
Abstract
Silica Nanoparticles (SNPs) have been found to exhibit higher cytotoxicity to various cancer cells than to normal cells, while the underlying mechanisms are not fully understood. Here, SNPs triggered much higher cytotoxicity and apoptosis rate in human hepatoma HepG2 cells than in their normal counterparts L-02 cells; we thus selected these two cell lines as the cell model to investigate the mechanisms involved in the SNP-induced selective toxicity to cancer cells. Although uptake pathways and cellular trafficking of SNPs in HepG2 and L-02 cells were similar, more SNPs were taken up and accumulated in the mitochondria of cancer cells. After the removal of free SNPs from the culture medium, nanoparticles were excreted from HepG2 cells more effectively in the first 24 h, but 72 h later more SNPs still remained in cancer cells, leading to the continuous drop in cell viability of HepG2 cells. SNPs triggered a higher ROS generation, along with a lower intracellular GSH content and CAT activity in HepG2 cells than in L-02 cells. This could be due to the fact that HepG2 cells showed a much lower tolerance to H2O2-induced oxidative stress and cell death. Thus, the selective cytotoxicity of SNPs towards cancer cells could probably be explained by the higher particle uptake efficiency and cell sensitivity to oxidative stress as observed in HepG2 cells.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Nanotechnology has become one of the most innovative technologies in the past few decades due to the unique physicochemical properties and biological effects of nanomaterials, and has been exploited to revolutionize the industry including electronics energy and medicine [1, 2]. Among various types of nanomaterials, SNPs have been widely used in agricultural field, food industry and various industrial applications [3, 4]. Recently, they have also widely been explored in the biomedical field, mainly as nanocarriers for cancer treatment [5]. Interestingly, it has been reported that SNPs could induce cytotoxicity to different cancer cells, including human lung cancer cells, glioblastoma, and hepatocellular carcinoma [6,7,8]. The exposure of SNPs caused cell cycle arrest, apoptosis or autophagy in these cancer cells mainly by inducing ROS generation and oxidative stress [9]. Our previous studies have shown that SNPs triggered oxidative stress and mitochondrial apoptosis in cancer cells, and exhibited much higher cytotoxicity to hepatocellular carcinoma HepG2 cells than to normal L-02 cells [10]. The observed selective cytotoxicity to cancer cells might be related to cellular uptake process and efficiency of SNPs. However, the mechanisms underlying the selective cytotoxicity of SNPs to tumor cells are not fully understood.
Cellular entry is an important step for imparting cytotoxicity by nanoparticles including SNPs [11]. The cellular uptake and cytotoxicity of SNPs are not only affected by their physiochemical properties including size, shape and surface chemistry, but also are highly cell-type–dependent [12,13,14]. SNPs were found to be taken up via the energy-dependent endocytosis process, and enclosed in membrane-bounded organelles [15]. In human cervical carcinoma HeLa cells, SNPs could be detected in the cytosol, and were preferentially localized in lysosomes [16]. In this process, the smaller particles could be more easily endocytosed and translocated into lysosomes of human breast cancer cells (MDA-MB-468), causing greater cytotoxicity to MDA-MB-468 cells [17]. After internalization, nanoparticles may be excreted from cells due to exocytosis. Slowing et al. demonstrated that exocytosis of mesoporous silica nanoparticles (MSNs) took place in different cells and observed variations in ability of different cells to expel these nanoparticles [18]. In addition, the exocytosis rate of phosphonate-modified MSNs (P-MSNs) was also found to be cell-type–dependent. P-MSNs were excreted from cells by lysosomal exocytosis, inhibitors of lysosomal exocytosis could significantly slow down exocytosis and enhance the efficacy of P-MSN drug delivery [19]. In our previous studies, we have already found that the differential cytotoxicity of SNPs to different normal and tumor cells was associated with the amount of internalized SNPs [10, 20], which might be related to the cellular uptake, intracellular trafficking and excretion of SNPs in different cells.
ROS generation has been considered as a major mechanism for the cytotoxicity of SNPs [21,22,23]. Actually, ROS in low concentration is required in normal cells for signal transduction. While in cancer cells, increased ROS production has been detected as a consequence of their accelerated metabolism to maintain the high proliferation rate. Thus, cancer cells have developed an increased antioxidant defense system to maintain ROS homeostasis, and such an altered redox environment of cancer cells may render them more sensitive to elevated ROS levels [24, 25]. Based on these findings, a number of pro-oxidant strategies have been proposed for effective cancer therapies [26,27,28,29]. It has been reported that ROS overproduction that resulted from SNPs exposure could trigger cell death via apoptosis and autophagy in various cancer cells [9, 30, 31]. Similarly, our previous studies also showed that SNPs induced intrinsic apoptosis and increased ROS in human hepatoma, gastric and cervical cancer cells [10, 20]. These results suggested that cancer cells might be more sensitive to elevated ROS levels than normal cells, leading to enhanced cytotoxicity of SNPs in cancer cells, which need to be further investigated.
In this study, we used human hepatic cancer cells (HepG2) and their normal counterparts (L-02 cells) as models to explore the mechanism of the selective toxicity of SNPs to cancer cells from two aspects. On the one hand, cellular uptake pathway, trafficking behavior and excretion dynamics of SNPs by different cells were checked using fluorescein isothiocyanate-labeled SNPs (FITC-SNPs). On the other hand, to provide a better understanding of the role of elevated ROS production in cancer-cell-specific cytotoxicity of SNPs, SNP-induced ROS production, intracellular level of antioxidant molecule GSH and enzyme activity of catalase (CAT) were determined in HepG2 and L-02 cells, and the sensitivity of these two cells to H2O2-induced cytotoxicity was compared as well.
Materials and methods
Chemicals and reagents
SNPs were purchased from Shanghai Cabot Chemical Co Ltd. Fluorescein isothiocyanate (FITC), N,N-dimethylformamide, 3-aminopropyltriethoxysilane (APTES), 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT), and methyl-β-cyclodextrin (MβCD), chlorpromazine and 4′,6-diamidino-2-phenylindole (DAPI) were supplied by Sigma-Aldrich (St. Louis, MO, USA). LysoTracker™ Red DND-99 and MitoTracker™ Red were purchased from Invitrogen (Carlsbad, CA, USA).
Characterization of SNPs
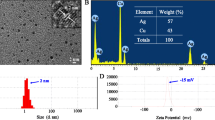
Particle size and morphology were characterized using transmission electron microscopy (TEM; JEM-2100; JEOL, Tokyo, Japan). For TEM observation, the particle samples were dispersed in ethanol, deposited on a carbon-coated copper grid, and dried at room temperature. The SNP samples were suspended in water or Dulbecco’s Modified Eagle Medium with 10% serum (cDMEM) at concentration of 1 mg/ml, and used for determination of hydrodynamic size and zeta potential via Dynamic light scattering (DLS; Zetasizer Nano ZS; Malvern Instruments, Worcestershire, UK).
Synthesis of FITC-labeled SNPs
Fluorescein isothiocyanate (FITC) (1 mg) was dissolved in 5 ml of N,N-dimethylformamide solution. After that, 500 μl of APTES was added and stirred for 2 h in the dark at room temperature. The reaction mixture was then added to a solution of 50 mg of SNPs in 50 ml of ultrapure water under stirring for 8 h in the dark at room temperature. The FITC-labeled SNPs were collected by centrifugation at 10,000 rpm for 5 min.
Cell culture
HepG2 cells and L-02 cells were purchased from the Cell Bank of Type Culture Collection of the Chinese Academy of Sciences (Shanghai, China). Cells were cultured in DMEM (Gibco, Carlsbad, CA, USA) containing 10% fetal bovine serum, 100 U/ml penicillin and 0.1 mg/ml streptomycin, in a humidified 5% CO2 incubator at 37 °C.
Cell viability assay
MTT assay was performed to evaluate the cell viability of HepG2 and L-02 cells upon the nanoparticle and H2O2 treatments. Cells at a density of 5 × 103 cells per well were seeded into 96-well plates. After cell adhesion (12 h), the culture medium was replaced with the fresh medium containing SNPs at different concentrations. Following that, 30 μl of MTT solution (5 mg/ml) was added to each well. After incubation for 4 h, the culture medium was aspirated, cells were washed with PBS to remove nanoparticles, and 200 μl of DMSO (Beyotime Bioengineering Institute, Nantong, Jiangsu, China) was added for dissolution of formazan crystal for 10 min at 37 °C. The absorbance was measured at the wavelength of 492 nm using a microplate reader (Molecular Devices SpectraMax M2, Sunnyvale, USA).
To evaluate cytotoxicity of H2O2 to HepG2 and L-02 cells, cells were plated at a density of 7 × 103 cells per well and incubated overnight. The culture medium was then replaced with the fresh medium with H2O2 at different concentrations. After H2O2 treatment for 24 h, cell viability was determined by MTT assay as described above.
Cell apoptosis assay
The Annexin V-FITC Apoptosis Detection Kit (Beyotime Bioengineering Institute, Nantong, Jiangsu, China) was used to detect cell apoptosis induced by SNPs. HepG2 and L-02 cells were plated in six-well culture plates at a concentration of 1 × 106 cells per well overnight before being treated with 160 or 320 µg/ml of SNP particles for 24 h. After treatment, cells were collected, washed with PBS, and resuspended in 400 µl of binding buffer for staining, according to the manufacturer’s instructions. Finally, at least 1 × 104 cells were collected and analyzed with the flow cytometer (FACSAria™, BD Biosciences, CA, USA).
Measurement of cellular uptake and excretion of SNPs
The uptake kinetics, pathways and excretion of SNPs were measured using the FITC-SNPs by flow cytometry. HepG2 and L-02 cells were seeded into six-well culture plates at a density of 2 × 105 cells per well overnight. Thereafter, the culture medium was replaced with the fresh medium containing FITC-SNPs (320 μg/ml). At the indicated time point, the medium was removed, cells were washed twice with ice-cold PBS, harvested after trypsinization and centrifugation. Cells were then treated with trypan blue solution (400 µg/ml) for 1 min to quench the fluorescence of FITC-SNPs adsorbed onto cell membrane, collected, wash with PBS, and resuspended in PBS buffer for flow cytometry analysis. The intracellular FITC fluorescence was measured using flow cytometry (FACSAria™, BD Biosciences, CA, USA) at an excitation wavelength of 488 nm and emission wavelength of 525 nm. To determine the uptake pathways, cells were pretreated with the clathrin-mediated endocytosis inhibitor chlorpromazine (0.0627 mM) for 1 h, or with the caveolin-mediated endocytosis inhibitor MβCD (10 mM) for 30 min, respectively. The cells were then incubated with FITC-SNPs (320 μg/ml). After 2 and 3 h, cells were harvested for fluorescence determination following the same procedure as described above.
In order to evaluate excretion of nanoparticles, cells were treated with FITC-SNPs (320 µg/ml) under the same conditions as for the uptake experiments. After exposure to cells for 24 h, the medium with SNPs was removed. Cells were washed with PBS and incubated in fresh medium without nanoparticles for additional 24, 48 or 72 h, and then were harvested for flow cytometry analysis following the same procedure as the uptake experiments.
Detection of intracellular localization of SNPs
The lysosomal probe LysoTracker™ Red DND-99 and mitochondrial probe MitoTracker™ Red was used to determine the intracellular localization of SNPs. HepG2 and L-02 cells were plated in 20 mm glass bottom culture dishes and incubated with FITC-SNPs (320 µg/ml) for 12 h. The treated cells were subsequently washed twice with PBS, stained with the LysoTracker™ Red DND-99 or MitoTracker™ Red, fixed with 0.25% glutaraldehyde, and then stained with DAPI immediately before observation using a confocal laser scanning microscope (CLSM; Nikon, Tokyo, Japan).
Measurement of reactive oxygen species (ROS), GSH and CAT activity
Intracellular ROS was detected using the fluorescent probe 2′,7′-dichlorofluorescin diacetate (DCFH-DA; Beyotime Institute of Biotechnology, Nantong, China). HepG2 and L-02 cells were plated in 6 cm culture dishes and treated with SNPs (320 µg/ml) for 24 h. After treatment, cells were washed with PBS and then incubated with DCFH-DA (10 µM) diluted in serum-free medium for 30 min at 37 °C in the dark. Cells were then washed three times with PBS, harvested and resuspended in PBS. The fluorescence intensity was measured using the flow cytometer (FACSAria™, BD Biosciences, CA, USA) at an excitation wavelength of 488 nm and an emission wavelength of and 535 nm. The measured fluorescence intensity was normalized to that of the control cells without SNPs treatment.
Cellular GSH content and CAT activity were measured after the same treatment for ROS assay, using the kits (Jiancheng Bioengineering Institute, Nanjing, China) according to the manufacturer’s instructions. Briefly, after 24 h exposure to SNPs, cells were harvested, washed with PBS, and homogenized. Cell lysates were centrifuged at 12,000 rpm for 10 min to obtain supernatants for GSH and CAT activity assay. Protein concentration of the sample was determined utilizing the BCA protein assay kit (Beyotime Institute of Biotechnology, Nantong, China). The GSH content was expressed in terms of µM/mg protein, and normalized to that of the control cells without SNPs treatment.
Statistical analysis
All experiments were conducted three times unless otherwise indicated. Results are presented as means ± standard deviation. Student’s t-test was performed to compare two means of one variable. Differences were considered to be significant if p < 0.05.
Results
Characterization of SNPs
The transmission electron microscopy (TEM) image showed that the amorphous silica nanoparticles (SNPs) were in a spherical shape with a diameter of about 20 nm (Fig. 1). When dispersed in ultrapure water and complete cell culture medium DMEM with 10% serum (cDMEM), SNPs were negatively charged, exhibited the hydrodynamic size of approximately 238 and 330 nm, respectively (Table 1). The larger diameter in aqueous solutions revealed the aggregation of SNPs, and the reduced surface charge in cDMEM might decrease the repulsive force between particles leading to an increased diameter.

Transmission electron microscopy image showed nanoparticles of SNPs were sphere-like and ~ 20 nm in diameter
SNPs induced selective cytotoxicity and apoptosis in HepG2 cells
In our previous studies, it was found that SNPs selectively inhibited cell proliferation and induced apoptosis in human hepatoma HepG2 cells, but appeared to be less toxic to normal liver L-02 cells. Here, we confirmed such cancer-specific cytotoxic effects of SNPs using MTT assays and annexin V-FITC/PI double staining. As shown in Fig. 2A, SNPs were more toxic to HepG2 cells. For HepG2 cells, cell viability decreased to around 24% after exposure to SNPs at the concentration of 320 μg/ml for 48 h, while cell viability of L-02 cells only slightly decreased by 14% after the same treatment. With the increase of SNP concentration, the induction of apoptosis was much more prominent in HepG2 cells than in L-02 cells (Fig. 2B). Following the exposure of SNPs at the concentration of 320 μg/ml for 24 h, the percentage of early and late apoptosis reached 50% and 23%, respectively (Fig. 2C), suggesting that early apoptosis might mostly affect the viability of HepG2 cells. On the contrary, about 94% of the L-02 cells were alive after the same treatment.

Selective cytotoxicity and apoptosis induced by SNPs in HepG2 cells. (A) Cell viability of HepG2 and L-02 cells treated with SNPs for 48 h. Error bars represent the standard deviation calculated from three independent experiments. *p < 0.05 vs control. Representative dot plots (B) and apoptotic rate (C) obtained from annexin V-FITC/PI double staining of HepG2 and L-02 cells assessed with flow cytometry after treatment with SNPs for 24 h
Cellular uptake and excretion of SNPs
As FITC has been commonly and widely used to label nanoparticles for analysis of their uptake and intracellular localization [32,33,34,35], we used FITC-SNPs to investigate cellular uptake and removal of SNPs in HepG2 and L-02 cells using flow cytometry. We studied the dynamics of uptake firstly. As shown in Fig. 3A, cellular uptake was a time-dependent process in both HepG2 and L-02 cells. However, internalization of nanoparticles was more efficient in HepG2 cells, reaching a maximum after cells were exposed to SNPs for 4 h. While for L-02 cells, a maximum fluorescence was detected at 8 h, which was only 26% of the maximal value observed in HepG2 cells. With the incubation temperature dropped from 37 to 4 °C, the uptake of SNPs was inhibited significantly, suggesting that energy-dependent endocytic mechanisms were involved in particle internalization.

Cellular internalization of SNPs in HepG2 and L-02 cells. (A) Time course of SNPs uptake. HepG2 (square) and L-02 cells (triangle) were incubated with FITC-SNPs (320 μg/ml) for 0–12 h at 37 °C (solid) or 4 °C (open), and the cellular fluorescence was detected using flow cytometry. (B) Uptake pathways of FITC-SNPs. Cells were pretreated with chlorpromazine (0.0627 mM) for 1 h or MβCD (10 mM) for 30 min, respectively, and then incubated with 320 μg/ml SNPs for 2 and 3 h. All of the fluorescence intensities were normalized to the percentage of maximum fluorescence value. Error bars represent the standard deviation calculated from three independent experiments. *p < 0.05 vs control
We next investigated the endocytotic pathways for internalization of SNPs in HepG2 and L-02 cells with specific inhibitors of endocytosis. Chlorpromazine is known to inhibit clathrin-dependent endocytosis, and methyl-β-cyclodextrin (MβCD) has been extensively used to inhibit caveolae-mediate uptake. Cells were preincubated with the inhibitors, and then exposed to FITC-SNPs for 2 and 3 h to evaluate the pathway of uptake. Cellular uptake of particles was sensitive to these two inhibitors, suggesting that SNPs were internalized within HepG2 and L-02 cells through both clathrin-dependent and caveolae-dependent endocytosis (Fig. 3B). However, SNPs uptake was much more significantly inhibited by MβCD in the two cell lines, implying a major role for caveolin-mediated pathway in cellular uptake of SNPs.
In addition to cellular uptake, removal of nanoparticles can also decide their fate and cytotoxicity. We thus examined cellular excretion of SNPs in HepG2 and L-02 cells. Cells were treated with FITC-SNPs for 24 h to allow particle internalization. After that, the cells were washed with PBS to remove FITC-SNPs and incubated in fresh culture medium without nanoparticles for additional 24, 48, or 72 h. Flow cytometry was performed to monitor nanoparticles that still remained inside the cells. As shown in Fig. 4A, the fluorescence intensity inside the HepG2 cells was 2.6-fold higher than that in L-02 cells after incubation with FITC-SNPs for 24 h due to an enhanced cellular uptake. Thereafter, removal of the particles from both cells could be observed for the following 72 h, as indicated by the decreased fluorescence. Clearance of SNPs was more prominent in HepG2 cells, especially during the first 24 h. However, a relatively high fluorescence could be detected in cancer cells throughout the whole excretion process with relative fluorescence intensity of 21% and 13% after 48 and 72 h of particle removal, while L-02 had 11% and 7%, respectively. Thus, more SNPs were retained in HepG2 cells, which could lead to the increased cytotoxicity of SNPs, as shown in Fig. 4B. Exposure of HepG2 and L-02 cells to SNPs for 24 h resulted in a similar cytotoxicity of 22% and 15%, respectively. However, after removal of SNPs, cell viability of L-02 cells recovered to 94% at the first 24 h and then was gradually increased to 98% after 48 and 72 h. In contrast to normal cells, HepG2 cells exhibited continuous drop in cell viability to 69%, 65% and 68% after 24, 48 and 72 h.

Excretion effect of SNPs on HepG2 and L-02 cells. (A) Excretion of SNPs by HepG2 and L-02 cells. (B) Cell viability of HepG2 and L-02 cells during the exclusion process. Cells were incubated with SNPs (320 μg/ml) for 24 h, then washed with PBS twice before adding fresh growth medium at 0 h time point for further incubation (24, 48 and 72 h). The cellular fluorescence was detected using flow cytometry, and normalized to the percentage of maximum fluorescence value to obtain the relative fluorescence of FITC-SNPs. Cell viability of HepG2 and L-02 cells was detected by MTT assay. Error bars represent the standard deviation calculated from three independent experiments. *p < 0.05 vs control
Intracellular trafficking and localization of SNPs
Intracellular trafficking and subcellular localization of nanoparticles can also decide their fate and cytotoxicity. We thus investigated and compared trafficking and localization of SNPs in HepG2 and L-02 cells via confocal microscopy and fluorescent probes for lysosomes and mitochondria, namely LysoTracker Red DND-99 and MitoTracker Red CMXRos, respectively. As shown in Fig. 5A, HepG2 cells took up much more of SNPs than L-02 cells, most nanoparticles (green) were localized in lysosomes (red) of both cancer and normal cells to yield yellow signals, indicating that SNPs were internalized and transported via the same lysosomal pathway in HepG2 and L-02 cells. We then examined the distribution of SNPs in mitochondria and found that some red signals from the mitochondria and green signals of SNPs were colocalized in both normal and cancer cells, whereas much more FITC signals were located in the mitochondria of HepG2 cells (Fig. 5B).

Localization of FITC-SNPs in lysosomes (A) and mitochondria (B) of HepG2 and L-02 cells. HepG2 and L-02 cells were treated with FITC-SNPs (320 μg/ml) for 24 h, then stained by LysoTracker™ Red DND-99 and MitoTracker™ Red CMXRos for 30 min respectively and observed by CLSM. Scale bar: 25 μm
SNPs induced enhanced oxidative stress and weakened antioxidant defense in HepG2 cells
Oxidative stress has been reported to be a primary mechanism for SNP-induced cytotoxicity. Here, we further investigated the role of oxidative stress in selective cytotoxicity of SNPs to cancer cells. It could be found that SNPs induced a much severe oxidative stress in HepG2 cells than in L-02 cells. As shown in Fig. 6A, after 24 h of SNPs treatment at the concentration of 160 and 320 μg/ml, ROS formation increased by 20 ± 6.8% and 110 ± 2.6% in HepG2 cells, while only by 9 ± 2.5% and 20 ± 2.1% in L-02 cells. On the contrary, a stronger decrease in GSH level in HepG2 cells was observed under the same treatment. With the SNP concentration increased to 160 and 320 μg/ml, the GSH content in HepG2 cells decreased to 85.3 ± 5.5% and 67.1 ± 3.3%, while it only dropped to 91.1 ± 5% and 82.9 ± 6.4% in L-02 cells (Fig. 6B). Although HepG2 and L-02 cells exhibited a similar level of CAT activity without SNP treatment, exposure to SNPs induced a dramatic decrease in CAT activity in HepG2 cells, cellular CAT activity decreased from 76 to 59 and 45 U/mg at doses of 160 and 320 μg/ml SNPs, respectively. Conversely, in L-02 cells, the same SNP treatment resulted in a slightly increase of CAT activity from 77 to 80 and 80 U/mg (Fig. 6C). As both GSH and CAT play the key role in cellular antioxidant defenses, these results indicated that SNPs stimulated a significantly weaker antioxidant activity for scavenging excess ROS in cancer cells, suggesting that HepG2 cells might be more sensitive to oxidative stress. We thus evaluated the cytotoxicity of the peroxide (H2O2) to normal and cancer cells, as H2O2 is one of the three major types of ROS and is well known to elicit a variety of oxidative damage including cell death. Obviously, HepG2 cells exhibited much lower tolerance to H2O2-induced cytotoxicity, cell viability dropped to 61% at the H2O2 level of 100 μM and decreased to 31% at the 250 μM exposure. In contrast, significant loss of viability (61% of control) was only observed at the H2O2 level above 1000 μM for L-02 cells (Fig. 6D).

Oxidative stress and antioxidative response in HepG2 and L-02 cells. Effects of SNPs on ROS generation (A), GSH level (B) and catalase activity (C) in HepG2 and L-02 cells following SNP treatment for 24 h. (D) Cell viability of HepG2 and L-02 cells after 24 h exposure to hydrogen peroxide. Error bars represent the standard deviation calculated from three independent experiments. *p < 0.05 vs control
Discussion
Based on the selective cytotoxicity of SNPs to cancer cells as previously reported [7, 9, 10, 31] and we showed here (Fig. 7), the underlying mechanisms for SNPs to induce tumor-specific toxicity was investigated in this study using the human hepatoma HepG2 versus normal liver cell L-02 as cell models. Our results showed that although the uptake pathway and intracellular fate of SNPs were similar in both HepG2 and L-02 cells, more nanoparticles were taken up into HepG2 cells and transported to mitochondria. On the other hand, 72 h after SNP removal, the remaining SNPs in HepG2 cells were still about twice as many as those in L-02 cells, in spite of a more effective exclusion of SNPs in the first 24 h period in HepG2 cells. Overall, the increased long-term accumulation of SNPs continued to inhibit cell viability of cancer cells, mainly because SNPs could greatly improve the intracellular ROS level via depleting intracellular GSH and inhibiting CAT activity and thus elevated the oxidative stress in HepG2 cells. Such a difference in response to the SNP-induced oxidative stress could be explained by a reduced antioxidant capacity of HepG2 cells, as exposure to H2O2 triggered a much higher cytotoxicity in HepG2 cells than in L-02 cells. Therefore, compared to normal cell L-02, the higher particle uptake efficiency and the lower tolerance to oxidative stress made HepG2 cells more sensitive to SNP-induced oxidative damage, eventually leading to the selective cytotoxicity of SNPs towards cancer cells. Our results thus provided novel insights into tumor-specific cytotoxicity of SNPs and the underlying mechanisms, suggesting the potential to develop SNP-based biomodulators [5] for improving the anticancer efficacy of nanomedicines.

Schematic illustration of the mechanism of selective cytotoxicity of SNPs to cancer cells
It has been reported that increased cellular uptake of SNPs resulted in their higher cytotoxicity [32, 36]. Previously, we also found that SNPs caused differential cytotoxic effects in different human cancer cells, which might be related to the varying uptake process and efficiency of SNPs in these cancer cells [20]. Here, to explore the mechanisms involved in tumor-specific cytotoxicity of SNPs, we investigated the uptake, transport and excretion of SNPs in normal and tumor cells. SNPs may enter cells through passive mechanisms or various active endocytosis pathways, including pinocytosis, caveolae- and clathrin-mediated endocytosis, depending on particle physicochemical property and cell type [32, 36, 37]. In this study, SNPs were found to be taken up by HepG2 and L-02 cells both through energy-dependent pathways, mainly via caveolae-dependent endocytosis. It has also been reported that SNPs could be internalized via endocytosis by normal and tumor cells [37, 38]. Therefore, the internalization pathway was not likely responsible for the higher cytotoxicity of SNPs observed in tumor cells, and we then examined the excretion of SNPs from different cells. The excretion of SNPs from HepG2 cells has been confirmed [39], and mesoporous silica nanoparticles were observed to be exocytosed from cells [18], mainly through lysosomal exocytosis but with variations in the exocytosis rate among different cell lines [19]. In our study, we also observed the decrease of FITC-SNPs fluorescence intensity in both HepG2 and L-02 cells after the removal of the free particles, indicating the excretion of SNPs from cells. Additionally, a much higher rate of exocytosis was found in HepG2 cells during the first 24 h. This could be related to the high concentration of internalized SNPs in HepG2 cells rather than the effect of different cell type, as the exocytosis rate decreased remarkably with the reduction of intracellular particles 24 h later. Although SNPs were excreted from HepG2 and L-02 cells at a similar rate for 24–72 h, the higher initial concentration resulted in more residual amounts of particles in HepG2 after 72 h incubation in fresh medium without SNPs. Such a higher retention of nanoparticles in tumor cells than in normal fibroblasts was also found for gold nanoparticles [40].
Since intracellular location and translocation of nanoparticles is directly related to their cytotoxicity, the trafficking and localization of SNPs in HepG2 and L-02 cells were investigated and compared. SNPs have been reported to accumulate in the cytosol and endosome of HeLa cells, they were preferentially localized in lysosomes but not toxic to HeLa cells [16]. In contrast, cytotoxicity of mesoporous silica nanoparticles to human breast cancer cells MDA-MB-468 was suggested to be that more particles were endocytosed and located within lysosomes [17], while internalized SNPs induced cytotoxicity in L-02 cells by targeting mitochondria and their quality control process [41]. Here, we found that SNPs were transported via the same pathway in HepG2 and L-02 cells, translocated into lysosomes and mitochondria. So, it should not be the cellular trafficking of SNPs that caused the differential toxicity of the particles to normal and cancer cells. However, much more particles were finally located in the mitochondria of HepG2 cells, which should induce mitochondrial damage and selective cytotoxicity to cancer cells.
The above results demonstrated that it was not the uptake pathway, exclusion process and intracellular translocation, but the efficient uptake and mitochondria accumulation of SNPs that contributed to their selective toxicity to HepG2 cells. Similarly, more pristine mesoporous silica nanoparticles could be internalized into human breast adenocarcinoma MCF-7 cells compared to foreskin fibroblast BJ cells, leading to selective cytotoxic effects including cell cycle arrest and cell death [42]. In addition, gold nanorods were found to be taken up through similar pathways in human lung carcinoma A549 cells, normal bronchial epithelial 16HBE cells and primary adult stem cells, while selective mitochondria accumulation of the nanorods was thought to cause cancer cell-specific cytotoxicity to A549 cells [43]. Interestingly, these nanoparticles could be more efficiently internalized and accumulate in organelles such as mitochondria to cause selective cytotoxicity to cancer cells, thus being used to design highly specific nanomedicines for cancer therapy. Meanwhile, further work is needed to unravel the underlying mechanism.
ROS generation has been considered as an important mechanism responsible for the toxicity of SNPs [21, 44], ROS formation and buildup have been found to play a vital role in SNP-induced mitochondrial damage and intrinsic apoptosis in glioblastoma cells [45] and HepG2 cells [46]. Our previous study also revealed that the differential level of ROS generation in cancer cells HepG2 and MGC80-3 is closely related to the cytotoxic effects caused by SNPs [20]. Here, when we found that SNPs triggered a much higher level of ROS and cytotoxicity in tumor cells, we go further to explore the underlying mechanisms.
In comparison with normal cells, SNPs caused a significantly higher level of ROS and lower GSH content in HepG2 cells. In addition, CAT, which plays an important role in antioxidant defense system, decreased dramatically in HepG2 cells, while increased significantly in L-02 cells after SNP treatment. These results indicated that SNPs stimulated a weaker antioxidant activity for scavenging excess ROS in cancer cells, suggesting that HepG2 cells might be more sensitive to oxidative stress. Furthermore, enhanced cytotoxicity was observed in HepG2 cells as compared to L-02 cells after direct exposure of these cells to H2O2, demonstrating that HepG2 cells had much lower tolerance to H2O2-induced oxidative stress. Actually, increased ROS production is generally recognized as a hallmark of various cancers, contributing to proliferation and survival of cancer cells [47]. Such abnormal increases in ROS render cancer cells dependent on an increased antioxidant capacity to maintain a correct redox balance, thus may also provide a biochemical basis to preferentially induce cell death in cancer cells through a ROS-mediated mechanism by further increasing ROS or reducing antioxidant activity [25, 48, 49]. For instance, the natural compound with chemopreventive activity, β-phenylethyl isothiocyanate, was found to selectively kill cancer cells via depletion of GSH and inhibition of GPX enzyme activity [50]. Thus, SNPs triggered ROS generation in both HepG2 and L-02 cells, while cancer cells exhibited a decreased antioxidant capacity and could not cope with the higher intracellular ROS level, which eventually led to apoptosis and cytotoxicity in HepG2 cells. Similarly, bare iron oxide nanoparticles were reported to be significantly more toxic to human lung cancer cells than to the normal human lung fibroblast cells, by inducing ROS production [51]. In addition, ROS-mediated mechanism was also found to cause the selective killing of human cervical cancer cells (HeLa) by black phosphorus-based nanosheets [52].
However, it has been reported that human vascular endothelial cells are more sensitive to SNP-induced cell death than HeLa cells [13]. Moreover, although SNPs and H2O2 caused increase of oxidative stress and depletion of GSH in both A549 and pleural mesothelial cells (Met-5A), A549 cells were relatively more resilient [53]. Additionally, it was found that when comparing to poorly differentiated hepatocellular carcinoma SK-Hep-1cells, well-differentiated HepG2 cells showed a more robust antioxidant defense system against CuO nanoparticle-mediated oxidative stress [54]. These differences in cellular susceptibility to oxidative damage may be attributed to not only the properties of nanoparticles, but also the cell types [55]. Therefore, in order to understand these discrepancies and how designs of nanoparticles could be optimized for therapeutic use, careful and full characterization of SNPs, including protein corona formed under biological conditions, along with the cell type specific cellular response, will require further study.
Conclusions
In this study, we focused on understanding the mechanisms involved in the selective cytotoxicity of SNPs to cancer cells. We demonstrated that SNPs were internalized mainly via caveolae-dependent endocytosis, and transported to lysosomes and mitochondria, in both cancer and normal cells. However, SNPs were more effectively taken up by HepG2 cells, which resulted in a higher amount of nanoparticles in cytosol and mitochondria of cancer cells. Although both cancer and normal cells continued to excrete SNPs up to 72 h post incubation and HepG2 cells excreted SNPs more rapidly in the first 24 h, the retention of SNPs was found to be higher in HepG2 cells than in L-02 cells, contributing to continuous drop in cell viability of HepG2 cells. Moreover, SNPs induced a higher intracellular ROS level, along with a lower GSH level and CAT activity in HepG2 cells. Such an imbalance between ROS production and ROS scavenging in cancer cells led to the increased oxidative stress in HepG2 cells. As HepG2 cells were more sensitive to the H2O2-induced oxidative stress, they thus exhibited much lower tolerance to SNP-triggered oxidative stress, which may be crucial to understand the selective cytotoxicity of SNPs to cancer cells. Such a cancer-specific feature should be investigated in more types of cancer cells, and then be exploited to develop SNP based nanomedicines with improved efficacy and selectivity.
Data availability
All data that support the findings of this study are included within the article.
References
Kirtane AR, Verma M, Karandikar P (2021) Nanotechnology approaches for global infectious diseases. Nat Nanotechnol 16(4):369–384
Salamanca-Buentello F, Daar AS (2021) Nanotechnology, equity and global health. Nat Nanotechnol 16(4):358–361
Jeelani PGH, Mulay P, Venkat R, Ramalingam C (2020) Multifaceted application of silica nanoparticles. A review Silicon 12(6):1337–1354
Rastogi A, Tripathi DK, Yadav S, Chauhan DK, Zivcak M, Ghorbanpour M (2019) Application of silicon nanoparticles in agriculture. 3 Biotech 9(3):1–11
Yang Y, Zhang M, Song H, Yu C (2020) Silica-based nanoparticles for biomedical applications: from nanocarriers to biomodulators. Acc Chem Res 53(8):1545–1556
Tokgun O, Demiray A, Kaya B, Karagur E, Demir E, Burunkaya E, Akca H (2015) Silica nanoparticles can induce apoptosis via dead receptor and caspase 8 pathway on A549 cells. Adv Food Sci 37:65–70
Krętowski R, Kusaczuk M, Naumowicz M, Kotyńska J, Szynaka B, Cechowska-Pasko M (2017) The effects of silica nanoparticles on apoptosis and autophagy of glioblastoma cell lines. Nanomaterials 7(8):230
Niu Y, Tang E, Zhang Q (2019) Cytotoxic effect of silica nanoparticles against hepatocellular carcinoma cells through necroptosis induction. Toxicol Res 8(6):1042–1049
Yu Y, Duan J, Yu Y, Li Y, Liu X, Zhou X, Ho K, Tian L, Sun Z et al (2014) Silica nanoparticles induce autophagy and autophagic cell death in HepG2 cells triggered by reactive oxygen species. J Hazard Mater 270:176–186
Lu X, Qian J, Zhou H, Gan Q, Tang W, Lu J, Yuan Y, Liu C (2011) In vitro cytotoxicity and induction of apoptosis by silica nanoparticles in human HepG2 hepatoma cells. Int J Nanomedicine 6:1889–1901
Beddoes CM, Case CP, Briscoe WH (2015) Understanding nanoparticle cellular entry: a physicochemical perspective. Adv Colloid Interface Sci 218:48–68
Malugin A, Herd H, Ghandehari H (2011) Differential toxicity of amorphous silica nanoparticles toward phagocytic and epithelial cells. J Nanopart Res 13(10):5381–5396
Blechinger J, Bauer AT, Torrano AA, Gorzelanny C, Brauchle C, Schneider SW et al (2013) Uptake kinetics and nanotoxicity of silica nanoparticles are cell type dependent. Small 9(23):3970–3980
Perez-Garnes M, Gutierrez-Salmeron M, Morales V, Chocarro-Calvo A, Sanz R, Garcia-Jimenez C, Garcia-Munoz RA et al (2020) Engineering hollow mesoporous silica nanoparticles to increase cytotoxicity. Mater Sci Eng C 112:110935
Chu ZQ, Huang YJ, Tao Q, Li Q (2011) Cellular uptake, evolution, and excretion of silica nanoparticles in human cells. Nanoscale 3(8):3291–3299
Al-Rawi M, Diabate S, Weiss C (2011) Uptake and intracellular localization of submicron and nano-sized SiO2 particles in HeLa cells. Arch Toxicol 85(7):813–826
He Q, Zhang Z, Gao Y, Shi J, Li Y (2009) Intracellular localization and cytotoxicity of spherical mesoporous silica nano- and microparticles. Small 5(23):2722–2729
Slowing II, Vivero-Escoto JL, Zhao Y, Kandel K, Peeraphatdit C, Trewyn BG (2011) Exocytosis of mesoporous silica nanoparticles from mammalian cells: from asymmetric cell-to-cell transfer to protein harvesting. Small 7(11):1526–1532
Yanes RE, Tarn D, Hwang AA, Ferris DP, Sherman SP, Thomas CR (2013) Involvement of lysosomal exocytosis in the excretion of mesoporous silica nanoparticles and enhancement of the drug delivery effect by exocytosis inhibition. Small 9(5):697–704
Wu Y, Tang W, Wang P, Liu C, Yuan Y, Qian J (2015) Cytotoxicity and cellular uptake of amorphous silica nanoparticles in human cancer cells. Part Part Syst Charact 32(7):779–787
Lehman SE, Morris AS, Mueller PS, Salem AK, Grassian VH, Larsen SC (2016) Silica nanoparticle-generated ROS as a predictor of cellular toxicity: mechanistic insights and safety by design. Environ Sci Nano 3(1):56–66
Wang F, Gao F, Lan M, Yuan H, Huang Y, Liu J (2009) Oxidative stress contributes to silica nanoparticle-induced cytotoxicity in human embryonic kidney cells. Toxicol In Vitro 23(5):808–815
Ye Y, Liu J, Xu J, Sun L, Chen M, Lan M (2010) Nano-SiO2 induces apoptosis via activation of p53 and Bax mediated by oxidative stress in human hepatic cell line. Toxicol In Vitro 24(3):751–758
Sosa V, Moline T, Somoza R, Paciucci R, Kondoh H, LLeonart ME (2013) Oxidative stress and cancer: an overview. Ageing Res Rev 12(1):376–390
Moloney JN, Cotter TG (2018) ROS signalling in the biology of cancer. Semin Cell Dev Biol 80:50–64
Perillo B, Di Donato M, Pezone A, Di Zazzo E, Giovannelli P, Galasso G (2020) ROS in cancer therapy: the bright side of the moon. Exp Mol Med 52(2):192–203
Reczek CR, Chandel NS (2017) The two faces of reactive oxygen species in cancer. Annu Rev Biochem 1:79–98
Cho HD, Lee JH, Moon KD, Park KH, Lee MK, Seo KI (2018) Auriculasin-induced ROS causes prostate cancer cell death via induction of apoptosis. Food Chem Toxicol 111:660–669
Raza MH, Siraj S, Arshad A, Waheed U, Aldakheel F, Alduraywish S (2017) ROS-modulated therapeutic approaches in cancer treatment. J Cancer Res Clin 143(9):1789–1809
Inoue M, Sakamoto K, Suzuki A, Nakai S, Ando A, Shiraki Y (2021) Size and surface modification of silica nanoparticles affect the severity of lung toxicity by modulating endosomal ROS generation in macrophages. Part Fibre Toxicol 18(1):1–20
Lin W, Huang YW, Zhou XD, Ma Y (2006) In vitro toxicity of silica nanoparticles in human lung cancer cells. Toxicol Appl Pharmacol 217(3):252–259
Zhu J, Liao L, Zhu L, Zhang P, Guo K, Kong J (2013) Size-dependent cellular uptake efficiency, mechanism, and cytotoxicity of silica nanoparticles toward HeLa cells. Talanta 107:408–415
Shahabi S, Treccani L, Dringen R, Rezwan K (2015) Dual fluorophore doped silica nanoparticles for cellular localization studies in multiple stained cells. Acta Biomater 14:208–216
Veeranarayanan S, Poulose AC, Mohamed S, Aravind A, Nagaoka Y, Yoshida Y (2012) FITC labeled silica nanoparticles as efficient cell tags: uptake and photostability study in endothelial cells. J Fluoresc 22(2):537–548
Li J, Tian J, Yin H, Peng Y, Liu S, Yao S, Zhang L (2021) Chemical conjugation of FITC to track silica nanoparticles in vivo and in vitro: an emerging method to assess the reproductive toxicity of industrial nanomaterials. Environ Int 152:106497
Saikia J, Yazdimamaghani M, Hadipour Moghaddam SP, Ghandehari H (2016) Differential protein adsorption and cellular uptake of silica nanoparticles based on size and porosity. ACS Appl Mater Interfaces 8(50):34820–34832
Li L, Xi WS, Su Q, Li Y, Yan GH, Liu Y (2019) Unexpected size effect: the interplay between different-sized nanoparticles in their cellular uptake. Small 15(38):1901687
Glorani G, Marin R, Canton P, Pinto M, Conti G, Fracasso G (2017) Pegylated silica nanoparticles: cytotoxicity and macrophage uptake. J Nanopart Res 19(8):1–14
Hu L, Mao Z, Zhang Y, Gao C (2011) Influences of size of silica particles on the cellular endocytosis, exocytosis and cell activity of HepG2 cells. J Nanosci Lett 1:1–16
Ivosev V, Sanchez GJ, Stefancikova L, Haidar DA, Gonzalez Vargas CR, Yang X (2020) Uptake and excretion dynamics of gold nanoparticles in cancer cells and fibroblasts. Nanotechnology 31(13):135102
Qi Y, Ma R, Li X, Lv S, Liu X, Abulikemu A (2020) Disturbed mitochondrial quality control involved in hepatocytotoxicity induced by silica nanoparticles. Nanoscale 12(24):13034–13045
Menon N, Leong DT (2016) Cytotoxic effects of phosphonate-functionalized mesoporous silica nanoparticles. Acs Appl Mater Inter 8(3):2416–2422
Wang L, Liu Y, Li W, Jiang X, Ji Y, Wu X (2011) Selective targeting of gold nanorods at the mitochondria of cancer cells: implications for cancer therapy. Nano Lett 11(2):772–780
Guo CX, Yang M, Jing L, Wang J, Yu Y, Li Y (2016) Amorphous silica nanoparticles trigger vascular endothelial cell injury through apoptosis and autophagy via reactive oxygen species-mediated MAPK/Bcl-2 and PI3K/Akt/mTOR signaling. Int J Nanomedicine 11(2016):5257–5276
Kusaczuk M, Kretowski R, Naumowicz M, Stypulkowska A, Cechowska-Pasko M (2018) Silica nanoparticle-induced oxidative stress and mitochondrial damage is followed by activation of intrinsic apoptosis pathway in glioblastoma cells. Int J Nanomedicine 13:2279–2294
Ahmad J, Ahamed M, Akhtar MJ, Alrokayan SA, Siddiqui MA, Musarrat J (2012) Apoptosis induction by silica nanoparticles mediated through reactive oxygen species in human liver cell line HepG2. Toxicol Appl Pharmacol 259(2):160–168
Sabharwal SS, Schumacker PT (2014) Mitochondrial ROS in cancer: initiators, amplifiers or an Achilles’ heel? Nat Rev Cancer 14(11):709–721
Gorrini C, Harris IS, Mak TW (2013) Modulation of oxidative stress as an anticancer strategy. Nat Rev Drug Discov 12(12):931–947
Wang J, Yi J (2008) Cancer cell killing via ROS To increase or decrease, that is the question. Cancer Biol Ther 7(12):1875–1884
Trachootham D, Zhou Y, Zhang H, Demizu Y, Chen Z, Pelicano H (2006) Selective killing of oncogenically transformed cells through a ROS-mediated mechanism by beta-phenylethyl isothiocyanate. Cancer Cell 10(3):241–252
Khan MI, Mohammad A, Patil G, Naqvi SA, Chauhan LK, Ahmad I (2012) Induction of ROS, mitochondrial damage and autophagy in lung epithelial cancer cells by iron oxide nanoparticles. Biomaterials 33(5):1477–1488
Kong N, Ji X, Wang J, Sun X, Chen G, Fan T (2020) ROS-mediated selective killing effect of black phosphorus: mechanistic understanding and its guidance for safe biomedical applications. Nano Lett 20(5):3943–3955
Berg JM, Romoser AA, Figueroa DE, Spencer West C, Sayes CM (2013) Comparative cytological responses of lung epithelial and pleural mesothelial cells following in vitro exposure to nanoscale SiO2. Toxicol In Vitro 27(1):24–33
Kung ML, Hsieh SL, Wu CC, Chu TH, Lin YC, Yeh BW (2015) Enhanced reactive oxygen species overexpression by CuO nanoparticles in poorly differentiated hepatocellular carcinoma cells. Nanoscale 7(5):1820–1829
Kim IY, Joachim E, Choi H, Kim K (2015) Toxicity of silica nanoparticles depends on size, dose, and cell type. Nanomedicine 11(6):1407–1416
Funding
This study is supported by the National Key Research and Development Program of China (2020YFA0908900), the National Natural Science Foundation of China (31871011).
Author information
Authors and Affiliations
Contributions
Peng Wang: Investigation, Original draft.
Tao Shen: Formal analysis, Writing—original draft.
Yi Sun: Data Curation.
Xinhui Cui: Validation.
Changsheng Liu: Resources, Supervision, Funding acquisition.
Yuan Yuan: Methodology, Supervision.
Jiangchao Qian: Conceptualization, Methodology, Writing—review & editing, Funding acquisition.
Corresponding author
Ethics declarations
Conflict of interest
We declare that we do not have any commercial or associative interest that represents a conflict of interest in connection with the work submitted.
Additional information
Publisher's note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Springer Nature or its licensor (e.g. a society or other partner) holds exclusive rights to this article under a publishing agreement with the author(s) or other rightsholder(s); author self-archiving of the accepted manuscript version of this article is solely governed by the terms of such publishing agreement and applicable law.
About this article

Cite this article
Wang, P., Shen, T., Sun, Y. et al. Selective killing of cancer cells by silica nanoparticles due to increased nanoparticle internalization and cellular sensitivity to oxidative stress. J Nanopart Res 25, 13 (2023). https://doi.org/10.1007/s11051-022-05643-9
Received:
Accepted:
Published:
DOI: https://doi.org/10.1007/s11051-022-05643-9