
Abstract
Molecular dynamics simulation was employed to understand the thermodynamic behavior of cuboctahedron (cub) and icosahedron (ico) nanoparticles with 2–20 number of full shells. The original embedded atom method (EAM) was compared to the more recent highly optimized version as inter-atomic potential. The thermal stability of clusters were probed using potential energy and specific heat capacity as well as structure analysis by radial distribution function (G(r)) and common neighbor analysis (CNA), simultaneously, to make a comprehensive picture of the solid-state and melting transitions. The result shows ico is the only stable shape of small clusters (Pd55–Pd309 using original EAM and Pd55 using optimized version) those are melting uniformly due to their small diameter. An exception is cub Pd309 modeled via optimized EAM that transforms to ico at elevated temperatures. A similar cub to ico transition was predicted by original EAM for Pd923–Pd2075 clusters, while for the larger clusters both cub and ico are stable up to the melting point. As detected by \(G(r)\) and CNA, moderate and large cub clusters were showing surface melting by nucleation of the liquid phase at (100) planes and growth of liquid phase at the surface before inward growth. While diagonal (one corner to another) melting was dominating over ico clusters owing to their partitioned structure, which retarded the growth of the liquid phase. The large ico clusters, using optimized EAM, presented a combination of surface and diagonal melting due to the simultaneous diagonal melting started from different corners. Finally, the melting temperature as well as latent heat of fusion were calculated and compared with the available models and previous studies, which showed, unlike the present result, the models failed to predict size-dependent motif cross-over.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Palladium nanoparticles have received growing interest due to their novel properties, such as size (Chen et al. 2016) and shape (facet) (Wang et al. 2017) dependent catalytic activity, controlled oligomerization (Zhivonitko et al. 2016), and enhanced hydrogen storage (Rangel et al. 2016). However, due to the higher surface to volume ratio (Schmidt et al. 1998; Safaei et al. 2008), the nanoparticles are known as the most unstable structures among different nano-solids. Thus, it is necessary to predict their thermal stability for practical applications specifically when instability at elevated temperature can be considered as a failure. For instance, reshaping, melting, or aggregation of catalyst clusters might happen far below the bulk melting point (Tmb), which results in changing their properties as well as functionality.
The nanoparticles melting temperature (Tmp) and enthalpy (Hmp), among the other thermodynamic properties, have received considerable attention (Qi 2016). Several models have been developed to predict the nanoparticles size-dependent \(T_{\text {mp}}\) (Goldstein et al. 1992; Jiang et al. 1999; Safaei 2010) and \(H_{\text {mp}}\) (Zhang et al. 2000; Jiang et al. 2002; Attarian Shandiz and Safaei 2008; Fu et al. 2017). However, developing a universal model requires an extensive effort and each model has to be verified with experimental or simulation data (Liang et al. 2017), which to our knowledge, is not available for Pd clusters. In addition, these models fail to describe the melting dynamic and size-dependent melting mechanisms. For instance, quite recently, it has been shown that reshaping of Ag particles depends on the size and a transition from homogeneous to surface melting mechanism occurs by an increase in the particle size (Liang et al. 2017).
Alternatively, atomistic Monte Carlo (MC) (Westergren and Nordholm 2003) and molecular dynamics (MD) (Baletto et al. 2002; Pan et al. 2005; Miao et al. 2005; Schebarchov and Hendy 2006) simulations have proven to be an excellent tool for probing the nanoparticles stability and melting behavior. For instance, Baletto et al. (2002) have shown that for small Pd clusters icosahedron (ico) structure is more stable, while decahedron (dec) and cuboctahedron (cub) are more stable at the moderate and large clusters, respectively. In this regard, they compared Rosato-Guillopé-Legrand and embedded atom method (EAM) force fields those were in close agreement. However, they only focused on the energetics of the different structures rather than showing a dynamic transition between allotropes. Thus, it still remains unclear that under what condition an unstable shape transforms into a more stable counterpart. Later, Pan et al. (2005) reported in the case of Pd309 cluster both of the cub and ico shapes are stable in a wide range of temperatures using Sutton-Chen (SC) potential. They also reported a surface-melting mechanism, which was detected using radial distribution function, \(G(r)\). However, the \(G(r)\) has been defined separately for each shell of the cluster in their work, which can be misleading, i.e., the diffusion of the surface atoms, those have more degree of freedom, into inner shells causes a broadening of the \(G(r)\) peak for the outer shell, which can be misinterpreted as surface melting (Bertoldi et al. 2017). They regretted this drawback by reporting quasi-solid core at the melting point (Tmp) of clusters, while their \(G(r)\) patterns indicated broad peaks for all shells (Pan et al. 2005). They also observed a minor peak prior to the main melting peak in the specific heat capacity \(C_{p}\) of the ico cluster, which they attributed to the surface melting. Using a similar potential, Miao et al. (2005) reported the surface melting for a spherical Pd456 cluster based on bond-orientational order parameters (BOP). The BOP is a local structure characterization method obtained by determining the angle between a list of nearest neighbors (NN) (Steinhardt et al. 1983). Thus, it determines which atoms are in solid/liquid states and enables the study of the surface melting more precisely. Later, Schebarchov and Hendy (2006) pointed out the BOP is unable to distinguish between fcc and hcp (Steinhardt et al. 1983), and it is not efficient to detect solid-state transitions in the clusters. To maintain brevity, cub to ico transition can be detected by mapping twinning boundaries in ico cluster those can be characterized as hcp atoms. Thus, they suggested common neighbor analysis (CNA), instead of BOP, which is very sensitive to the angles between different pairs of NN (Tsuzuki et al. 2007). The efficiency of CNA for the structural transition has been demonstrated earlier by Baletto et al. (2000, 2001), and an interested reader is referred to Ref. (Rossi and Ferrando 2007) to learn about the potential of CNA for the detailed characterization of clusters. Schebarchov and Hendy (2006) successfully demonstrated dec to ico transition through a solid-liquid state for a Pd877 cluster. This is interesting since they used the EAM force field and obtained a different result than static energy calculation by Baletto et al. (2002) with similar potential. It is worth noting that for small clusters those have a high degree of freedom and might undergo several structure transformations at elevated temperatures, the Lindemann index (Lindemann 1910) (δL) gives scattered results as reported elsewhere (Alavi and Thompson 2006; Zhang and Douglas 2013).
In the present work, the phenomenological melting of symmetric shell Pd clusters of cub and ico structure is investigated. Since EAM potential predicts solid-state phase change, according to previous studies, original and highly optimized EAM were utilized in the present study. In addition, different methods of caloric curves, \(G(r)\) and CNA were utilized to make a comprehensive understanding of the nanoparticles melting. This accompanied with a brief discussion on the limitation of each method and the resulting misinterpretation. Finally, the size-dependent results were compared to state of the art models.
Method
MD simulation
MD simulations were performed by solving Newton’s equation of motion (Allen and Tildesley 1989) using large-scale atomistic/molecular massively parallel simulator (LAMMPS) open source codeFootnote 1 (Plimpton 1995; Plimpton and Thompson 2012).
The original (Foiles et al. 1986) and highly optimized (Sheng et al. 2011) EAM was used to depict the inter-atomic potential between Pd atoms. Equation 1 represents the original formulation of EAM potential:
where \(E_{i}\) and \(F_{i}\) are cohesive and embedding energies of atom i, respectively. \(\rho _{\text {ij}}(r_{\text {ij}})\) is the electron density of j atoms located around the i atom at distance \(r_{\text {ij}}\). Clearly, \(F_{i}\) is a many-body interaction term, while \(U_{\text {ij}}\) takes the pair interaction into account.
The EAM potential was confirmed for describing solid characteristics, such as cohesive energy and elastic constant as well as metals melting point (Foiles et al. 1986; Sheng et al. 2011). Moreover, it is reliable in determining the transitional properties especially the heat of fusion and heat capacities above room temperature. The EAM has also been verified for a quantitatively correct description of such nanoscale systems; for instance, surface energy and geometry of low index surfaces.
The time integration of the equation of the motion was performed regarding the Verlet algorithm (Verlet 1967; Kateb and Dehghani 2012) with a timestep of 3 fs. The temperature control was done using the Nose-Hoover thermostat with a damping of 30 fs. These conditions were designed to generate positions and velocities sampled from canonical (NVT) ensemble. The initial velocities of the atoms were defined randomly from a Gaussian distribution at the appropriate temperature of 300 K and relaxed for 300 ps in NVT ensemble. Since practical nanoparticle melting is mostly performed in the vacuum, the heat associated with the particle’s melting cannot be removed so efficiently by the surrounding medium. But, the particles are in contact with the substrate, which allows control over temperature, and hence, the NVT ensemble provides a realistic representation of such systems. The simulations were performed by starting at 300 K, and then the temperature was elevated at a heating rate of \(1.4\times 10^{12}\) K/s.
Cluster preparation
The Pd nanoparticles were considered to be in the cub and ico forms those found in experimental characterizations (José-Yacamán et al. 2001). For different sizes of given clusters, cub and ico were made considering the so-called magic number (Poole and Owens 2003) or a total number of cluster atoms (Nt), which is described as the function of the shell number (n):
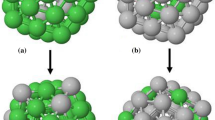
where \(n = 0\) denotes a mono-atomic system and \(n\geq 1\) defines the full shell clusters. Here, clusters with sizes \(\sim \)1.5–12 nm, including n \(=\) 2–20 shells (Nt = 55 − 28741 atoms) were considered. Figure 1 illustrates the 8-cub (n = 8) and 8-ico as an example. The figure also shows a slice of particles indicating 8-cub is made by fcc and surface atoms, while the 8-ico presents more complicated arrangement, i.e., 20 fcc tetrahedral (green) those surrounded an ico atom (indicated by yellow in Fig. 1d) separated by twinning grain boundaries detected as hcp (red).

Illustration of a 8-cub and b 8-ico particles with eight being the shell number, n. The crystal arrangement is shown through a slice of c 8-cub and d 8-ico indicating fcc, hcp, ico, and unknown using green, red, yellow, and blue colors, respectively
We chose to use the cluster radius (Rp) defined by Guinier formula (Miao et al. 2005), which is nearly constant during surface diffusion, solid-state transition, and at the beginning of melting.
where \(R_{g}\) is the particle gyration radius and \(r_{a}\) is atomic radius equal to 0.137 nm. The term particle size hereafter is referred to as \(D = 2R_{p}\) calculated at ambient temperature.
Melting criteria
The melting is commonly detected from isotherm transition in the so-called caloric curve(s), e.g., potential energy (U) plotted versus temperature (T) (Shim et al. 2002; Zhao et al. 2001). Qi et al. (2001) defined \(T_{\text {mp}}\) as the temperature with the maximum apparent heat capacity. In MD simulations, specific heat capacity at constant pressure (Cp) can be calculated by derivative of the U average (Uave) during heating:
here \(\Re \) stands for the universal gas constant.
It is worth noting that the precision of \(T_{\text {mp}}\) value calculated from \(C_{p}\) is dependent on the number available data points (U vs. T) and a limited number of sampling, which is usually the case in MC simulation, results in a huge error.
Several methods are used in MD simulations to identify the melting process based on atomic specifics. The first criterion proposed by Lindemann (Lindemann 1910) who stated the melting of crystals occurs when the average amplitude of atomic vibrations is higher than a threshold value. The global \(\delta _{L}\) is a system average of atomic quantity, which shows a linear increase with temperature increment in solid-state and a step change due to the melting. However, most of the vibrations of the surface atoms in the small clusters, which have more degree of freedom, were assumed as melting behavior by this model (Alavi and Thompson 2006; Zhang and Douglas 2013). This is a serious issue, since it may lead to misinterpreting of the surface melting instead of a solid-state transition. The most straightforward structure analysis is offered by \(G(r)\) or pair correlation function (Iida and Guthrie 1988). It describes how density varies as a function of distance in a system of particles from a reference particle.
where \(\rho _{a}\) is the atom numbers density, r is the distance from reference particle, and dr determines the bin size. The angle brackets, i.e., HCode \(\langle \rangle _{T}\) denote the time average at constant T.
Here, \(G(r)\) was extracted by 300 ps relaxation of clusters in NVT ensemble at desired T, i.e., 300–2500 K with 100 K steps. Then, the \(G(r)\) averaged out over the whole time with 10− 4 nm bin size without periodic boundary condition (PBC). This results in a pattern of several peaks corresponding to number and distance of NNs, which applies to a wide range of materials. The melting transition causes a variation in the density and can be detected by shifting and broadening of peaks in the \(G(r)\) pattern. However, complex solid-state transition at nanoscale, such as cub to ico with constant coordination number and even distance, is very hard to determine with \(G(r)\).
Recently, CNA has shown promising tool due to providing the possibility of a distinction between allotropic transitions and melting process. Briefly, the CNA determining local crystal structure based on the decomposition of first NNs obtained from \(G(r)\) into different angles (Faken and Jónsson 1994; Tsuzuki et al. 2007). It is quite sensitive technique to the symmetry of different pairs of bonds. Thus, a twinning grain boundary as the main difference of ico and cub cluster can be determined based on a slight angle difference between first NNs, while it holds entire properties of an fcc atom.
Visualization
The open visualization tool (OVITO) packageFootnote 2 was used to generate atomistic illustrations (Stukowski 2009).
Result and discussion
Potential
Table 1 summarizes important values reproduced by optimized EAM potential in comparison with original EAM and experimental or ab initio results. The fcc to hcp transformation energy, \({\Delta } E\), is optimized to be equal to 0.02 eV/atom in agreement with thermodynamically assessed value at room temperature (Dinsdale 1991) and more precise than 0.026 eV/atom obtained by original EAM (Foiles et al. 1986). This value is very important for realization of ico to cub transition and stability of ico structure. It is believed the vacancy formation energy, \(E_{v}\), is the elementary mechanism of melting, and thus, it is one of the most important parameter to capture realized melting. The optimized force field results in vacancy formation energy of 1.4 eV similar to value estimated from experimental melting point (Kraftmakher 1986) and better than 1.44 eV obtained from original potential (Foiles et al. 1986). The values for surface energy, \(\gamma _{\text {sv}}\), in the optimized EAM predicts higher values than original EAM regarding all plains. The experimental value of the \(\gamma _{\text {sv}}\) is estimated from contact angle representing an average over all planes, but it is still higher than both EAM values. Finally, the optimized EAM results in \(T_{\text {mb}}\) value closer to experimental value (Rao and Rao 1964).
Figure 2 demonstrates the result of both EAM potential in determining the cohesive energy, \(E_{c}\), of solid Pd in comparison with ab initio results. The minimum in fcc denotes equilibrium \(a_{0}\) and \(E_{c}\) equal to 0.389 nm and 3.911 eV/atom, respectively, which both EAM are in agreement with ab initio result. However, the \(a_{0}\) and \(E_{c}\) values for hcp determined by ab initio are smaller than both EAM by 0.005 nm and 0.06 eV, respectively. In general, compared to ab initio results, both EAM are slightly off in the repulsive range, but they are acceptable in attraction part. The optimized EAM, however, tend to zero more smoothly.

Validation of original and optimized EAM potentials for determining Ec in comparison with ab initio calculation
Further, the dynamic structure factor, \(S(q)\), was calculated, which is a conventional method to examine liquid structure (Iida and Guthrie 1988). The \(S(q)\) was calculated here using the Fourier transform of \(G(r)\).
here q is the wave-vector in reciprocal space.
The \(G(r)\) was obtained in the same way described in “Melting criteria” but with \(dr= 10^{-5}\) nm and PBC for 500 atoms in total. The wave-vector increment was chosen to be compatible with PBC, i.e., HCode \(2\pi /L\) with L being the cube side length equal to 5× a0 (1.945 nm).
Figure 3 shows \(S(q)\) for the liquid Pd calculated using both EAM in comparison with the tight-binding calculation (Alemany et al. 1999) and the experimental result obtained by x-ray Raman scattering (Waseda 1980). The figure clearly shows both EAM potentials are in close agreement with the tight-binding and experimental results. However, the main discrepancies are the peaks’ position predicted by both EAM those are slightly off. In addition \(S(q)\) of the main peak obtained by original EAM shows notably smaller value (12%).

Relaxation
Each cluster was relaxed as discussed in “MD simulation” to minimize the potential energy of the entire system at the beginning. For the small cub clusters, a transformation to ico is expected during relaxation at room temperature (Baletto et al. 2002). The optimized EAM only predicts 2-cub transformation to 2-ico, while relaxation using original EAM shows 2-cub and 4-cub are transforming to ico clusters in agreement with the previous result using the same potential (Schebarchov and Hendy 2006). This difference originates from higher energy barrier for the cub to ico transformation in optimized potential. As mentioned in the introduction the solid-state transition is expected to occur via an intermediate quasi-liquid state (Schebarchov and Hendy 2006), which seems to require higher energy using optimized EAM. In the following, we stick to ground-state notations of the cluster and still call them 2-cub and 4-cub. We would like to remark that the cub structure is not the best fcc structure with the lowest energy, since its (100) facets are too large compared to (111) facets.
The relaxation of higher shell number shows that there is no change in the shape and symmetry of the particle, indicating both potentials can successfully model the stable shapes of Pd clusters.
Melting criteria
Caloric curves
Figure 4 illustrates the variation of U and \(C_{p}\) versus T for the 8-cub particle (Pd2057) and corresponding snapshots of point A–F for the optimized EAM and G–L for the original EAM. Both caloric curves present typical melting behavior, including an isotherm transition due to \(H_{\text {mp}}\). The gradual melting transition (non-isotherm melting) is more drastic in the curve obtained by original EAM, which makes determining \(T_{\text {mp}}\) harder. Nevertheless, \(T_{\text {mp}}\) of 1277 and 1387 K were determined using main \(C_{p}\) peaks for the original and optimized EAM, respectively. The difference came from the fact that optimized potential predicts bulk melting point more precisely and higher than original EAM. Snapshots A–F belong to the optimized EAM, which in general present more red atoms with higher potential energy. This is expected since U for the optimized EAM is always stand above original EAM values. It can be clearly seen in snapshots A–D, the cub structure remains unchanged without any evidence of surface melting but slightly rounding in the corners. Snapshots E and F were taken at and after \(T_{\text {mp}}\) those are indicating a homogeneous melting mechanism. In snapshots G–I a solid-state cub to ico transition is evident (see the movie in the supplementary materials). This is associated with a clear step change in U and corresponding local minimum in \(C_{p}\) both at 1070 K from original EAM. It has been shown previously that, a solid-state transition is accompanied with a step change in U and a local minimum in \(C_{p}\), which was referred to as negative \(C_{p}\) (Zhang et al. 2010). The J–L snapshots were taken close to melting point those showing a diagonal (corner to corner) melting, which started at the bottom and moving up, where (111) facet on the top can still be seen. This is the reason for drastic non-isotherm melting in U of the original EAM. Finally, snapshot L shows a complete liquid state.

Variation of U and Cp with temperature for 8-cub particle using optimized and original EAM. a–f are the corresponding snapshots of points (A–F) obtained by optimized EAM, while g–l are snapshots of point (G–L) from original EAM. The color bar in the inset indicates potential energy level of each atom
It is worth noting that the minor \(C_{p}\) peak is sometimes misinterpreted as surface melting (Pan et al. 2005). However, as mentioned in the introduction, the improper use of \(G(r)\) was the real reason for the misinterpretation. No surface melting is detected here as can be seen in snapshot H, which is taken after minor \(C_{p}\) peak.
Radial distribution function
Figures 5 depicts the \(G(r)\) variation with temperature for 8-cub using both potentials. In both cases, the \(G(r)\) shows the same pattern for solid and liquid-like states in agreement with previously reported patterns (Pan et al. 2005). The solid-like state at the bottom of each figure can be interpreted from four main peaks indicated by dashed line corresponding to four shells (not to be confused with fourth NNs). The 1st shell is consisting of 12 equidistant atoms, and thus, presents a single peak, while further shells contain more peaks due to their geometrical complexity. The liquid-like state at top of each figure consisting of four broad peaks those are also indicated by dashed lines. It can be clearly seen that the increase in temperature causes peak broadening in both solid and liquid-like states. The melting occurs when there is step change in the peaks’ positions (dashed lines). Figure 5a shows homogeneous melting of all four shells at about 1400 K using the optimized EAM in agreement with 1387 K obtained from caloric curves. In Fig. 5b, there are two-step change in the peaks’ positions at about 1000 and 1400 K. It is shown earlier that the first transition belongs to cub to ico transformation. However, since the step change in peaks’ positions is more evident in the fourth peak (corresponding to fourth shell), it has been misinterpreted as shell-by-shell melting in the previous study (Pan et al. 2005).

Variation of normalized G(r) with T for 8-cub cluster obtained by a optimized and b original EAM force fields. The colorbar indicates normalized G(r) and dashed lines indicate its main peaks
Common neighbor analysis
Here, the optimized EAM is neglected, since original EAM present a more complicated case with a solid-state transition and drastic non-isotherm melting. Figure 6 presents the variation in the ratio of different structures with T according to the CNA for 8-cub cluster obtained by original EAM. The figure also includes corresponding snapshots of the particle cross-section at points A–F those indicated with dotted lines. The mirror change can be clearly seen in fcc and disordered structure, including surface atoms, up to \(\sim \) 940 K. The cross-section of the particle at point A in this region shows only nucleation of disordered atoms close to the surface and remaining fcc atoms in green. In the 940–1050 K range, some hcp atoms are randomly appearing as can be seen in the snapshot B. It is worth noting that disordered phase exists everywhere even in the core part before transition to ico. Then, the transition occurs at 1070 K, between point B and C, with a clear transition of fcc to disordered atoms (\(\sim \) 13%) followed by nucleation of hcp from disordered atoms (\(\sim \) 7%). Snapshot C shows an ico atom in the center (yellow), and the same portion of fcc and hcp (in red). It is worth noting that there are no fcc and hcp atoms in the small area at the bottom of the snapshot C, which is associated with \(\sim \) 6% left disordered during the transition. Thus, CNA predicts an incomplete transition, while we could not detect such thing using \(G(r)\) and caloric curves. Thereafter, both fcc and hcp transform to disordered atoms as shown in the D–F snapshots. In specific, the diagonal melting is evident in D–F with a disordered lower half growing towards crystalline aggregates at the upper half until total disorder is achieved.

variation of structure ratios with T and cross-section of the corresponding snapshots at A – F indicated in figure by dashed lines
The local maximum in the ratio of disordered atoms, between B–C, was previously interpreted as “coexistence of solid-liquid phase” by focusing only on the CNA criteria (Schebarchov and Hendy 2006). However, our \(G(r)\) result shows no liquid-like state during this transition. Thus, the transition occurs with a so-called military diffusion mechanism, which is very fast and short ranged, and the term “transition quasi-liquid state” is more preferred over solid-liquid coexistence. It is worth noting that the transition is limited to 6 and 8-cub clusters possibly because for the larger clusters, the activation energy required for this aim is higher than melting.
Size and shape-dependent melting mechanism
Table 2 summarizes melting mechanisms utilizing the abovementioned criteria. Below 3 nm (n = 2–4), the ico is the only stable shape of clusters before melting those are melting uniformly due to the small diameter of particles. For the larger diameters, cub clusters are generally melting with surface melting and ico clusters present diagonal melting. Optimized EAM predicts combined surface and diagonal melting for 16–20-ico clusters (10–12 nm in diameter). It is worth noting that there is also a difference in the surface melting of cub and ico clusters. The surface melting of the cub clusters is associated with nucleation of the liquid phase at (100) planes followed by its growth at the surface, which deforms polyhedral to a sphere with a lower surface area and surface energy. This is interesting since it is shown experimentally that the (100) planes present an incomplete surface melting in the bulk state, while (110) and (111) planes present complete and no surface melting, respectively (Vanselow and Howe 1988). Thus, the so-called two-stage melting is found to be associated with faster growth of liquid phase at the surface before moving towards the core. While in the case of large ico clusters, 16–20-ico using optimized EAM, surface melting is in fact simultaneous diagonal melting starting from different corners rather than formation of liquid shell followed by melting in the core. This difference can be explained by more resistance of (111) planes against surface melting compared to (100) ones as detected in many experiments. The interested reader is referred to reference (Vanselow and Howe 1988) and references therein. Thus, ico clusters with totally (111) planes at the surface present almost no surface melting. While cub cluster with both (100) and (111) planes at the surface shows surface melting.
Size-dependent melting temperature
Safaei (2010) developed a model considering the effects of the first and second NNs atomic interactions. An approximation of the formula without considering of second NNs atomic interaction is as follows:
here, \(N_{s}\) stands for the number of surface atoms, q is the coordination number ratio with \(Z_{v}\) equal to 12 for interior atoms, and \(\bar {Z}_{s}\) as the average coordination numbers of surface atoms. The \(\epsilon _{v}\) and \(\bar {\epsilon }_{s}\) respectively are bond energies of interior and surface atoms, which the latter consists of cluster faces, edges, and corners.
Figure 7 compares normalized \(T_{\text {mp}}\) obtained from caloric curves in comparison with previous MD (Pan et al. 2005; Miao et al. 2005) and MC (Lee et al. 2001) simulations. The figure also contains values calculated from Eq. 7 assuming the bond strength for the surface and interior atoms to be equal. It is evident that \(T_{\text {mp}}\) values obtained by optimized EAM always stands between original EAM and Safaei model. In addition, previous MD results using SC force field are predicting slightly higher values than original EAM but lower than optimized EAM. The original EAM data set shows a clear change around 5 nm for the cub due to the fact that 2- and 4-cub already transformed to ico during relaxation; and 4–5 nm cub (corresponding to 6- and 8-cub) are transforming to ico before melting. In the case of optimized EAM, \(T_{\text {mp}}\) is higher for ico until 5 nm and is lower for \(D>\) 5 nm. This is a clear indication of the stability of ico clusters up to 5 nm and cub clusters for 6 nm and larger diameters. Thus, the deficiency of the models is assuming a static lattice without showing cross-over of different structure. It can be seen that the model always predicts higher \({T}_{\text {mp}}\) for the ico clusters. This is because the model is focused on coordination number, which is slightly lower for cub due to lower coordination number of (100) planes at the surface. While ico surface is only made out of (111) planes with higher coordination number.

The original EAM overestimates \(T_{\text {mp}}\) for ico particles larger than 6 nm, which means they are more stable than cub clusters. This might be associated with the \(\gamma _{\text {sv}}\) difference in two potentials.
Size-dependent melting enthalpy
Attarian Shandiz and Safaei (2008) proposed the following model for the size-dependent \(H_{\text {mp}}\) of clusters.
with \(D_{0}\) being specific diameter, which the entire atoms are located at the surface (i.e., HCode \(N_{s}=N_{t}\)) and \(H_{\text {mb}}\) is the bulk melting enthalpy.
The variation of \(H_{\text {mp}}\) with particle size is shown in Fig. 8 in comparison with Attarian Shandiz and Safaei (2008) model and previous MD results (Pan et al. 2005; Miao et al. 2005). It is worth noting that, the model shows a negligible difference between ico and cub, and thus, it is plotted for different \(\bar {Z}_{s}\) of 6 and 3 corresponding to \(q = 0.5\) and 0.25, respectively. As shown in the figure, the model predicts an increase in the \(H_{\text {mp}}\) with the particle size in agreement with the result of the optimized EAM. While the original EAM underestimates \(H_{\text {mp}}\) for particles larger than 5 nm. It can be seen SC potential (Pan et al. 2005) fails to predict correct values for cub and ico by a huge error (\(\sim \) 100%). While quantum corrected SC results in more accurate value for the spherical cluster (Miao et al. 2005). However, the model determines \(H_{\text {mp}}<0\) below 0.5754 and 1.1984 nm respectively for \(\bar {Z}_{s}\) of 6 and 3 meaning that melting smaller particles are exothermic and favorable. This failure originated from the crystalline basis of the model, which is not defined for a few atoms. Both EAM potentials predict a unified trend for ico particles. For the cub clusters, however, there is step change at \(\sim \)4 nm (corresponding to 6-cub), which is due to the transition to ico before melting. Thus, the accuracy of models depends on knowing the stable structure of the cluster, which they fail to predict.

Variation of Hmp with the diameter of Pd clusters obtained by optimized and original EAM potential, in comparison with previous MD results (Pan et al. 2005; Miao et al. 2005) as well as Attarian Shandiz and Safaei (2008) model using D0 = 0.6712 nm and the bulk value 16.7 kJ/mol (Iida and Guthrie 1988)
Conclusion
In conclusion, the stability and melting behavior of Pd55–Pd28741 clusters of cub and ico shape studied by molecular dynamics simulation using two EAM force field, i.e., original and highly optimized one. Different melting criteria were discussed those make a clear picture of the melting process. The result shows small cub clusters (Pd55– \(\text {Pd}_{309}\)) are unstable those are melting uniformly due to their small diameter. The only exception is Pd309 using optimized EAM, which transforms to ico at elevated temperatures before melting. A similar cub to ico transition is predicted by original EAM for Pd923 – Pd2075 cluster and for larger clusters both cub and ico are stable up to the melting point. In general, as detected by \(G(r)\) and CNA, the cub clusters present surface melting while ico clusters are melting diagonally (corner to corner) thanks to their partitioned structure. However, large ico cluster also present surface melting due to simultaneous diagonal melting from different corners. This is entirely different from surface melting observed in cub clusters. The later is associated with nucleation of liquid phase at the (100) planes and its growth at the surface before moving inward.
Notes
version 14 Apr 2013 available at http://lammps.sandia.gov
Version 2.7.1 available at http://ovito.org/
References
Alavi S, Thompson DL (2006) Molecular dynamics simulations of the melting of aluminum nanoparticles. The J Phys Chem A 110(4):1518–1523
Alemany MMG, Diéguez O, Rey C, Gallego LJ (1999) Molecular-dynamics study of the dynamic properties of fcc transition and simple metals in the liquid phase using the second-moment approximation to the tight-binding method. Physical Review B: Condensed Matter 60(13):9208–9211. https://doi.org/10.1103/PhysRevB.60.9208
Allen MP, Tildesley DJ (1989) Computer simulation of liquids. Oxford university press, Oxford
Ashcroft NW, Mermin ND (1976) Solid state physics holt. Rinehart and Winston, New York
Attarian Shandiz M, Safaei A (2008) Melting entropy and enthalpy of metallic nanoparticles. Mater Lett 62(24):3954–3956
Baletto F, Mottet C, Ferrando R (2000) Reentrant morphology transition in the growth of free silver nanoclusters. Phys Rev Lett 84(24):5544
Baletto F, Mottet C, Ferrando R (2001) Microscopic mechanisms of the growth of metastable silver icosahedra. Phys Rev B 63(15):155,408
Baletto F, Ferrando R, Fortunelli A, Montalenti F, Mottet C (2002) Crossover among structural motifs in transition and noble-metal clusters. The J Chem Phys 116(9):3856–3863
Bertoldi DS, Millán EN, Guillermet AF (2017) Thermodynamics of the melting process in au nano-clusters: phenomenology, energy, entropy and quasi-chemical modeling. J Phys Chem Solids 111:286–293
Chen T, Zhang Y, Xu W (2016) Size-dependent catalytic kinetics and dynamics of Pd nanocubes: a single-particle study. Phys Chem Chem Phys 18(32):22,494–22,502
Dinsdale AT (1991) SGTE Data for pure elements. Calphad 15(4):317–425
Faken D, Jónsson H (1994) Systematic analysis of local atomic structure combined with 3d computer graphics. Comput Mater Sci 2(2):279–286
Foiles S, Baskes M, Daw M (1986) Embedded-atom-method functions for the fcc metals Cu, Ag, Au, Ni, Pd, Pt, and their alloys. Phys Rev B 33(12):7983
Fu Q, Zhu J, Xue Y, Cui Z (2017) Size- and shape-dependent melting enthalpy and entropy of nanoparticles. J Mater Sci 52(4):1911–1918. https://doi.org/10.1007/s10853-016-0480-9
Goldstein AN, Echer CM, Alivisatos AP (1992) Melting in semiconductor nanocrystals. Science 256(5062):1425–1427
Iida T, Guthrie RIL (1988) The physical properties of liquid metals. Clarendon Press, Oxford
Jiang Q, Shi HX, Zhao M (1999) Melting thermodynamics of organic nanocrystals. The J Chem Phys 111(5):2176–2180
Jiang Q, Yang CC, Li JC (2002) Melting enthalpy depression of nanocrystals. Mater Lett 56 (6):1019–1021
José-Yacamán M, Marín-Almazo M, Ascencio JA (2001) High resolution TEM studies on palladium nanoparticles. J Mol Catal A Chem 173(1):61–74
Kateb M, Dehghani K (2012) Comparison of fracture behavior of sharp with blunt crack tip in nanocrystalline materials by molecular dynamics simulation. Int J Mod Phys: Conference Series 5:410–417
Kraftmakher YA (1986) Equilibrium concentration of vacancies in metals. In: Seeger A, Schumacher D, Schilling W, Diehl J (eds) Vacancies and interstitials in metals: international conference proceeding, North Holland, Amsterdam, pp 59, held in jülich, Germany September pp 23-28
Lee YJ, Lee EK, Kim S, Nieminen RM (2001) Effect of potential energy distribution on the melting of clusters. Phys Rev Lett 86(6):999
Liang T, Zhou D, Wu Z, Shi P (2017) Size-dependent melting modes and behaviors of Ag nanoparticles: a molecular dynamics study. Nanotechnology 28(48):485,704
Lindemann FA (1910) The calculation of molecular vibration frequencies. Physikalische Zeitschrift 11:609–612
Miao L, Bhethanabotla VR, Joseph B (2005) Melting of Pd clusters and nanowires: a comparison study using molecular dynamics simulation. Phys Rev B 72(13):134,109
Pan Y, Huang S, Liu Z, Wang W (2005) Molecular dynamics simulation of shell-symmetric Pd nanoclusters. Mol Simul 31(14-15):1057–1061
Plimpton S (1995) Fast parallel algorithms for short-range molecular dynamics. J Comput Phys 117 (1):1–19
Plimpton SJ, Thompson AP (2012) Computational aspects of many-body potentials. MRS Bull 37(5):513–521
Poole CP Jr, Owens FJ (2003) Introduction to nanotechnology. Wiley, New Jersey
Qi W (2016) Nanoscopic thermodynamics. Acc Chem Res 49(9):1587–1595
Qi Y, Çagin T, Johnson WL, Goddard WA III (2001) Melting and crystallization in Ni nanoclusters: the mesoscale regime. The J Chem Phys 115(1):385–394
Rangel E, Sansores E, Vallejo E, Hernández-Hernández A, López-Pérez P (2016) Study of the interplay between N-graphene defects and small Pd clusters for enhanced hydrogen storage via a spill-over mechanism. Phys Chem Chem Phys 18(48):33, 158–33,170
Rao CN, Rao KK (1964) Effect of temperature on the lattice parameters of some silver-palladium alloys. Can J Phys 42(7):1336–1342
Rossi G, Ferrando R (2007) Freezing of gold nanoclusters into poly-decahedral structures. Nanotechnology 18(22):225,706
Safaei A (2010) The effect of the averaged structural and energetic features on the cohesive energy of nanocrystals. J Nanoparticle Res 12(3):759–776
Safaei A, Shandiz MA, Sanjabi S, Barber ZH (2008) Modeling the melting temperature of nanoparticles by an analytical approach. The J Phys Chem C 112(1):99–105. https://doi.org/10.1021/jp0744681
Schebarchov D, Hendy S (2006) Solid-liquid phase coexistence and structural transitions in palladium clusters. Physical Review B 73(12):121,402
Schmidt M, Kusche R, von Issendorff B, Haberland H (1998) Irregular variations in the melting point of size-selected atomic clusters. Nature 393(6682):238–240
Sheng HW, Kramer MJ, Cadien A, Fujita T, Chen MW (2011) Highly optimized embedded-atom-method potentials for fourteen fcc metals. Phys Rev B 83(13):134,118. https://doi.org/10.1103/PhysRevB.83.134118
Shim JH, Lee BJ, Cho YW (2002) Thermal stability of unsupported gold nanoparticle: a molecular dynamics study. Surf Sci 512(3):262–268
Steinhardt PJ, Nelson DR, Ronchetti M (1983) Bond-orientational order in liquids and glasses. Phys Rev B 28(2):784
Stukowski A (2009) Visualization and analysis of atomistic simulation data with OVITO–the open visualization tool. Model Simul Mater Sci Eng 18(1):015,012
Tsuzuki H, Branicio PS, Rino JP (2007) Structural characterization of deformed crystals by analysis of common atomic neighborhood. Comput Phys Commun 177(6):518–523
Tyson WR, Miller WA (1977) Surface free energies of solid metals: estimation from liquid surface tension measurements. Surf Sci 62(1):267–276
Vanselow R, Howe RF (1988) Chemistry and physics of solid surfaces VII, vol 10. Springer, Berlin
Verlet L (1967) Computer “experiments” on classical fluids. I. Thermodynamical properties of lennard-Jones molecules. Phys Rev 159(1):98
Wang W, Xu J, Zhao Y, Qi G, Wang Q, Wang C, Li J, Deng F (2017) Facet dependent pairwise addition of hydrogen over Pd nanocrystal catalysts revealed via NMR using para-hydrogen-induced polarization. Phys Chem Chem Phys 19(14):9349–9353
Waseda Y (1980) The structure of non-crystalline materials: liquids and amorphous solids. McGraw-Hill International Book Co., New York
Westergren J, Nordholm S (2003) Melting of palladium clusters–density of states determination by Monte Carlo simulation. Chem Phys 290(2):189–209
Zhang H, Douglas JF (2013) Glassy interfacial dynamics of Ni nanoparticles: part I colored noise, dynamic heterogeneity and collective atomic motion. Soft matter 9(4):1254–1265
Zhang M, Efremov MY, Schiettekatte F, Olson EA, Kwan AT, Lai SL, Wisleder T, Greene JE, Allen LH (2000) Size-dependent melting point depression of nanostructures: nanocalorimetric measurements. Phys Rev B 62(15): 10,548
Zhang Y, Wen YH, Zhu ZZ, Sun SG (2010) Structure and stability of fe nanocrystals: an atomistic study. The J Phys Chem C 114(44):18,841–18,846
Zhao SJ, Wang SQ, Cheng DY, Ye HQ (2001) Three distinctive melting mechanisms in isolated nanoparticles. The J Phys Chem B 105(51):12,857–12,860
Zhivonitko VV, Skovpin IV, Crespo-Quesada M, Kiwi-Minsker L, Koptyug IV (2016) Acetylene oligomerization over Pd nanoparticles with controlled shape: a parahydrogen-induced polarization study. The J Phys Chem C 120(9):4945–4953. https://doi.org/10.1021/acs.jpcc.5b12391
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
The authors declare that they have no conflict of interest.
Electronic supplementary material
Below is the link to the electronic supplementary material.
(MP4 5.49 MB)
(MP4 2.89 MB)
(MP4 5.45 MB)
Rights and permissions
About this article

Cite this article
Kateb, M., Azadeh, M., Marashi, P. et al. Size and shape-dependent melting mechanism of Pd nanoparticles. J Nanopart Res 20, 251 (2018). https://doi.org/10.1007/s11051-018-4355-7
Received:
Accepted:
Published:
DOI: https://doi.org/10.1007/s11051-018-4355-7