
Abstract
Hepatocellular carcinoma (HCC) is the most common type of liver malignant tumor, which is often diagnosed in advanced stages, resulting in low survival rate. The sensitive diagnosis of early HCC presents a great interest. Herein, a novel superparamagnetic contrast agent composed of iron oxide nanoparticles is reported. Firstly, polyethyleneimine-coated iron oxide (Fe3O4@PEI) nanoparticles (NPs) were synthesized via a mild reduction route, followed by their modification of polyethylene glycol monomethyl ether (mPEG-COOH) via 1-ethyl-3-(3-(dimethylamino)propyl) carbodiimide hydrochloride coupling chemistry. After acetylation of the remaining PEI amines, the PEGylated Fe3O4 (Fe3O4@PEI.Ac-mPEG-COOH) NPs were successively characterized via different techniques. The Fe3O4@PEI.Ac-mPEG-COOH probes with an Fe3O4 NP size of 9 nm are water dispersible and cytocompatible within the given concentration range. The percentages of PEI and m-PEG-COOH on the particles surface are calculated to be 15.5 and 7.2%, respectively. Prior to the administration of Fe3O4@PEI.Ac-mPEG-COOH NPs of ultrahigh r 2 relaxivity (461.29 mM−1 s−1) via tail intravenous injection for MR imaging of HCC, the orthotopic model of HCC was established in the nude mice by surgical transplantation with HCCLM3 cells. The analysis of MR signal intensity (SI) in the orthotopic tumor model demonstrated that the developed Fe3O4@PEI.Ac-mPEG-COOH NPs were able to infiltrate into the tumor area through the enhanced permeability and retention (EPR) effect reaching the bottom at 2 h postinjection. The developed Fe3O4@PEI.Ac-mPEG-COOH NPs may be further applied for theranostics of different diseases through combing various therapeutic agents.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Hepatocellular carcinoma (HCC) is acknowledged as a typical type of liver cancer worldwide. Meanwhile, it is hard to be diagnosed early and cured in time, leading to patients’ low survival rate (Liu et al. 2011; Villanueva and Llovet 2011). Therefore, building a tumor model in corresponding environment to imitate the physiological conditions is crucial for accurately studying the mechanism of HCC. In this case, the accurate diagnosis of orthotopic HCC at an early stage will have more practical meanings through the operation of advanced molecular imaging techniques.
Magnetic resonance imaging (MRI) has the merits of non-ionizing radiation, high sensitivity, good spatial resolution, and offering tomographic information and three-dimensional detail of the soft tissue (Krishnamurthy et al. 2015; Parker et al. 2015; Pierre et al. 2015; Riederer et al. 2015). Nowadays, enhanced MR scan is used for the differential diagnosis of benign and malignant disease and the commercially available MR contrast agents used in clinical settings for cancer diagnosis are generally T 1 contrast media (Gd or Mn/chelator complexes) (Salaa et al. 2010). However, with respect to the short blood circulation time and potential renal toxicity of Gd/chelator complexes, the application of T 2 contrast agents is expected to provide additional pathological information in cancer diagnosis. In recent years, magnetic iron oxide (Fe3O4 and Fe2O3) nanoparticles (NPs) with excellent biocompatibility have gained great attention in various biomedical applications (Chen et al. 2010; Cheng et al. 2012; Guo and Shi 2012; Pan et al. 2007), particularly in MR imaging of cancer due to their high r 2 relaxivity (Krishnamurthy et al. 2015; Kumar et al. 2010). As is well known, the model of orthotopic transplanted tumors can approach the real physiopathologic processes better than the model of subcutaneous transplanted tumors. Herein, the iron oxide contrast agents for T 2-weight MR imaging has been employed in the orthotopic tumor model of HCC.
Fe3O4 NPs are liable to aggregate because of their inherent magnetic property and large surface energy (Krishnamurthy et al. 2015; Kumar et al. 2010). Likewise, the Fe3O4 NPs modified with reactive surface groups have an ability to be further functionalized to have improved colloidal stability and biocompatibility and to have specificity for MR imaging of different biological systems. A prior work has demonstrated that polyethyleneimine (PEI) can be used as an effective stabilizer to synthesize PEI-coated Fe3O4 (Fe3O4@PEI) NPs (Cai et al. 2013; Li et al. 2014). Hence, the synthesis of stable Fe3O4@PEI NPs with ultrahigh r 2 relaxivity is available via a mild reduction route. It is also known that PEGylation of NPs can alleviate the uptake of NPs by the reticuloendothelial system (RES) and prolong their blood circulation time (Peng et al. 2012; Wen et al. 2013). Furthermore, it has been demonstrated that NPs can be retained in tumor sites via a passive targeting strategy based on the enhanced permeability and retention (EPR) effect in the tumor tissues with leaky vasculature and poor lymphatic drainage (Barreto et al. 2011; Maeda 2010; Minelli et al. 2010).
In this work, Fe3O4@PEI NPs were synthesized via a mild reduction route, according to the previous work (Hu et al. 2015; Li et al. 2015). Then, the NPs were modified with polyethylene glycol monomethyl ether (mPEG-COOH) via 1-ethyl-3-(3-(dimethylamino)propyl) carbodiimide hydrochloride (EDC) coupling chemistry. After acetylation of the remaining PEI surface amines, PEGylated Fe3O4 NPs (Fe3O4@PEI.Ac-mPEG) were prepared and characterized via different techniques. Then, their cytotoxicity was evaluated by 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT) viability assay and cell morphology observation. Furthermore, the developed Fe3O4@PEI.Ac-mPEG NPs were used for MR imaging of HCCLM3 cells in vitro and the orthotopic HCC tumor model in vivo. The developed Fe3O4@PEI.Ac-mPEG NPs are expected to become a nanoplatform for MR imaging of various tumors.
Experimental
Materials
PEG monomethyl ether with terminal end of carboxyl group (mPEG-COOH, Mw = 2000) was acquired from Shanghai Yanyi Biotechnology Corporation (Shanghai, China). EDC and N-hydroxysuccinimide (NHS) were purchased from J&K Chemical Ltd. (Shanghai, China). Ferric chloride hexahydrate (FeCl3·6H2O > 99%), branched PEI (Mw = 25,000), ammonia (25–28%), triethylamine, acetic anhydride, sodium sulfite, dimethyl sulfoxide (DMSO), and all the other chemicals and solvents were obtained from Sigma-Aldrich (St. Louis, MO). All the chemicals were used without further purification. MTT was acquired from Shanghai Sangon Biological Engineering Technology & Services Co., Ltd. (Shanghai, China). HCCLM3 cells (a HCC cell line) were from Liver Cancer Institute, Zhongshan Hospital, Fudan University (Shanghai, China). Dulbecco’s modified Eagle’s medium (DMEM), fetal bovine serum (FBS), penicillin, and streptomycin were obtained from Hangzhou Jinuo Biomedical Technology (Hangzhou, China). The water employed in all experiments was purified from a Milli-Q Plus 185 water purification system with resistivity higher than 18.2 MΩ cm (Millipore, Bedford, MA).
PEGylated Fe3O4@PEI NPs
Fe3O4@PEI NPs were synthesized via a mild reduction route (Hu et al. 2015; Li et al. 2015). Briefly, FeCl3·6H2O dissolved in water was placed into a 250-mL three-necked flask and then vigorously stirred in N2 atmosphere for 15 min. And then, the prepared sodium sulfite solution was added slowly in it. After 30 min, PEI and ammonia were successively added in it. The reaction mixture was vigorously stirred for 30 min at 60∼70 °C and then for another 1.5 h at room temperature. The Fe3O4@PEI NPs were gained after necessary magnetically collection, washing by water, and centrifugation (8000 rpm, 10 min) to remove the aggregated larger particles. A total of 10 mL of Fe3O4@PEI NPs (80 mg) aqueous solution was washed with DMSO for three times and then dissolved into 10 mL DMSO. The Fe3O4@PEI NPs were conjugated with mPEG according to protocols reported in the literature (Hu et al. 2015). The DMSO solution of PEG preactivated by NHS and EDC was added into the aforementioned Fe3O4@PEI solution to form the raw Fe3O4@PEI-mPEG-COOH product after vigorous magnetic stirring for 1 day. The remaining PEI amines were then acetylated according to the literature (Li et al. 2015). Briefly, triethylamine was added into the aforementioned Fe3O4@PEI-mPEG-COOH solution under vigorous vibrating at room temperature. After 30 min, acetic anhydride was added into the above mixture solution and the reaction was continued for 1 day. After 3 cycles of magnetic separation/washing/redispersion, the formed Fe3O4@PEI.Ac-mPEG-COOH NPs were obtained.
Although the complete acetylation is unable to screen the positive surface potential of the particles due to the nature of the acetylation reaction, it is believed that the steric hindrance of the PEG spacer is able to largely avoid the electrostatic interaction-based non-specific cellular uptake of the particles (Cai et al. 2013). Finally, the formed PEGylated Fe3O4 (Fe3O4@PEI.Ac-mPEG-COOH) NPs were either dispersed in 10 mL water or lyophilized to get dry powder for further use.
Characterization techniques
Dynamic light scattering (DLS) and zeta potential were performed using a Malvern Zetasizer Nano ZS model ZEN3600 (Worcestershire, UK) with a standard 633-nm laser. Prior to measurements, samples were dispersed in water (0.1 mg/mL). The organic component percentage of samples was determined by thermal gravimetric analysis (TGA) using a TG 209 F1 (NETZSCH Instruments Co., Ltd., Selb/Bavaria, Germany) thermogravimetric analyzer. The analysis process was operated under flowing N2 atmosphere with a setting procedure (temperature range 25–700 °C, heating rate 10 °C/min). The morphology of the NPs was characterized by transmission electron microscopy (TEM, JEOL 2010F analytical electron microscope, Tokyo, Japan) with an accelerating voltage of 200 kV. Samples were prepared by depositing a diluted NPs suspension (6 μL) onto a carbon-coated copper grid and dried in air before measurements. TEM images were randomly selected and the average size and size distribution were analyzed using an ImageJ software (National Institutes of Health, Bethesda, MD). At least 300 NPs were countered and analyzed. The Fe concentration in the NPs suspension was determined by Leeman Prodigy inductively coupled plasma-optical emission spectroscopy (ICP-OES, Hudson, NH). Transverse relaxation time, T 2, of NPs was measured by using a 0.5-T NMI20-Analyst nuclear magnetic resonance (NMR) analyzing system (Shanghai Niumag Corporation, Shanghai, China). Samples were diluted in water with Fe concentration in the range of 0.0025–0.04 mM. The acquisition parameters of T 2-weighted sequence were set as follows: TR = 5800 ms, TE = 60 ms, FOV = 60 mm × 60 mm, echo train length = 1, echo spacing = 23.5 ms, slice thickness = 0.6 mm, and matrix = 156 × 156. T 2 relaxivity (r 2) was determined by linearly fitting 1/T 2 (s−1) as a function of Fe concentration (mM).
Cytotoxicity assay and cell morphology observation
HCCLM3 cells were routinely cultured and passaged in 25 cm2 plates in DMEM supplemented with 10% heat-inactivated FBS, penicillin (100 U/mL), and streptomycin (100 μg/mL) at 37 °C and 5% CO2.
The MTT viability assay was carried out to evaluate in vitro cytotoxicity of the PEGylated Fe3O4 NPs under a standard manufacturer instruction (Cai et al. 2013; Shen et al. 2012). Briefly, HCCLM3 cells were seeded into a 96-well plate with 200 μL of DMEM at a density of 1 × 104 cells per well. After incubation overnight to allow the cells to become about 75% confluence, the adherent cells were cultured with 200-μL fresh medium containing PBS (control) and PEGylated Fe3O4 NPs in an Fe concentration range of 0.25–2.0 mM for an additional 24 h, followed by adding 20 μL MTT solution (5 mg/mL in PBS) into each well. The HCCLM3 cells were then incubated for another 4 h at 37 °C. After that, the mixture was carefully discarded and replaced by 200 μL DMSO to dissolve the insoluble formazan crystals. Finally, a Thermo Scientific Multiskan MK3 enzyme-linked immunosorbent assay (ELISA) reader (Thermo scientific, Hudson, NH) was employed to record the absorbance of each well at 570 nm. Mean and standard deviation (SD) of five samples for each experimental group were reported. To further assess the cytotoxicity of PEGylated Fe3O4 NPs, the morphology of HCCLM3 cells treated with the particles at different Fe concentrations ranging from 0.25 to 2.0 mM for 24 h was observed by phase contrast microscopy (Leica DM IL LED inverted phase contrast microscope, Leica Microsystems, Wetzlar, Germany) with a magnification of ×200 for each sample.
In vitro MR imaging of HCCLM3 cells
HCCLM3 cells with 2 mL DMEM at a density of 1 × 105 per well were seeded into 24-well plate and cultured overnight at 37 °C and 5% CO2. Then, the adherent cells were cultured in a 2-mL fresh medium containing PBS (control) and PEGylated Fe3O4 NPs at different Fe concentrations (0.1, 0.2, 0.4 and 0.8 mM, respectively). After that, the cells incubated at 37 °C and 5% CO2 for an additional 6 h were washed with PBS for five times, trypsinized, centrifuged, and resuspended in 1 mL of PBS (containing 0.5% agarose) in a 2-mL of Eppendorf tube before MR imaging. T 2-weighted MR imaging of the cell suspension was conducted by a 3.0-T Verio MR system (Siemens Healthcare, Munich, Germany) with TR/TE = 2500/69 ms, FOV = 60 mm × 60 mm, matrix = 416 × 416, and slice = 1.5 mm.
In vivo MR imaging of orthotopic HCC tumor model
All animal experiments were carried out in accordance with protocols approved by the ethical committee of Zhongshan Hospital. To establish the orthotopic HCC tumor models, HCCLM3 cells (1 × 107) were subcutaneously inoculated into the right flanks of 6-week-old BALB/c nu/nu male mice (Shanghai Slac Laboratory Animal Center, Shanghai, China). The HCCLM3 cell line was established at Liver Cancer Institute of Fudan University from the lung metastatic lesions of BALB/c nude mice bearing human hepatocellular carcinoma (HCC) from the metastatic HCC cell line MHCC97-H. It has been shown to have a high potential for lung metastases and extensive metastases when the cells are inoculated subcutaneously or orthotopically in nude mice.(Yang et al. 2005) The HCCLM3 cell line is very suitable to establish the orthotopic tumor model of HCC because of the high metastasis and little necrotic tissue.(Li et al. 2004) After 3–4 weeks, non-necrotic tumor tissue was cut into 1-mm3 pieces and orthotopically implanted into the livers of other 6-week-old BALB/c nu/nu male mice. After 3–4 weeks, the mice were anesthetized via intraperitoneal injection of pentobarbital sodium solution (2%, 0.1 mL), and PEGylated Fe3O4 NPs ([Fe] = 500 μg, 20 mg/kg, 0.1 mL in PBS) were injected into each mouse via the tail vein. After placing the mice into a wrist receiver coil, MR images of the mice at different time points (0.5, 1.0, 2.0, 4.0, and 6.0 h) postinjection were obtained using a 3.0-T Verio MR system under the same parameters for MR imaging of cells in vitro. n = 3 for each time point.
Histology studies
For histology studies, the PEGylated Fe3O4 NPs were delivered to tumor-bearing mice ([Fe] = 500 μg, 20 mg/kg, 0.1 mL PBS for each mouse) via the tail vein. The major organs of mouse (heart, liver, liver tumor, spleen, lung, and kidney) were harvested at 15 days postinjection, and then all organs were fixed in formaldehyde, embedded in paraffin, sectioned, and stained with hematoxylin and eosin (H&E) according to protocols described in the literature (Peng et al. 2013). Organs from healthy mouse were used as control. Finally, the sections were imaged with a phase contrast microscope (Leica DM IL LED inverted phase contrast microscope) with a magnification of ×200 for each sample.
In vivo biodistribution
To assess the biodistribution of the Fe3O4@PEI-mPEG-COOH NPs, HCCLM3 tumor-bearing mice were euthanized after intravenous injection of Fe3O4@PEI-mPEG-COOH NPs ([Fe] = 500 μg, 20 mg/kg, 0.1 mL in PBS) via tail vein at 24 h postinjection, and the heart, liver, spleen, lung, kidney, and tumor were extracted and weighed. The organs were then cut into small pieces and digested by aqua regia (nitric acid/hydrochloric acid, v/v = 1:3) for 1 day. The tumor-bearing mice without injection were used as control, and the Fe content in different organ pieces was determined by ICP-OES. For each experiment, a group of 3 mice was used to assess the mean and standard deviation of the data.
Statistical analysis
Student-Newman-Keuls test and one-way ANOVA were performed to evaluate the significance of the experimental data. A p value of 0.05 was selected as the significance level, and the data were indicated with one asterisk for p < 0.05, two asterisks for p < 0.01, and three asterisks for p < 0.001, respectively.
Results and discussion
Synthesis and characterization of PEGylated Fe3O4 NPs
In this study, Fe3O4@PEI NPs were synthesized via a mild reduction route according to the previous work (Hu et al. 2015; Li et al. 2015). The aminated PEI-enabled Fe3O4 NPs were modified with mPEG-COOH. After acetylation of the remaining PEI surface amines, the PEGylated Fe3O4 NPs were obtained (Scheme 1).

Schematic representation of the synthesis of the Fe3O4@PEI.Ac-mPEG-COOH NPs
Various techniques were employed to characterize the intermediate products and the prepared PEGylated Fe3O4 NPs. To quantify the percentage of PEI coating, TGA was carried out (Fig. 1). Obviously, the Fe3O4@PEI NPs had a weight of 84.5% at the temperature of 700 °C (Fig. 1, curve a). As naked Fe3O4 NPs do not show any significant weight loss (Li et al. 2015), the percentage of PEI coated on the particle surface was calculated to be 15.5%. Next, the prepared Fe3O4@PEI NPs were conjugated with mPEG-COOH via EDC coupling chemistry, and the degree of PEGylation was also quantitatively analyzed by TGA. According to the previous research results (Cai et al. 2013; Li et al. 2015), the initial weight loss of Fe3O4 is 0. Because of the thorough loss of PEI and PEG without carbonaceous mass remaining (Luo et al. 2015), clearly, after further modification with mPEG-COOH, the weight loss of Fe3O4@PEI-mPEG-COOH NPs was determined to be 22.7% (Fig. 1, curve b). Thus, the mPEG-COOH modified onto the particle surface was calculated to be 7.2%.

TGA curves of the Fe3O4@PEI (a) and Fe3O4@PEI-mPEG-COOH NPs (b)
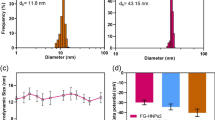
Zeta potential and hydrodynamic size of products synthesized by each step surface modification were recorded (Table 1). Obviously, the Fe3O4@PEI-COOH NPs have a quite positive surface potential of +44.8 mV due to the existence of numerous PEI amines. After modification of mPEG-COOH, the surface potential of Fe3O4@PEI-mPEG-COOH NPs displayed a slight decrease (+42.5 mV). To impart them with improved cytocompatibility as well as to avoid non-specific cell membrane binding, the positive charges of Fe3O4@PEI-mPEG-COOH NPs are needed to be further neutralized. Apparently, the surface potential of the formed Fe3O4@PEI.Ac-mPEG-COOH NPs dramatically decreased (+18.7 mV), suggesting the successful acetylation reaction. It should be mentioned that the positive surface charges of the Fe3O4@PEI.Ac-mPEG-COOH NPs were unable to be completely neutralized via acetylation reaction due to the fact that a number of PEI amines used to stabilize the Fe3O4 NPs are unable to be acetylated (Hu et al. 2015; Li et al. 2013). The hydrodynamic size of the formed NPs dispersed in ultrapure water was checked by dynamic light scattering (DLS) (Table 1). Clearly, the Fe3O4@PEI, Fe3O4@PEI-mPEG-COOH, and Fe3O4@PEI.Ac-mPEG-COOH NPs displayed a hydrodynamic size of 220.4, 231.1, and 242.9 nm, respectively. In order to evaluate the stability of these NPs, we measured the hydrodynamic size of the NPs again after 7 days of storage at room temperature just like the previously study (Cai et al. 2015). The hydrodynamic diameter of the NPs was 256.5 nm, which was not significantly different from the value observed before storage.
The morphology and size of the prepared PEGylated Fe3O4 NPs were characterized by TEM (Fig. 2). Obviously, the majority of the NPs have a quasi-spherical shape (Fig. 2a) and exhibit a narrow size distribution with a mean diameter of 9.0 ± 2.1 nm (Fig. 2b). The measured size by TEM is smaller than that measured by DLS, which can be attributed to the fact that TEM reveals the size of a single core NP, while DLS characterizes the size of NP clusters in aqueous solution (Calatayud et al. 2014; Liu et al. 2013).

TEM micrographs (a) and size distribution histogram (b) of the PEGylated Fe3O4 NPs
FTIR spectroscopy was next applied to qualitatively demonstrate the PEI coating on the surfaces of the Fe3O4@PEI-mPEG-COOH NPs (Fig. 3). Coated by PEI, the Fe3O4@PEI NP spectrum shows very strong bands at 3450 and 1630 cm−1, however, that naked Fe3O4 NPs only exhibit weak bands at 3450 and 1630 cm−1 (Cai et al. 2013; Li et al. 2015). Moreover, the strong absorption bands at 2930 and 2850 cm−1 in the spectrum of the Fe3O4@PEI NPs should be due to the −CH2− groups of PEI. The FTIR spectroscopic results qualitatively confirmed the successful coating of PEI on the surface of Fe3O4 NPs.

FTIR spectroscopy demonstration of the PEI coating on the surfaces of the Fe3O4@PEI-mPEG-COOH NPs
T 2 relaxivity measurements
From the T 2-weighted MR images (Fig. 4a), it can be clearly seen that PEGylated Fe3O4 NPs are able to decrease the MR signal intensity with the increase of Fe concentration. By linear fitting of the relaxation rate (1/T 2) versus the Fe concentration, the r 2 relaxivity of the PEGylated Fe3O4 NPs was calculated to be 461.29 mM−1 s−1 (Fig. 4b). Fe3O4 NPs prepared via the mild reduction route display a much higher r 2 value than those synthesized via co-precipitation method, hydrothermal route, or thermal decomposition approach (Cai et al. 2012; Shen et al. 2012; Yang et al. 2011), indicating their great potential for T 2-weighted MR imaging. The ultrahigh r 2 relaxivity should be due to the advantage of the mild reduction method, allowing for the generation of Fe3O4 NPs with high magnetic dipole interactions, in agreement with the literature (Hu et al. 2015).

T 2-weighted MR image (a) and linear fitting of 1/T 2 (b) of the PEGylated Fe3O4 NPs at an Fe concentration of 0.0025, 0.005, 0.01, 0.02, and 0.04 mM, respectively
In vitro cytotoxicity assay
However, experimental evidence in vitro has shown that the toxicity of iron oxide NPs (Mahmoudi et al. 2011). Then, in vitro cytotoxicity of the PEGylated Fe3O4 NPs was evaluated by the MTT cell viability assay (Fig. 5). Apparently, the viability of the HCCLM3 cells is higher than 80% after the treatment of PEGylated Fe3O4 NPs at different Fe concentrations (0.25–2.0 mM) for 24 h, confirming their good cytocompatibility. Furthermore, the cytocompatibility of the PEGylated Fe3O4 NPs was evaluated using phase contrast microscope to visualize the morphologies of cells treated with NPs at different Fe concentrations ranging from 0.25 to 2.0 mM (Fig. 6). Obviously, PEGylated Fe3O4 NPs do not change the morphologies of HCCLM3 when compared with those of control cells treated with PBS (Fig. 6a). The cell morphology observation result corroborates the aforementioned data of the MTT assay, indicating that the PEGylated Fe3O4 NPs are cytocompatible in the studied Fe concentration range. However, the iron oxide nanoparticles are having their toxicity when they are higher than the 100 mg/L, and no cytotoxic effect was found in the studied Fe concentration range. We can include a point that it’s a dependent cytotoxicity of the iron oxide nanoparticles (Parivar et al. 2016).

MTT viability assay of HCCLM3 cells after treatment with PBS (control) and PEGylated Fe3O4 NPs in an Fe concentration range of 0.25–2.0 mM for 24 h

Phase contrast microscopic images of HCCLM3 cells treated with PBS (a) and PEGylated Fe3O4 NPs at an Fe concentration of 0.25 (b), 0.5 (c), 1.0 (d), 1.5 (e), and 2.0 mM (f), respectively for 24 h. The scale bar in each panel represents 100 μm
MR imaging of cancer cells in vitro
With the confirmed ultrahigh r 2 relaxivity and excellent cytocompatibility, the PEGylated Fe3O4 NPs were next investigated to be used for MR imaging of cancer cells in vitro. Figure 7a shows the T 2-weighted MR images of HCCLM3 cells treated with the PEGylated Fe3O4 NPs at different Fe concentrations (0.1, 0.2, 0.4, and 0.8 mM, respectively). Obviously, a prominent decreasing trend of MR signal intensity of HCCLM3 cells is shown with the increasing Fe concentration, which is ascribed to the increasing cellular uptake of the NPs (Li et al. 2015). Furthermore, the decreasing trend was further verified by quantitatively analyzing the MR signal intensity of cell images (Fig. 7b). Apparently, the measured MR signal intensity of the HCCLM3 cells treated with the PEGylated Fe3O4 NPs is in a concentration-dependent manner, and the signal intensity of HCCLM3 cells treated with the NPs at a higher concentration is much lower than that of HCCLM3 cells treated with the NPs at a lower concentration. These results suggest that the formed PEGylated Fe3O4 NPs can be uptaken by HCCLM3 cells and enable MR imaging of HCCLM3 cells in vitro.

T 2-weighted MR imaging (a) and MR signal intensity analysis (b) of HCCLM3 cells treated with PBS and PEGylated Fe3O4 NPs at an Fe concentration of 0.1, 0.2, 0.4, and 0.8 mM, respectively, for 6 h
In vivo MR imaging of orthotopic HCC tumor model
Then, the developed PEGylated Fe3O4 NPs were utilized for in vivo MR imaging of an orthotopic HCC tumor model. Figure 8a reveals the contrast enhancement of T 2-weighted MR image of tumors before and after the treatment of the PEGylated Fe3O4 NPs. Apparently, from the time point of before injection to the time point of 2 h postinjection, the MR signals of tumors gradually decrease. This is attributed to the fact that the PEGylated NPs are able to be accumulated into the tumor region via the EPR effect. The MR signal intensity of tumors becomes the weakest at 2 h postinjection. At 4 h postinjection, the NPs begin to be metabolized, leading to the recovery of the MR signal. The quantitative MR signal intensity versus the time points of postinjection of the NPs (Fig. 8b) reveals that the tumor MR signal intensity becomes the lowest at 2 h postinjection and slightly recovers at 4 h postinjection. By statistical analysis, the MR signal intensity at 2 h postinjection is significantly lower than that at 0.5 and 6 h postinjection, but the comparisons with that at 1 or 4 h postinjection show no difference. Throughout these times, the MR signal intensity of the normal liver tissue is significantly lower than that of the tumor tissue. These results demonstrate that the developed Fe3O4@PEI.Ac-mPEG-COOH NPs are able to infiltrate into the tumor area through the EPR effect reaching the bottom at 2 h postinjection, and the MR signal intensity of the normal liver tissue is obviously lower than that of the tumor tissue because of the phagocytosis of Kupffer cells. When the lesions are highlighted via T 2-weighted negative contrast enhanced MR imaging, the biological characteristics (blood supply pattern) of HCC are also represented by the developed Fe3O4@PEI.Ac-mPEG-COOH NPs.

In vivo MR imaging (a) and signal intensity analysis (b) of tumors (asterisk) after intravenous injection of the PEGylated Fe3O4 NPs (500 μg Fe, 0.1 mL PBS for each mouse) at different time points postinjection
Histology examinations
An MTT cell viability assay had been applied to evaluate the cytotoxicity of the formed Fe3O4@PEI.Ac-mPEG-COOH NPs in the previous study (Hu et al. 2015), so in this study, we had not repeated it. Histology examinations were performed to assess the potential in vivo toxicity of the PEGylated Fe3O4 NPs. As shown in Fig. 9, the cell structure and morphology of all organs of the mouse treated with the PEGylated Fe3O4 NPs exhibit no apparent damages compared with those of the corresponding organs acquired from the control mice without treatment. This confirms that the injected PEGylated Fe3O4 NPs do not exert any obvious in vivo toxicity to the mouse and, hence, hold great potential to be used for tumor MR imaging applications.

H&E stained tissue sections from the mice at 15 days postintravenous injection of the PEGylated Fe3O4 NPs ([Fe] = 500 μg, 0.1 mL PBS for each mouse) and the mouse without treatment (control). The scale bar in each panel represents 100 μm
In vivo biodistribution
To evaluate the biodistribution of the Fe3O4@PEI-mPEG-COOH NPs, ICP-OES was performed to analyze the Fe concentration in several major organs including heart, liver, spleen, lung, kidney, and tumor (Fig. 10). It is clear that after 24 h postinjection of Fe3O4@PEI-mPEG-COOH NPs, the Fe concentration in all the organs and tumor tissue of the tumor-bearing mice is higher than that of the control mice without injection. But only the Fe concentration in spleen and liver is significantly higher. The majority of the Fe is uptaken by the spleen and liver, i.e., preferentially accumulation occurs in liver and spleen (Edge et al. 2016), and a quite small amount of Fe remains in the other organs, such as heart, lung, kidney, and tumor. The Fe accumulation in spleen and liver is typical due to the clearance effect of reticuloendothelial system (RES) located in these organs (Hanini et al. 2011; Kumar et al. 2010). The Fe content determined in main organs of control mice group is ascribed to that Fe element is one of the essential elements in the living body.

Biodistribution of the major organs and tissue of the mice, i.e., heart, liver, spleen, lung, kidney, and tumor, at 24 h postintravenous injection of a PBS solution containing the PEGylated Fe3O4 NPs (500 μg Fe, 0.1 mL PBS)
Conclusion
In summary, PEGylated Fe3O4 NPs with an ultrahigh r 2 relaxivity were prepared via a mild reduction route for T 2-weighted MR imaging of an orthotopic tumor model of HCC. The mild reduction endowed the synthesized Fe3O4@PEI NPs to be covalently conjugated with mPEG-COOH via EDC coupling chemistry. The thus formed PEGylated Fe3O4 NPs possessed good water dispersibility and cytocompatibility in the given Fe concentration range. No hematology studies were performed because the hemolysis assay of the same nanoparticles had been carried out, suggesting their negligible hemolytic activity (Hu et al. 2015; Li et al. 2013). Therefore, the developed PEGylated Fe3O4 NPs could be used for MR imaging of HCCLM3 cells in vitro and an orthotopic tumor model of HCC in vivo with the proven ultrahigh r 2 relaxivity (461.29 mM−1 s−1). Given the PEI amine-enabled conjugation chemistry, the PEGylated Fe3O4 NPs may be further integrated with some anticancer drugs into one unique nanoplatform for theranostic of various types of cancers.
References
Barreto JA, O’Malley W, Kubeil M, Graham B, Stephan H, Spiccia L (2011) Nanomaterials: applications in cancer imaging and therapy. Adv Mater 23:H18–H40. doi:10.1002/adma.201100140
Cai H et al (2012) Facile assembly of Fe3O4@Au nanocomposite particles for dual mode magnetic resonance and computed tomography imaging applications. J Mater Chem 22:15110–15120
Cai H et al. (2013) Facile hydrothermal synthesis and surface functionalization of polyethyleneimine-coated iron oxide nanoparticles for biomedical applications ACS Appl Mater Interfaces 5:1722–1731
Cai H et al. (2015) Dendrimer-assisted formation of Fe3O4/Au nanocomposite particles for targeted dual mode CT/MR imaging of tumors Small 11:4584–4593
Calatayud MP, Sanz B, Raffa V, Riggio C, Ibarra MR, Goya GF (2014) The effect of surface charge of functionalized Fe3O4 nanoparticles on protein adsorption and cell uptake Biomaterials 35:6389–6399 doi:10.1016/j.biomaterials.2014.04.009
Chen Y, Chen H, Zeng D, Tian Y, Chen F, Feng J, Shi J (2010) Core/shell structured hollow mesoporous nanocapsules: a potential platform for simultaneous cell Imaging and anticancer drug delivery. ACS Nano 4:6001–6013. doi:10.1021/nn1015117
Cheng L, Yang K, Li Y, Zeng X, Shao M, Lee S-T, Liu Z (2012) Multifunctional nanoparticles for upconversion luminescence/MR multimodal imaging and magnetically targeted photothermal therapy. Biomaterials 33:2215–2222. doi:10.1016/j.biomaterials.2011.11.069
Edge D et al (2016) Pharmacokinetics and bio-distribution of novel super paramagnetic iron oxide nanoparticles (SPIONs) in the anaesthetized pig. Clin Exp Pharmacol Physiol 43:319–326. doi:10.1111/1440-1681.12533
Guo R, Shi X (2012) Dendrimers in cancer therapeutics and diagnosis. Curr Drug Metab 13:1097–1109
Hanini A, Schmitt A, Kacem K, Chau F, Ammar S, Gavard J (2011) Evaluation of iron oxide nanoparticle biocompatibility. Int J Nanomedicine 6:787–794. doi:10.2147/IJN.S17574
Hu Y et al (2015) Facile synthesis of RGD peptide-modified iron oxide nanoparticles with ultrahigh relaxivity for targeted MR imaging of tumors. Biomater Sci 3:721–732. doi:10.1039/c5bm00037h
Krishnamurthy R, Hernandez A, Kavuk S, Annam A, Pimpalwar S (2015) Imaging the central conducting lymphatics: initial experience with dynamic MR lymphangiography. Radiology 274:871–878. doi:10.1148/radiol.14131399
Kumar R, Roy I, Ohulchanskky TY, Vathy LA, Bergey EJ, Sajjad M, Prasad PN (2010) In vivo biodistribution and clearance studies using multimodal organically modified silica nanoparticles ACS Nano 4:699–708 doi:10.1021/nn901146y
Li Y et al (2004) Stepwise metastatic human hepatocellular carcinoma cell model system with multiple metastatic potentials established through consecutive in vivo selection and studies on metastatic characteristics. J Cancer Res Clin Oncol 130:460–468. doi:10.1007/s00432-004-0564-9
Li J, Zheng L, Cai H, Sun W, Shen M, Zhang G, Shi X (2013) Polyethyleneimine-mediated synthesis of folic acid-targeted iron oxide nanoparticles for in vivo tumor MR imaging. Biomaterials 34:8382–8392
Li J et al (2014) Hyaluronic acid-modified hydrothermally synthesized iron oxide nanoparticles for targeted tumor MR imaging. Biomaterials 35:3666–3677
Li J et al (2015) Facile synthesis of folic acid-functionalized iron oxide nanoparticles with ultrahigh relaxivity for targeted tumor MR imaging. J Mater Chem B 3:5720–5730. doi:10.1039/c5tb00849b
Liu Y, Chen Z, Liu C, Yu D, Lu Z, Zhang N (2011) Gadolinium-loaded polymeric nanoparticles modified with anti-VEGF as multifunctional MRI contrast agents for the diagnosis of liver cancer. Biomaterials 32:5167–5176. doi:10.1016/j.biomaterials.2011.03.077
Liu H et al (2013) Facile hydrothermal synthesis of low generation dendrimer-stabilized gold nanoparticles for in vivo computed tomography imaging applications. Polym Chem 4:1788–1795. doi:10.1039/c2py20993d
Luo Y, Yang J, Li J, Yu Z, Zhang G, Shi X, Shen M (2015) Facile synthesis and functionalization of manganese oxide nanoparticles for targeted T1-weighted tumor MR imaging. Colloids Surf B Biointerfaces 136:506–513. doi:10.1016/j.colsurfb.2015.09.053
Maeda H (2010) Tumor-selective delivery of macromolecular drugs via the EPR effect: background and future prospects. Bioconjug Chem 21:797–802. doi:10.1021/bc100070g
Mahmoudi M, Laurent S, Shokrgozar MA, Hosseinkhani M (2011) Toxicity evaluations of superparamagnetic iron oxide nanoparticles: cell “vision” versus physicochemical properties of nanoparticles. ACS Nano 5:7263–7276
Minelli C, Lowe SB, Stevens MM (2010) Engineering nanocomposite materials for cancer therapy. Small 6:2336–2357. doi:10.1002/smll.201000523
Pan B et al (2007) Dendrimer-modified magnetic nanoparticles enhance efficiency of gene delivery system. Cancer Res 67:8156–8163. doi:10.1158/0008-5472.can-06-4762
Parivar K, Malekvand FF, Bayat M, Alavian SM, Motavaf M (2016) Evaluation of iron oxide nanoparticles toxicity on liver cells of BALB/c rats. Iran Red Crescent Med J:18
Parker RA, 3rd et al. (2015) MR Imaging of the penis and scrotum radiographics: a review publication of the Radiological Society of North America, Inc 35:1033–1050 doi:10.1148/rg.2015140161
Peng C et al (2012) PEGylated dendrimer-entrapped gold nanoparticles for in vivo blood pool and tumor imaging by computed tomography. Biomaterials 33:1107–1119
Peng C et al (2013) Targeted tumor CT imaging using folic acid-modified PEGylated dendrimer-entrapped gold nanoparticles. Polym Chem 4:4412–4424. doi:10.1039/c3py00521f
Pierre EY, Ma D, Chen Y, Badve C, Griswold MA (2015) Multiscale reconstruction for MR fingerprinting magnetic resonance in medicine doi:10.1002/mrm.25776
Riederer SJ, Haider CR, Borisch EA, Weavers PT, Young PM (2015) Recent advances in 3D time-resolved contrast-enhanced MR angiography. Journal of Magnetic Resonance Imaging: JMRI 42:3–22. doi:10.1002/jmri.24880
Salaa E, Rockall A, Rangarajan D, Kubik-Huch RA (2010) The role of dynamic contrast-enhanced and diffusion weighted magnetic resonance imaging in the female pelvis. Eur J Radiol 76:367–385. doi:10.1016/j.ejrad.2010.01.026
Shen M et al. (2012) Facile one-pot preparation, surface functionalization, and toxicity assay of APTS-coated iron oxide nanoparticles Nanotechnology 23 doi:105601331088/0957–4484/23/10/105601
Villanueva A, Llovet JM (2011) Targeted therapies for hepatocellular carcinoma Gastroenterology 140:1410–1426 doi:10.1053/j.gastro.2011.03.006
Wen S et al (2013) Multifunctional dendrimer-entrapped gold nanoparticles for dual mode CT/MR imaging applications. Biomaterials 34:1570–1580
Yang J et al (2005) Molecular cytogenetic characteristics of the human hepatocellular carcinoma cell line HCCLM3 with high metastatic potential: comparative genomic hybridization and multiplex fluorescence in situ hybridization. Cancer Genet Cytogenet 158:180–183. doi:10.1016/j.cancergencyto.2004.05.010
Yang H et al (2011) Targeted dual-contrast T1-and T2-weighted magnetic resonance imaging of tumors using multifunctional gadolinium-labeled superparamagnetic iron oxide nanoparticles. Biomaterials 32:4584–4593
Acknowledgements
This research is financially supported by the National Natural Science Foundation of China (21273032, 81101150, 81341050, and 81371623), the Sino-German Center for Research Promotion (GZ899), the Program for Professor of Special Appointment (Eastern Scholar) at Shanghai Institutions of Higher Learning, and the Fundamental Research Funds for the Central Universities (EG2015021).
Author information
Authors and Affiliations
Corresponding authors
Ethics declarations
All animal experiments were carried out in accordance with protocols approved by the ethical committee of Zhongshan Hospital.
Competing fnancial interests
The authors declare no competing fnancial interests.
Additional information
Ruizhi Wang, Yong Hu, and Yuchan Yang contributed equally to this work.
Rights and permissions
About this article

Cite this article
Wang, R., Hu, Y., Yang, Y. et al. Using PEGylated iron oxide nanoparticles with ultrahigh relaxivity for MR imaging of an orthotopic model of human hepatocellular carcinoma. J Nanopart Res 19, 39 (2017). https://doi.org/10.1007/s11051-016-3718-1
Received:
Accepted:
Published:
DOI: https://doi.org/10.1007/s11051-016-3718-1