the case of S6.

Reflective SEFT-NIR spectra of horizontally immersed S7 showing a general increase in reflectivity compared to the vertical cases due to more GNU binding at the surface. a Glass + APTES, b glass + APTES + MUA, and c glass + APTES + MUA + GNU
SERS
In the horizontal case, the SERS signals are less prevalent compared to the vertical, with only two main peaks at 641 cm−1 and 3187 cm−1. The first intense peak as seen in Fig. 11a appears in the same glass region as Figs. 8a and 9a, showing consistency with S1–S6. Similarly, Fig. 11b shows little activity in the 3000–3500 cm−1 region which is also consistent. The signal is overall dampened from S7 likely due to the varying dispersion pattern of the GNU compared to the vertical case.

a SERS spectra of horizontally immersed S7, and b is the magnified second peak. It is noted that no signal was observed between 1000 and 3100 cm−1 compared to the vertical case
SEM
Figure 12a–c shows the SEM images of S7 from the top view near the edge of the substrate. It was observed that there is a more even monolayer compared to the vertical immersion, and a multilayer of GNU was found near the edges of the substrate. It is around the edges of the substrate where the concave bottom of the staining jar was closer throughout immobilization. Surface tension is made between the small distance from the jar to the substrate, and the smaller the distance, the more GNU solution is pulled toward the glass interfaces. There is also more excess ligand at the edges. This was similar to the capillary action that occurred when the glass forks were previously immersed vertically in the jar. Note the multilayer formation on the surface, i.e., it is thicker near the edge, and as we go toward the center it gets thinner. An ideal sensor would be the one with the least void space between the GNUs, and horizontal immersion is found to be better than the vertical in that respect, i.e., less void space. Some clusters seen as white spots are formed and distributed randomly at the surface, for example, in Fig. 12b.

SEM images of S7 from a top view a–c near the edge of the substrate showing less void space compared to the horizontal case, and d–g from the center of the substrate observed at different magnifications. The spiky features of GNU are more delineated in g
The SEMs in Fig. 12d–g are the top view of the immobilized surface at the center of the substrate observed at various magnifications. While a large area was covered by the immobilized GNU network, some small, isolated colonies across the center of the substrate and void spaces in between were observed. To have a greater distribution of colonies, i.e., smaller void space, a larger volume of GNU solution must be used.
Figure 13 shows the SEM images from the front view where a monolayer of GNU, Fig. 13a is formed near the edge due to densely distributed sharp GNU corners, tips, and nanogaps between the adjacent parts. In contrast to the single GNU layer densely packed in a two-dimensional plane, the clusters not only exist at the top layer but also at the crossing areas due to the spatially overlapping of the dual-layer clusters. Figure 13b indicates a stack of agglomerate consisting of four GNU, it is noted that some of the immobilized tips of GNU contacting the functionalized glass are blunted in the image. This may be due to the glass surface heating up during sonication, thereby heating bound GNU and causing them to morph to the surface. Figure 13c demonstrates the immobilization of some GNU on the side surface of the glass. However, since the sides of the glass do not play a role in sensing, it can be considered a waste of materials and an economical disadvantage. Therefore, the thinner the glass, the less the number of nanoparticles on the side surface.

SEM images of S7 from a frontal view illustrating a monolayer formation, b formation of GNU clusters, which could affect the SERS signal, and c immobilization on the sides of the glass, which do not contribute to the signal detection in the sensing process
AFM
Imaging of GNU-immobilized substrate by AFM indicates monolayers of GNU arranged randomly on the glass surface where Fig. 14a and b represent the 2D and 3D images of the surface morphology. Figure 14c and d show the corresponding images at a higher magnification where the separation between GNUs is in the order of or less than the diameter with an approximate roughness of ≈10 nm. Since in our case, the diameter of GNU was constant (neglecting the clustering) thus, the ratio of center-to-center distance (D) between GNUs to the radius (r) of the GNU, i.e., D/r, decreases by decreasing D (or increasing the particle size). Therefore, it is expected that the greater plasmon resonance coupling, and consequently a larger and positive shift of LSPR wavelength occurs when the interparticle distance decreases. It is suggested that the coupling hence the resonance shift becomes negligible at a larger separation distance of ≈2.5 times the nanoparticle size [51]. Using the experimental values, the fractional coverage was determined as ≈0.27 by calculating the area covered by the 2D projection of the GNU particles on the glass surface divided by the total area of the active surface.

AFM images of S7 with a 2D and b 3D images of the surface morphology, along with higher magnification c 2D and d 3D images. The roughness is 10 nm with a calculated fractional coverage of 0.27
Principal component analysis
Based on the data given in Table 1, the common lines between the sample (i.e., up to the MUA grafting) and substrates (i.e., after immersion in GNU) were identified, then the ratio of sample to substrate peak intensity was calculated and presented in Table 2. The ratio was calculated by dividing the intensity value of the peaks of the sample by the intensity value of the same peaks of the substrate. The average of the ratios within a sample was taken, as well as the standard deviation, to compare against the average ratio of the other samples. A plot was made of the average ratio as well as the standard deviations as shown in Fig. 15a. Figure 15b shows the percentage of the substrate covered by GNU based on Table 2. All substrates showed more than 90% GNU coverage with S3 ≈ 96% being the highest with a variation of ≈5% in the first set (S1–S3), and S4 ≈ 97% as the highest with less than 1% variation in the second set (S4–S6). The highest GNU coverage belonged to S7 ≤ 97.8%.

To visualize differences between the spectra files, principal component analysis (PCA) was used. This is a form of dimensionality reduction, simplifying a multidimensional dataset to a smaller set of dimensions that hold important information while discarding less important information. In our case, we reduced the Raman spectra with 3000 intensity values that correspond to 3000 wavenumbers and SEFT-NIR spectra with approximately 4000 intensity values. These intensity values are the original dimensions that were reduced into 3 dimensions, called the 3 principal components. These intensity values served as the independent variables, while the identity of the sample (S1–S7) served as the dependent variable. The form of PCA used by this program is partial least squares regression (PLS regression), using sklearn. cross_decomposition.PLSRegression, a function written by Scikit-Learn. Instead of calculating the principal components in a way such that they explain the variance among the X (independent variables) like normal PCA, PLS regression calculates the principal components in the X dimensions such that they explain the relation of the X to the Y values (dependent variable). In our case, this means the intensity values are reduced to a set of principal components that explain the correlation of the intensity to the identity of the spectrum. Therefore, the closer the group (i.e., substrate) members (i.e., the scan files of each substrate), the less variation between them, i.e., are more accurate, and the further the groups from each other are, the more distinguishable they are, i.e., significantly more meaningful.
The results of the PC plots in Fig. 16 can be interpreted as showing significant variance in the SEFT-NIR data, while the PC plots of the Raman data show that the spectra all look very similar. In the SEFT-NIR PC plot, several distinct groups can be seen, with groups S3, S5, S6, and S7 having groupings that are clearly distinct from all other groups, while S1, S2, and S4 all form clear groupings between sample groups, but the groups themselves are close together. One possible explanation for this is that the SEFT-NIR spectra of each substrate have different major peaks. They share some peaks but the peaks with the highest intensity are different per substrate. This is interpreted as major differences between the spectra files, allowing for clear differentiation via PCA due to the significant separation between the groups. This is the opposite for the Raman spectra, where the major peaks all remain the same in scans of different substrates and only change in intensity. This results in a PC plot where differentiation between sample groups is unclear, i.e., there is less distance between the groups.

Principal component plots of the a SEFT-NIR and b SERS data of all seven substrates along with the standard deviation. The SEFT-NIR data shows more distinction between substrates than SERS
Conclusion
In order to better understand the effect of immersion direction of substrates and hence the concentration and time of the immobilization, the glass surface was functionalized by –NH2 terminated APTES followed by covalent bonding of MUA to APTES vis thiol groups. GNU were covalently self-assembled on the glass as SERS substrate via APTES-MUA linkage in vertical and horizontal immersion configurations. All the substrates showed a very high GNU coverage of about 96% and more depending on the time and direction of immersion. In the vertical configuration, the highest GNU coverage was observed for S3 ≈ 96% in the first set, i.e., longer immersion time and lower concentration, and S4 ≈ 97% in the second set (i.e., lower immersion time at higher concentration). The horizontal immersion produced the highest GNU coverage at high concentration and longest immersion time (i.e., S7 ≈ 97.8%). The findings were evaluated by SERS, reflective FT-NIR spectroscopy, SEM, and AFM, and confirmed by PCA. However, higher GNU coverage does not necessarily produce the maximum SERS signal due to variation in clustering, aggregation, and hot spots. To summarize, we believe that (a) an optimized SERS substrate with high efficiency can be obtained when it is immersed horizontally at higher concentration and longer immersion time for more uniform GNU distribution and less void glass space. Thus, it is expected that this approach improves SERS signal with a higher intensity and enhancement factor compared to unoptimized substrates, and (b) statistical analysis using data files produces more reliable evidence for conclusion than SEM alone since the former includes all the data points whereas the latter shows the partial morphology and not the entire surface. We believe this work can evolve into investigations of different types of substrates and nanostructures with variety of sizes and shapes to deepen our understanding both theoretically and experimentally of ideal practical SERS substrates.
Data Availability
The data that support the findings of this study are available from the corresponding author upon reasonable request.
References
Zhou W, Gao X, Liu D, Chen X (2015) Gold nanoparticles for in vitro diagnostics. Chem Rev 115:10575–10636
Khosroshahi ME, Tajabadi M (2017) Multifunctional nanoplatform for targeted laser-induced hyperthermia and microscopy of breast cancer cells using SPION-based gold and folic acid conjugated nanodendrimers: an in vitro assay. J Nanomed Nanotechnol 8:1–11
Pankhurst Q, Connolly J, Jones S, Dobson J (2003) Applications of magnetic nanoparticles in biomedicine. J Phys D: Appl Phys 36:167–181
He H, Xie C, Ren J (2008) Nonbleaching fluorescence of gold nanoparticles and its applications in cancer cell imaging. Anal Chem 80:5951–6595
Khosroshahi ME, Tajabadi M (2016) Characterization and cellular fluorescence microscopy of superparamagnetic nanoparticles functionalized with third generation nanomolecular dendrimers: in-vitro cytotoxicity and uptake study. J Nanomater Mol Nanotechnol 5:1–11
Pattani V, Tunnell J (2012) Nanoparticle-mediated photothermal therapy: a comparative study of heating for different particle types. Lasers Surg Med 44:675–684
Liao SH, Liu CH, Bastakoti BP, Suzuki N, Chang Y, Yamauchi Y, Lin FH, Wu K (2015) Functionalized magnetic iron oxide/alginate core-shell nanoparticles for targeting hyperthermia. Int J Nanomed 10:3315–3328
Kim M, Lee JH, Nam JM (2019) Plasmonic photothermal nanoparticles for biomedical applications. Adv Sci 6:1–23
Gormley A, Larson N, Sadekar S, Robinson R, Ray A, Ghandehari H (2012) Guided delivery of polymer therapeutics using plasmonic photothermal therapy. Nano Today 7:158–167
Khosroshahi ME, Ghazanfari L, Hasannejad Z, Lenhert S (2015) Dynamics study and analysis of laser-induced transport of nanoferrofluid in water using fluorescein isothiocyanate (FITC) as fluorescence marker. Nanomed Nanotech 6:1–9
Licciardi M, Volsi AL, Mauro N, Scialabba C, Cavallaro G, Giammona G (2007) Preparation and characterization of inulin coated gold nanoparticles for selective delivery of doxorubicin to breast cancer cells. J Nanomater 2016:2078315
Willets K, Van Duyne R (2007) Localized surface plasmon resonance spectroscopy and sensing. An Phys Rev Chem 58:267–297
Petryayeva E, Krull U (2011) Localized surface plasmon resonance: nanostructures, bioassays and biosensing—a review. Anal Chim Acta 706:8–24
Jain P, Lee K, El-Sayed I, El-Sayed M (2006) Calculated absorption and scattering properties of gold nanoparticles of different size, shape, and composition: applications in biological imaging and biomedicine. J Phys Chem B 110:7238–7248
Noguez C (2007) Surface plasmons on metal nanoparticles: the influence of shape and physical environment. J Phys Chem 111:3806–3819
Hassannejad Z, Khosroshahi ME (2013) Synthesis and evaluation of time dependent optical properties of plasmonic–magnetic nanoparticles. Opt Mat 35:644–651
Khlebtsov B, Zharov V, Melnikov A, Tuchin V, Khlebtsov N (2006) Optical amplification of photothermal therapy with gold nanoparticles and nanoclusters. Nanotechnology 17:5167–5179
Nath N, Chilkoti A (2004) Label-free biosensing by surface plasmon resonance of nanoparticles on glass: optimization of nanoparticles size. Anal Chem 76:5370–5378
Wang C, Meloni M, Wu X, Zhuo M, He T, Wang J, Wang C, Dong P (2019) Magnetic plasmonic particles for SERS-based bacteria sensing: a review. AIP Adv 9:1–18
Ong T, Blanch E, Jones O (2020) SERS in environmental analysis monitoring and assessment. Sci Total Environ 720:1–12
Alvarez-Puebla R, Liz- Marzán L (2010) SERS-based diagnosis and biodetection. Small 6:604–610
Masson J (2017) Surface plasmon resonance clinical biosensors for medical diagnosis. ACS Sens 2:16–30
Tian Z, Ren B, Wu D (2002) Surface-enhanced Raman scattering: from noble to transition metals and from rough surfaces to ordered nanostructures. J Phys Chem B 106:9463–9483
López-Lorente ÁI (2021) Recent development on gold nanoparticles nanostructures for surface enhanced Raman spectroscopy: particle shape, substrates and analytical applications. A review. Anal Chimica Acta 1168:1–21
Bruzas I, Lum W, Gorunmez Z, Sagle L (2018) Advances in SERS substrates for lipid and protein characterization: sensing and beyond. Analyst 143:3990–4400
Wang Y, Wang E (2010) Nanoparticles SERS substrates. In: Schlucker S (ed) Surface enhanced Raman spectroscopy: analytical, biophysical and life science applications. Wiley-VCH, Weinheim, pp 39–69
Fortuni B, Inose T, Uezono S, Toyouchi S, Umemoto K, Sekine S, Fujita Y, Ricci M, Lu G, Masuhara A, Hutchison J, Latterini L, Uji-I H (2017) In situ synthesis of Au-shelled Ag nanoparticles on PDMS for flexible, long-life, and broad spectrum-sensitive SERS substrates. Chem Commun 53:1298–11301
Gu HY, Yu AM, Chen HY (2001) Direct electron transfer and characterization of hemoglobin immobilized on a Au colloid–cysteamine-modified gold electrode. J Electroanal Chem 516:119–126
Toderas F, Baia M, Baia L, Astilean S (2007) Controlling gold nanoparticle assemblies for efficient surface-enhanced Raman scattering and localized surface plasmon resonance sensors. Nanotechnology 18:25502
Grabar KC, Freeman RG, Hommer MB, Natan MJ (1995) Preparation and characterization of Au colloid monolayers. Anal Chem 67:735–743
Lee J, Shim S, Kim B, Shin HS (2011) Surface-enhanced Raman scattering of single-and few-layer graphene by the deposition of gold nanoparticles. Chem Eur J 17:2381–2387
Read CG, Steinmiller EM, Choi KS (2009) Atomic plane-selective deposition of gold nanoparticles on metal oxide crystals exploiting preferential adsorption of additives. J Am Chem Soc 131:12040–12041
Okumura M, Nakamura S, Tsubota S, Nakamura T, Azuma M, Haruta M (1998) Chemical vapor deposition of gold on Al2O3, SiO2, and TiO2 for the oxidation of CO and of H2. Cat Lett 51:53–58
Dharanivasan G, Rajamuthuramalingam T, Jesse D, Rajendiran N, Kathiravan K (2015) Gold nanoparticles assisted characterization of amine functionalized polystyerene multiwell plate and glass slide surfaces. Appl Nanosci 5:39–50
Sasmal HS, Halder A, Kunjattu S, Dey K, Nadol A, Ajithkumar T, Bedadur P, Banerjee R (2019) Covalent self-assembly in two dimensions: connecting covalent organic framework nanospheres into crystalline and porous thin films. J Am Chem Soc 141:20371–20379
Vandenberg E, Bertilsson L, Leidberg B, Uvdal K, Erlandsson R, Elwing H, Lundstrom I (1991) Structure of 3-aminopropyl triethoxy silane on silicon oxide. J Colloid Interf Sci 147:103–118
Weinrib H, Meiri A, Duadi H, Fixler D (2012) Uniformly immobilizing gold nanorods on a glass substrate. J Atomic Mol Opt Phys 2012:683830
Indrasekara AS, Meyers S, Shubeita S, Feldman LC, Gustafsson T, Fabris L (2014) Gold nanostar substrates for SERS-based chemical sensing in the femtomolar regime. Nanoscale 6:8891–8899
He S, Kang M, Khan F, Tan E, Reyes M, Kah J (2015) Optimizing gold nanostars as a colloid-based SERS substrate. J Opt 17:1–13
Lai C, Wang G, Ling T, Wang T, Chiu P, Chau Y, Huang C, Chiang H (2017) Near infrared SERS based on star shaped gold/silver nanoparticles and hyperbolic metamaterial. Sci Rep 7:1–8
Freeman R, Grabar K, Alison K, Bright R, Davis J, Guthrie A, Hommer M, Jackson M, Smith P, Walter D, Natan M (1995) Self-assembled metal colloid monolayers: an approach to SERS substrates. Science 267:1629–1632
Zhang W, Fang Z, Zhu X (2017) Near field Raman spectroscopy with aperture tips. Chem Rev 117:5095–5109
Zhu J, Gao J, Li J, Zhao J (2014) Improve the surface-enhanced Raman scattering from rhodamine 6G adsorbed gold nanostars with vimineous branches. Appl Surf Sci 322:136–142
Pallavicini P, Donà A, Casu A, Chirico G, Collini M, Dacarro G, Falqui A, Milanese C, Sironi L, Taglietti A (2013) Triton X-100 for three-plasmon gold nanostars with two photothermally active NIR (near IR) and SWIR (short-wavelength IR) channels. Chem Commun 49:6265–6267
Fan X, Zhang W, Singh D (2014) Light scattering and surface plasmons on small spherical particles. Light Sci Appl 3:1–14
Yu M, Huang Z, Liu Z, Chen J, Liu Y, Tang L, Liu G (2018) Annealed gold nanoshells with highly-dense hotspots foe large-area efficient Raman scattering substrates. Sens Actuators B 262:845–851
Hrelescu C, Sau T, Rogach A, Jackel F, Laurent G, Douillard L, Charra F (2011) Selective excitation of individual plasmonic hotspots at the tips of single gold nanostars. Nano Lett 11:402–407
Du L, Tang D, Yuan G, Wei S, Yuan X (2013) Emission pattern of surface-enhanced Raman scattering from single nanoparticle-film junction. Appl Phys Lett 102:1–14
McNay G, Eustace D, Smith W, Faulds K, Graham D (2011) SERS and SERRS: a review of applications. Appl Spect 65:825–837
Moskovits M (1982) Surface selection rules. J Chem Phys 77:4408–4416
Su K, Wei Q, Zhang X, Mock J, Smith D, Schultz S (2003) Interparticle coupling effects on plasmon resonances of nanogold particles. Nano Lett 3:1087–1090
Acknowledgements
The authors would like to thank MIS Electronics Inc. for supporting and providing funding for this research and Mr. H. Zereshkian from Circuit Plus Inc. for use of their oven for the experiment. We also thank York University for use of its SEM and AFM services.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
The authors declare no competing interests.
Additional information
Publisher's note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Below is the link to the electronic supplementary material.
Rights and permissions
Springer Nature or its licensor (e.g. a society or other partner) holds exclusive rights to this article under a publishing agreement with the author(s) or other rightsholder(s); author self-archiving of the accepted manuscript version of this article is solely governed by the terms of such publishing agreement and applicable law.
About this article

Cite this article
Khosroshahi, M.E., Chabok, R., Chung, N. et al. Optimization of immersion direction and time of covalently self-assembled monolayer gold nanourchins on glass as SERS substrate. J Nanopart Res 25, 97 (2023). https://doi.org/10.1007/s11051-023-05741-2
Received:
Accepted:
Published:
DOI: https://doi.org/10.1007/s11051-023-05741-2