
Abstract
A series of CuO/CeO2 catalysts were prepared using a solvothermal method and a subsequent activation process. The influences of activation temperature (300–600 °C) on physicochemical properties and catalytic behavior of the prepared CuO/CeO2 catalysts have been investigated by XRD, SEM, Raman spectroscopy, S BET measurement, XPS surface analysis, H2-TPR, CO-TPD techniques, and CO oxidation testing. The catalyst activated at 450 °C was found to have the highest catalytic activity, which can be ascribed to its higher dispersion of Cu species, higher concentration of oxygen vacancies, and larger amount of more active lattice oxygen. The lower activity of the CuO/CeO2 catalysts activated at lower (<450 °C) and higher (>450 °C) temperatures are attributed to the weaker interaction between CuO and CeO2, and to the sintering of CuO nanoparticles, respectively.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
As a major air pollutant, CO is emitted from many industrial processes, transportation, and domestic activities. In order to reduce or even eliminate the toxic emission, catalytic oxidation of CO is considered to be the most promising method (Prasad and Rattan 2010; Chuang et al. 2012).
Currently, there are two main types of catalysts for catalytic oxidation of CO: one is the noble metal catalysts such as Pd, Pt, and Au; the other is base metal catalysts such as Cu, Co, and Mn. Although the noble metal catalysts exhibit a very high activity for CO oxidation, the high price and limited availability restrain their large-scale industrial applications (Prasad and Rattan 2010). Hence, more and more attention has been paid to base metal catalysts in order to find an alternative catalytic component to minimize using or even replace the noble metals. Among them, CuO/CeO2 catalyst is considered to be the most effective due to the synergistic effect between CuO and CeO2 (Ayastuy et al. 2009, 2010; Jia et al. 2010, 2012; Zagaynov 2015).
It has been well illustrated that the activity of CuO/CeO2 catalyst for CO oxidation is greatly influenced by its preparation methods (Luo et al. 1997; Tang et al. 2004; Zhang et al. 2002). Currently, supported CuO/CeO2 catalysts are usually prepared by impregnation (Jia et al. 2012; Luo et al. 1997; Tang et al. 2004) and deposition–precipitation (Tang et al. 2004, 2005) methods. Impregnation method possesses the merit of simplicity and convenience (Luo et al. 1997; Manzoli et al. 2005), but the CuO particles over the prepared CuO/CeO2 catalysts tend to aggregate and lead to the poor dispersion of CuO, thus resulting in lower activity for CO oxidation (Tang et al. 2004). The CuO species of samples prepared by deposition–precipitation method are uniformly dispersed, but the preparation process is complex and it is easy for the impurities such as precipitants to get into the catalysts (Tang et al. 2004). Consequently, a variety of methods have been developed to obtain highly active CuO/CeO2 catalyst including solvated metal atom impregnation (Zhang et al. 2002), laser vaporization and controlled condensation (Sundar and Deevi 2006), template method (Luo et al. 2007a; Qi et al. 2012; Zhu et al. 2008), modified sol–gel method (Luo et al. 2007b, c), combustion method (Avgouropoulos and Ioannides 2007), and flame spray pyrolysis (Kydd et al. 2009). However, these methods generally suffer from some harsh conditions, such as the use of bis (toluene) copper complex (Zhang et al. 2002), laser (Sundar and Deevi 2006), surfactants (Luo et al. 2007a; Qi et al. 2012), templates (Zhu et al. 2008), and hyperthermal treatment (Avgouropoulos and Ioannides 2007; Kydd et al. 2009; Luo et al. 2007b, c). So it is highly desirable to explore new simple and effective preparation methods (Qin et al. 2010). Compared with the above methods, the solvothermal process has advantages such as simple preparation and inexpensive operation conditions. Besides, the size and shape of the resulted nanoparticles are easily controlled by adjusting reaction temperature, time, and pH in this process (Lai et al. 2015).
Herein, a series of CuO/CeO2 catalysts were prepared via a solvothermal method using ethylene glycol (EG) as a solvent and a subsequent activation process. The effect of activation temperature on the catalytic performance of the CuO/CeO2 catalysts for CO oxidation was investigated. In order to clearly elucidate the influence of activation temperature, the physicochemical properties of the prepared catalysts were characterized in depth by means of XRD, SEM, Raman, BET surface area measurement, XPS surface analysis, H2-TPR, and CO-TPD techniques. Furthermore, the catalytic performance of the catalysts was correlated with the results of physicochemical characterization.
Experimental
Catalyst preparation
All chemicals were purchased from Shanghai Chemical Reagent Corporation (PR China) and used as received without further purification in this work. CuO/CeO2 catalysts were prepared by a solvothermal method using EG as a solvent and a subsequent activation process. Commercial CeO2 support was pretreated at 120 °C for 4 h before use to remove the impurity adsorbed on the surface. After that, the treated CeO2 was added into an EG solution of Cu(NO3)2·3H2O and then reacted at 180 °C for 3 h in a Teflon-lined autoclave with continuous stirring. The suspension was allowed to cool down to room temperature (RT). The precipitate was separated by filtration, then repeatedly washed with absolute ethanol, and dried at 120 °C for ca. 12 h. Finally, the precursor was activated in a furnace under static air atmosphere at different temperatures (300–600 °C) for 4 h at a rate of 10 °C min−1. The obtained CuO/CeO2 catalysts were named CC-300, CC-400, CC-450, CC-500, and CC-600. The CuO loading in all the catalysts expressed as the CuO/(CuO + CeO2) wt% ratio maintains as constant (10 wt%).
Catalyst characterization
XRD measurements were conducted with a Rigaku Ultima IV using Cu Ka radiation source generated at 30 kV and 25 mA. Two theta angles ranged from 10 to 90° with a speed of 4° per minute. The crystallite size of CuO was determined from full widths at half maxima of CuO (002) and CuO (111) peaks at 35.5° and 38.7°. The cell parameter values were calculated by standard cubic indexation method using the intensity of the CeO2 (111) peak.
The surface morphology was observed by scanning electron microscopy (SEM, FEI, Quanta 200 FEG).
Raman spectra (at 4 cm−1 resolution) was obtained with a DXR-Raman instrument (Thermo Fisher Scientific, American) using the 532 nm exciting line (200 mW beam), five scans for each spectrum.
N2 adsorption–desorption isotherms were obtained at −196 °C on a Micrometrics ASAP-2020 adsorption apparatus, after all samples were degassed under vacuum at 200 °C for 10 h. The specific surface areas were calculated from the linear part of the Brunauer–Emmett–Teller plot.
The chemical states of the elements were investigated by X-ray photoelectron spectroscopy (XPS, kratos Axis Ultra DLD spectrometer), using Al Ká radiation. All the binding energy values were calibrated using C 1 s = 284.6 eV as a reference.
Temperature-programmed reduction with H2 (H2-TPR) was carried out using 10 vol% H2/N2 as a reducing gas in a quartz microreactor. Approximately 0.02 g of a freshly calcined catalyst was placed on top of glass wool in the reactor. The outlet of the reactor was connected to a glass column packed with molecular sieve 5 Å in order to remove the moisture produced from reduction. The flow rate of the reducing gas was kept at 50 mL min−1, and the temperature was raised from 50 to 500 °C at a rate of 10 °C min−1. The consumption of H2 was monitored by a thermal conductivity detector (TCD).
The CO adsorption property was measured by temperature-programmed desorption of CO. The catalyst (0.1 g) was pretreated under He flow (50 mL·min−1) with the temperature ramping from RT to 500 °C at a rate of 10 °C min−1, and then held at 500 °C for 40 min before being cooled down to RT in He flow. The next step was CO adsorption at RT for 30 min, and then the catalyst was swept again with He for 3 h. Lastly, the sample was heated in flowing He (50 mL min−1) up to 500 °C at a rate of 10 °C min−1, while the desorbed species was detected with a quadrupole mass spectrometer (QMS, Balzers OmniStar 200).
Catalytic testing
The catalytic activity for CO oxidation was tested in a stainless steel tube microreactor (i.d. = 6 mm). Typically, 0.2 g of catalyst was placed between two layers of quartz wool. Gas mixtures containing 4 vol. % CO and 10 vol% O2 in N2 were passed through the catalyst bed at a flow rate of 30 mL min−1, corresponding to a space velocity of 9000 mL g−1 h−1. The samples were heated in N2 (50 mL min−1) at a rate of 10 °C min−1 from RT to 200 °C and held at this temperature for 40 min in order to remove possible impurities. After cooling to RT in N2, the feed gas was introduced into the system. The temperature was increased stepwise (by 5–10 °C), and the catalyst was held for about 30 min at each fixed temperature to achieve steady-state activity. In this case, changes in the catalyst temperature due to heat release because of the possible occurrence of exothermic reactions of CO oxidation were not detected. To allow for the detection of CO and CO2 with a flame ionization detector (FID), a methanator was inserted between one GC column and the FID. The gas compositions before and after the reaction were analyzed by an on-line gas chromatography with an FID. The activity was expressed by the conversion of CO.
Results and discussion
Characterization of catalysts
Structural and textural properties
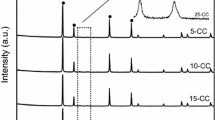
XRD patterns of the CuO/CeO2 catalysts are shown in Fig. 1. All samples exhibit strong diffraction peaks at 2θ = 29.2°, 33.6°, 48.6°, and 57.8°, corresponding to cubic fluorite CeO2 phase (JCPDS 34-0394). The narrow diffraction lines indicate better crystallinity of the CeO2 support. Weak diffraction peaks corresponding to CuO appear at 2θ of 35.5° and 38.7° [JCPDS 41-0254] for all the catalysts, and the intensity of these peaks increases progressively with the increase in the activation temperature. The result indicates that the crystal size of CuO becomes larger as a result of particle sintering during the activation process at higher temperatures (Mai et al. 2011).

XRD patterns of different CuO/CeO2 catalysts
Table 1 shows the crystal sizes of CuO and CeO2 on different CuO/CeO2 catalysts estimated by the Scherrer equation. It can be seen that the crystal sizes of CuO enlarge with the increase in activation temperature from 300 to 600 °C, which can be connected to sample sintering. In addition, the crystal size of CeO2 is essentially constant (39.0–40.6 nm) in all of the catalysts, which can be attributed to the commercial CeO2 used in these experiments through the high-temperature roasting process.
The lattice parameters obtained from the CeO2 (111) plane are also shown in Table 1. It is clear that lattice constriction has happened on all the CuO/CeO2 catalysts comparing with support CeO2, indicating that some Cu2+ ions have been incorporated into the CeO2 lattice to form Cu–Ce–O solid solution in our catalysts (Cao et al. 2008; Guo et al. 2014). From Table 1, on the other hand, it is shown that the lattice parameter of CeO2 varies in an irregular fashion as the activation temperature increases from 300 to 600 °C. This phenomenon can be attributed to the coexistence of two opposite effects (Fu et al. 2015): (1) the substitution of larger Ce4+ (0.097 nm) by smaller Cu2+ (0.072 nm) contracts the CeO2 cell; (2) the formation of oxygen vacancies caused by the aliovalent doping (Ce4+ by Cu2+) or creation of Ce3+ (0.103 nm) sites expands the CeO2 cell.
The SEM images of different CuO/CeO2 samples calcined under different temperatures are shown in Fig. 2. From Fig. 2, we can see that the sizes of the CuO particles on the support surface increase with the increase in calcination temperature from 300 to 600 °C. In addition, the particles gradually agglomerate when the calcination temperature rises from 300 to 600 °C.

SEM images of different samples: a CC-300, b CC-450, and c CC-600
Oxygen vacancies are considered as an indicator for the promotion of CO oxidation. Raman spectroscopy can provide direct evidence of oxygen vacancies and defects owing to changes in the vibrational structure of the CeO2 lattice (Yen et al. 2012). Raman spectra of CeO2 support and different CuO/CeO2 catalysts are shown in Fig. 3. It can be seen that all the samples exhibit a strong vibration band at about 460 cm−1, corresponding to the F 2g Raman vibration mode of fluorite CeO2 (Kydd et al. 2009). Clearly, a red-shift of the F 2g band is observed on all the CuO/CeO2 catalysts compared with support CeO2 (466 cm−1), and the reason may be the presence of oxygen vacancies, which is related to structural defects derived from partial incorporation of Cu2+ into the CeO2 lattice (Gnanakumar et al. 2014). This result is in good agreement with the decrease of lattice parameters obtained by XRD analysis.

Raman patterns of the CeO2 support and different CuO/CeO2 catalysts
More specifically, the F 2g bands shift from 462 to 455 cm−1 when the activation temperature increases from 300 to 450 °C. However, the bands shift from 455 to 463 cm−1 when the activation temperature further increases from 450 to 600 °C. Moreover, the width of the F 2g band for the catalyst CC-450 is remarkably larger than those of the other catalysts. These results suggest that the catalyst CC-450 has the largest amount of oxygen vacancies among the investigated CuO/CeO2 catalysts (Gnanakumar et al. 2014). The less amounts of oxygen vacancies over the catalysts activated at lower temperatures (<450 °C) can be attributed to the fact that lower temperature is unfavorable to the interaction between CuO and the support CeO2 (Liu et al. 2013), while the less amounts of oxygen vacancies over the catalysts calcined at higher temperatures (>450 °C) can be attributed to the larger CuO particles formed at higher temperatures (Sun et al. 2015).
The S BET values of the CeO2 support and CuO/CeO2 catalysts are listed in Table 1. All catalysts exhibit lower S BET compared to CeO2 support, most probably due to the blocking of the pores of CeO2 by Cu species (Tang et al. 2004).
Chemical state of elements
In order to illuminate the chemical states of the elements existing in the CuO/CeO2 catalysts, XPS experiments were conducted. The Cu 2p3/2, Ce 3d, and O 1 s XPS spectra of the representative CuO/CeO2 catalysts are shown in Fig. 3a–c.
As shown in Fig. 4a, the broad peak of Cu 2p3/2 binding energy at 930–937 eV can be deconvoluted into two peaks, centered at 934.4 and 932.7 eV, respectively. According to the literature (Avgouropoulos and Ioannides 2003), the presence of higher Cu 2p3/2 binding energy (about 934 eV) and the shake-up peak (about 938–945 eV) indicates the existence of Cu2+ species in these catalysts. Meanwhile, the lower Cu 2p3/2 binding energy (932.7 eV) suggests the presence of reduced Cu species in these catalysts. On the other hand, the reduction degree of Cu species can be investigated by calculating the ratio of the intensities of the satellite peaks to those of the principal peaks (I sat/I pp), which is 0.55 for pure Cu2+ (Avgouropoulos and Ioannides 2006). As shown in Table 2, the I sat/I pp values of these CuO/CeO2 catalysts are lower than 0.55, confirming once again the presence of low-valence Cu species in these CuO/CeO2 catalysts. The formation of the reduced Cu species may result from strong interaction of CuO with the support CeO2 (Kundakovic and Flytzani-Stephanopoulos 1998), though the reduction of Cu2+ under the procedure of XPS measurement cannot be completely discarded (Zhu et al. 2004).

XPS spectra of different CuO/CeO2 catalysts: a Cu 2p3/2, b Ce 3d, and c O1 s
The Ce 3d XPS data obtained from different catalysts are shown in Fig. 4b. Two sets of spin-orbital multiplets relating to the 3d3/2 and 3d5/2 are labeled as u and v, respectively. According to previous research (Qi et al. 2012), the complex spectrum of Ce 3d is decomposed into ten components. Lines v 0 and v′ are contributed to the Ce 3d9 4f2 O2 p5 and the Ce 3d9 4f1 O2 p6 of Ce(III), respectively, and the same assignation can also be applied to the u structures. However, u 0 and v 0 are invisible in our samples, and the same phenomenon was also found over the CuO/CeO2 catalyst prepared by the conventional impregnation method (Sun et al. 2015). Thus, an acceptable way to calculate the reduction degree of CeO2 is to consider the relative intensities of the u′ and v′ peaks to the total Ce 3d region.
The calculated percentages of Ce(III) for different CuO/CeO2 catalysts are listed in Table 2. We can see that all the catalysts have the ability to carry out the reduction of CeO2, and the Ce(III) % value of the CC-450 catalyst is the largest. As reported, the existence of Ce3+ in CeO2 suggests the formation of oxygen vacancies (Li et al. 2011; Chang et al. 2006). Therefore, it can be inferred that the CC-450 catalyst has the highest level of oxygen vacancies, consistent with the above Raman results. Additionally, the values of Ce(III) % for these CuO/CeO2 catalysts are approximately two times higher than that of pure CeO2 reported in the literature (Ce(III) % = 6.75 %) (Cai et al. 2010), but the lattice parameters of these samples are similar to the reported values. This may be due to the fact that the sampling depth of XPS is lower than 10 nm, while the XRD analysis, from which the lattice parameters are determined, is a “bulk” technique. Therefore, there is a very large difference between the two kinds of analytical tools. In fact, almost all of the CeO2 in bulk phase is still fully oxidized, thus the CeO2 reduction level observed from XPS has a negligible effect on the lattice parameters. On the contrary, these CuO/CeO2 catalysts show slightly lower lattice parameters caused by the incorporation of Cu2+ into the CeO2 lattice (Sun et al. 2015).
The XPS spectra of the O1 s region (Fig. 4c) show the contribution of two kinds of surface oxygen species. The low binding energy of 529–530 eV is characteristic of the lattice oxygen (denoted as Olat) associated with metal oxides, while the high binding energy in the region of 531–533 eV may be assigned to adsorbed oxygen (denoted as Oads) (Hu et al. 2009; Sun et al. 2014). The ratios of the peak area of lattice oxygen to total areas of lattice oxygen and absorbed oxygen for these catalysts are presented in Table 2. It can be seen that the ratios for these CuO/CeO2 catalysts increase with the increase in activation temperature, suggesting that a part of adsorbed oxygen has been transformed into lattice oxygen during the high-temperature activation process (Li et al. 2010).
Reducibility of catalysts
H2-TPR experiments were conducted to investigate the reducibility of the CuO/CeO2 catalysts; the results are presented in Fig. 5. As shown, there are three peaks (denoted as α, β, and γ, respectively) being detected in these catalysts. A qualitative attribution of the TPR peaks to different Cu species over CeO2 has been proposed by many researchers. For example, Luo and Avgouropoulos et al. (Avgouropoulos and Ioannides 2003; Luo et al. 1997) attributed the lower and higher temperature peaks to finely dispersed CuO strongly interacting with CeO2 and larger CuO particles, respectively. Yen and Zhang et al. (Yen et al. 2012; Zhang et al. 2015) reported two reductive peaks which are attributed to the reduction of well-dispersed surface Cu species and Cu2+ in the CeO2 lattice, respectively. Zou and Moretti et al. (Moretti et al. 2011; Zou et al. 2006) suggested, for the more complex H2-TPR profiles, the contribution of both copper clusters and isolated Cu2+ ions. In this work, based on these literature data and the above XRD results, the α, β, and γ peaks are attributed to the reduction of finely dispersed CuO strongly interacting with CeO2, Cu species doped into the support CeO2, and small two- and three-dimensional clusters of CuO weakly associated with the CeO2 or the isolated bulk CuO without interaction with the CeO2, respectively.

TPR profiles of different CuO/CeO2 catalysts
As seen in Fig. 5, with the increase of activation temperature from 300 to 450 °C, the intensity of α and β peaks increases slightly and then decreases noticeably with further increase of activation temperature from 450 to 600 °C. This result indicates that more Cu species is finely dispersed on the surface of CeO2 or incorporated into the CeO2 lattice for the CC-450 catalyst. On the other hand, the temperature of α peak for the CC-450 catalyst is noticeably lower than that for the CC-300 catalyst. This result suggests that the interaction between the finely dispersed Cu species and CeO2 for the former is stronger than that for the latter, since the stronger interaction leads to the lower reduction temperature (Scirè et al. 2010). Furthermore, the temperature of γ peak for the CC-450 catalyst is the lowest among these catalysts. This result suggests that the size of CuO clusters or bulk CuO particles on the CC-450 catalyst is the smallest, because the higher the reduction temperature is, the larger the CuO particle will be (Zhang et al. 2006). These results are in good agreement with those obtained by the above XRD and Raman analyses.
CO-TPD study
TPD profiles of CO2 after CO adsorption on different CuO/CeO2 catalysts are shown in Fig. 6. Almost all the adsorbed CO has been released after washing the sample under flowing He at RT. In the heating process, majority of adsorbed CO desorbs as CO2 resulting from different carbonate species produced by the reaction of absorbed CO with lattice oxygen (Caputo et al. 2008). From Fig. 6, it can be seen that there are two CO2 desorption peaks, including a main peak at 100–200 °C and a shoulder peak at >200 °C, which is consistent with the result of Avgouropoulos and Ioannides (Avgouropoulos and Ioannides 2008). According to the literature (Caputo et al. 2008), these two peaks may be related to different CO adsorption approaches: a fraction of CO has formed CO2 at RT, which probably reacts with CeO2 surface and adsorbs as carbonate, and releases at lower temperatures (100–200 °C); another fraction of the CO probably develops into bidentate carbonate on the reactive sites, which may evolve across intermediate species (may be as COx) when the temperature increases and finally desorbs as CO2 at higher temperatures (200–400 °C). More specifically, with the increase of activation temperature of the CuO/CeO2 catalysts from 300 to 450 °C, the temperature of CO2 desorption peak decreases noticeably from about 170 to 120 °C, implying that the produced carbonate species can desorb more easily (Luo et al. 1997). However, with further rise of the activation temperature, the CO2 desorption peak temperature no longer decreases and the corresponding intensity of CO2 desorption peak decreases slightly. As a result, it can be inferred that the CC-450 catalyst has the largest amount of more active lattice oxygen among the investigated CuO/CeO2 catalysts.

TPD profiles of CO2 after CO adsorption at ambient temperature over different CuO/CeO2 catalysts
Activity of catalysts
Figure 7 shows CO oxidation activities of CuO/CeO2 catalysts (CC-400 and CC-500 have been removed for clarity). The T50 (temperature for the CO conversion of 50 %) values are 86, 81, 71, 80, and 80 °C, for the catalysts CC-300, CC-400, CC-450, CC-500, and CC-600, respectively. It can be seen that the activity of CuO/CeO2 catalysts for CO oxidation increases first with the increase of activation temperature, passing through a maximum at 450 °C, then decreases at higher activation temperatures.

CO oxidation activities of different CuO/CeO2 catalysts
There are many factors that can affect the activity of the CuO/CeO2 catalyst for CO oxidation. On the basis of the above characterization results, the reason for the highest activity of the CC-450 catalyst can be explained as follows: (1) It is well known that the Cu species that are well dispersed and strongly interacting with the support CeO2 are active toward CO oxidation, while larger bulk CuO particles contribute only a little or even negatively to the activity owing to the coverage of active species on the catalyst surface (Li et al. 2015; Luo et al. 1997; Mrabet et al. 2012). The above XRD result suggests that fewer larger bulk CuO particles are formed on the surface of CC-450 catalyst. In addition, the TPR result indicates that there are large amounts of well-dispersed Cu species strongly interacting with CeO2 on the CC-450 catalyst than on the other catalysts. These structural features of the Cu species on the CC-450 catalyst are responsible for its higher activity. (2) CO oxidation on CuO/CeO2 catalysts is considered to be a redox reaction involving oxygen vacancies. So, the higher the relative concentration of oxygen vacancies is, the better the performance of the catalyst will be (Jia et al. 2012; Li et al. 2010; Liu et al. 2010). Therefore, the highest activity of the CC-450 catalyst can be reasonably attributed to the highest concentration of oxygen vacancies obtained by Raman and XPS analyses. (3) It was reported that the oxidation of CO on metal oxides conforms to the following mechanism: (1) CO + Olat. → CO2 + Ovac., (2) O2 + 2Ovac. → 2Olat., where the Olat. and Ovac. represent lattice oxygen and oxygen vacancy, respectively (Li et al. 2010). Obviously, the largest amount of more active lattice oxygen of the CC-450 catalyst as revealed by CO-TPD analysis accords well with its highest CO oxidation. Similar results were also reported by other researchers (Li et al. 2010; Liu et al. 2010; Zou et al. 2011).
The CO oxidation activities of the CuO/CeO2 catalyst prepared by the present solvothermal method and those from the literature are presented in Table 3. It can be seen that our best-performing catalyst (CC-450) shows activities comparable to or even better than those of the CuO/CeO2 catalysts prepared by other methods reported in the literature. It suggests that the use of simple solvothermal method can lead to appreciable activities, comparable to or even better than those achieved by other complex methods.
Conclusions
A series of CuO/CeO2 catalysts were prepared using a solvothermal method and subsequently activated at different temperatures. The catalyst activated at 450 °C shows the highest activity due to the better dispersion of CuO species as well as enhanced interaction between CuO and CeO2. The strong interaction between CuO and CeO2 can induce the formation of large amounts of oxygen vacancies and activate the lattice oxygen, which is beneficial to the higher activity for CO oxidation. However, higher activation temperatures (>450 °C) lead to the aggregation and growth of CuO nanoparticles on CeO2 surface due to the sintering, and the redundant larger bulk CuO species weaken the catalytic activity. On the other hand, the lower activity of CuO/CeO2 catalysts activated at lower temperatures (<450 °C) can be attributed to the weaker interaction between CuO and the support CeO2.
References
Avgouropoulos G, Ioannides T (2003) Selective CO oxidation over CuO–CeO2 catalysts prepared via the urea–nitrate combustion method. Appl Catal A 244:155–167
Avgouropoulos G, Ioannides T (2006) Effect of synthesis parameters on catalytic properties of CuO–CeO2. Appl Catal B 67:1–11
Avgouropoulos G, Ioannides T (2007) Adsorption and reaction of CO on CuO–CeO2 catalysts prepared by the combustion method. Catal Lett 116:15–22. doi:10.1007/s10562-007-9114-9
Avgouropoulos G, Ioannides T (2008) TPD and TPSR study of CO interaction with CuO–CeO2 catalysts. J Mol Catal A 296:47–53. doi:10.1016/j.molcata.2008.09.013
Ayastuy JL, Gurbani A, González-Marcos MP, Gutiérrez-Ortiz MA (2009) Kinetics of carbon monoxide oxidation over CuO supported on nanosized CeO2. Ind Eng Chem Res 48:5633–5641
Ayastuy JL, Gurbani A, González-Marcos MP, Gutiérrez-Ortiz MA (2010) CO oxidation on CeXZr1−XO2-supported CuO catalysts: correlation between activity and support composition. Appl Catal A 387:119–128. doi:10.1016/j.apcata.2010.08.015
Cai W, Chen F, Shen X, Chen L, Zhang J (2010) Enhanced catalytic degradation of AO7 in the CeO2–H2O2 system with Fe3+ doping. Appl Catal B 101:160–168
Cao J-L, Wang Y, Zhang T-Y, Wu S-H, Yuan Z-Y (2008) Preparation, characterization and catalytic behavior of nanostructured mesoporous CuO/Ce0.8Zr0.2O2 catalysts for low-temperature CO oxidation. Appl Catal B 78:120–128
Caputo T, Lisi L, Pirone R, Russo G (2008) On the role of redox properties of CuO/CeO2 catalysts in the preferential oxidation of CO in H2-rich gases. Appl Catal A 348:42–53. doi:10.1016/j.apcata.2008.06.025
Chang L-H, Sasirekha N, Chen Y-W, Wang W-J (2006) Preferential oxidation of CO in H2 stream over Au/MnO2–CeO2 catalysts. Ind Eng Chem Res 45:4927–4935. doi:10.1021/ie0514408
Chuang K-H, Shih K, Wey M-Y (2012) The influences of microwave irradiation and polyol precursor pH on Cu/AC catalyst and its CO oxidation performance. J Nanopart Res 14:1178–1185. doi:10.1007/s11051-012-1178-9
Cui X, Wang Y, Chen L, Shi J (2014) Synergetic catalytic effects in tri-component mesostructured Ru–Cu–Ce oxide nanocomposite in CO oxidation. ChemCatChem 6:2860–2871. doi:10.1002/cctc.201402392
Fu G-Y, Mao D-S, Sun S-S, Yu J, Yang Z-Q (2015) Preparation, characterization and CO oxidation activity of Cu–Ce–Zr mixed oxide catalysts via facile dry oxalate-precursor synthesis. J Ind Eng Chem 31:283–290. doi:10.1016/j.jiec.2015.06.038
Gnanakumar ES, Naik JM, Manikandan M, Raja T, Gopinath CS (2014) Role of nanointerfaces in Cu-and Cu + Au-based near-ambient-temperature CO oxidation catalysts. ChemCatChem 6:3116–3124
Guo R-T et al (2014) Effect of Cu doping on the SCR activity of CeO2 catalyst prepared by citric acid method. J Ind Eng Chem 20:1577–1580
Hu C-Q, Zhu Q-S, Chen L, Wu R-F (2009) CuO-CeO2 binary oxide nanoplates: synthesis, characterization, and catalytic performance for benzene oxidation. Mater Res Bull 44:2174–2180
Jia A-P, Jiang S-Y, Lu J-Q, Luo M-F (2010) Study of catalytic activity at the CuO–CeO2 interface for CO oxidation. J Phys Chem C 114:21605–21610
Jia A-P, Hu G-S, Meng L, Xie Y-L, Lu J-Q, Luo M-F (2012) CO oxidation over CuO/Ce1−xCuxO2−δ and Ce1−xCuxO2−δ catalysts: synergetic effects and kinetic study. J Catal 289:199–209. doi:10.1016/j.jcat.2012.02.010
Kundakovic L, Flytzani-Stephanopoulos M (1998) Reduction characteristics of copper oxide in cerium and zirconium oxide systems. Appl Catal A 171:13–29
Kydd R et al (2009) Flame-synthesized ceria-supported copper dimers for preferential oxidation of CO. Adv Funct Mater 19:369–377. doi:10.1002/adfm.200801211
Lai J, Niu W, Luque R, Xu G (2015) Solvothermal synthesis of metal nanocrystals and their applications. Nano Today 10:240–267. doi:10.1016/j.nantod.2015.03.001
Li J, Zhu P, Zuo S, Huang Q, Zhou R (2010) Influence of Mn doping on the performance of CuO–CeO2 catalysts for selective oxidation of CO in hydrogen-rich streams. Appl Catal A 381:261–266. doi:10.1016/j.apcata.2010.04.020
Li J, Han Y, Zhu Y, Zhou R (2011) Purification of hydrogen from carbon monoxide for fuel cell application over modified mesoporous CuO–CeO2 catalysts. Appl Catal B 108–109:72–80
Li Y, Cai Y, Xing X, Chen N, Deng D, Wang Y (2015) Catalytic activity for CO oxidation of Cu–CeO2 composite nanoparticles synthesized by a hydrothermal method. Anal Methods. 7:3238–3245
Liu Y, Wen C, Guo Y, Lu G, Wang Y (2010) Modulated CO oxidation activity of M-doped ceria (M=Cu, Ti, Zr, and Tb): role of the Pauling electronegativity of M. J Phys Chem C 114:9889–9897. doi:10.1021/jp101939v
Liu Z-G, Chai S-H, Binder A, Li Y-Y, Ji L-T, Dai S (2013) Influence of calcination temperature on the structure and catalytic performance of CuOx–CoOy–CeO2 ternary mixed oxide for CO oxidation. Appl Catal A 451:282–288
Luo M-F, Zhong Y-J, Yuan X-X, Zheng X-M (1997) TPR and TPD studies of CuO/CeO2 catalysts for low temperature CO oxidation. Appl Catal A 162:121–131. doi:10.1016/S0926-860X(97)00089-6
Luo M-F, Ma J-M, Lu J-Q, Song Y-P, Wang Y-J (2007a) High-surface area CuO–CeO2 catalysts prepared by a surfactant-templated method for low-temperature CO oxidation. J Catal 246:52–59. doi:10.1016/j.jcat.2006.11.021
Luo M-F, Song Y-P, Lu J-Q, Wang X-Y, Pu Z-Y (2007b) Identification of CuO species in high surface area CuO–CeO2 catalysts and their catalytic activities for CO oxidation. J Phys Chem C 111:12686–12692
Luo M-F, Song Y-P, Wang X-Y, Xie G-Q, Pu Z-Y, Fang P, Xie Y-L (2007c) Preparation and characterization of nanostructured Ce0.9Cu0.1O2−δ solid solution with high surface area and its application for low temperature CO oxidation. Catal Commun 8:834–838. doi:10.1016/j.catcom.2006.09.017
Mai H, Zhang D, Shi L, Yan T, Li H (2011) Highly active Ce1−xCuxO2 nanocomposite catalysts for the low temperature oxidation of CO. Appl Surf Sci 257:7551–7559
Manzoli M, Monte RD, Boccuzzi F, Coluccia S, Kašpar J (2005) CO oxidation over CuOx–CeO2–ZrO2 catalysts: transient behaviour and role of copper clusters in contact with ceria. Appl Catal B 61:192–205. doi:10.1016/j.apcatb.2005.05.005
Moretti E et al (2011) Effect of thermal treatments on the catalytic behaviour in the CO preferential oxidation of a CuO–CeO2–ZrO2 catalyst with a flower-like morphology. Appl Catal B 102:627–637. doi:10.1016/j.apcatb.2011.01.004
Mrabet D, Abassi A, Cherizol R, Do T-O (2012) One-pot solvothermal synthesis of mixed Cu–Ce–Ox nanocatalysts and their catalytic activity for low temperature CO oxidation. Appl Catal A 447–448:60–66. doi:10.1016/j.apcata.2012.09.005
Prasad R, Rattan G (2010) Preparation methods and applications of CuO–CeO2 catalysts: a short review. Bull Chem React Eng Catal 5:7–30
Qi L et al (2012) Influence of cerium precursors on the structure and reducibility of mesoporous CuO-CeO2 catalysts for CO oxidation. Appl Catal B 119–120:308–320. doi:10.1016/j.apcatb.2012.02.029
Qin J, Lu J, Cao M, Hu C (2010) Synthesis of porous CuO–CeO2 nanospheres with an enhanced low-temperature CO oxidation activity. Nanoscale 2:2739–2743
Scirè S, Riccobene P-M, Crisafulli C (2010) Ceria supported group IB metal catalysts for the combustion of volatile organic compounds and the preferential oxidation of CO. Appl Catal B 101:109–117
Sun X-L, Gong C-R, Lv G, Bin F, Song C-L (2014) Effect of Ce/Zr molar ratio on the performance of Cu–Cex–Zr1-x/TiO2 catalyst for selective catalytic reduction of NOx with NH3 in diesel exhaust. Mater Res Bull 60:341–347
Sun S, Mao D, Yu J, Yang Z, Lu G, Ma Z (2015) Low-temperature CO oxidation on CuO/CeO2 catalysts: the significant effect of copper precursor and calcination temperature. Catal Sci Technol 5:3166–3181
Sundar RS, Deevi S (2006) CO oxidation activity of Cu–CeO2 nano-composite catalysts prepared by laser vaporization and controlled condensation. J Nanopart Res 8:497–509. doi:10.1007/s11051-005-9030-0
Tang X, Zhang B, Li Y, Xu Y, Xin Q, Shen W (2004) Carbon monoxide oxidation over CuO/CeO2 catalysts. Catal Today 93–95:191–198. doi:10.1016/j.cattod.2004.06.040
Tang X, Zhang B, Li Y, Xu Y, Xin Q, Shen W (2005) CuO/CeO2 catalysts: redox features and catalytic behaviors. Appl Catal A 288:116–125. doi:10.1016/j.apcata.2005.04.024
Yen H, Seo Y, Kaliaguine S, Kleitz F (2012) Tailored mesostructured copper/ceria catalysts with enhanced performance for preferential oxidation of CO at low temperature. Angew Chem Int Ed 51:12032–12035
Zagaynov IV (2015) Catalytic activity of CuO–CeO2 in oxidation of CO high content gas mixture. Mater Res Bull 61:36–39. doi:10.1016/j.materresbull.2014.09.087
Zhang S-M, Huang W-P, Qiu X-H, Li B-Q, Zheng X-C, Wu SH (2002) Comparative study on catalytic properties for low-temperature CO oxidation of Cu/CeO2 and CuO/CeO2 prepared via solvated metal atom impregnation and conventional impregnation. Catal Lett 80:41–46
Zhang Y, Fei J, Yu Y, Zheng X (2006) Methanol synthesis from CO2 hydrogenation over Cu based catalyst supported on zirconia modified γ-Al2O3. Energ Convers Manag 47:3360–3367. doi:10.1016/j.enconman.2006.01.010
Zhang J-C, Guo J-X, Liu W, Wang S-P, Xie A-R, Liu X-F, Wang J, Yang Y-Z (2015) Facile preparation of Mn+ -doped (M=Cu Co, Ni, Mn) hierarchically mesoporous CeO2 nanoparticles with enhanced catalytic activity for CO oxidation. Eur J Inorg Chem 6:969–976
Zhu H et al (2004) Characterization of copper oxide supported on ceria-modified anatase. J Mol Catal A 219:155–164
Zhu J, Gao Q, Chen Z (2008) Preparation of mesoporous copper cerium bimetal oxides with high performance for catalytic oxidation of carbon monoxide. Appl Catal B 81:236–243. doi:10.1016/j.apcatb.2007.12.017
Zou H, Dong X, Lin W (2006) Selective CO oxidation in hydrogen-rich gas over CuO/CeO2 catalysts. Appl Surf Sci 253:2893–2898. doi:10.1016/j.apsusc.2006.06.028
Zou H, Chen S, Liu Z, Lin W (2011) Selective CO oxidation over CuO–CeO2 catalysts doped with transition metal oxides. Powder Technol 207:238–244. doi:10.1016/j.powtec.2010.11.005
Acknowledgments
This project was financially supported by the National Natural Science Foundation of China (21273150) and the ‘‘ShuGuang’’ Project (10GG23) of Shanghai Municipal Education Commission and Shanghai Education Development Foundation.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of Interest
The authors declare that they have no conflict of interest.
Rights and permissions
About this article

Cite this article
Zheng, Y., Mao, D., Sun, S. et al. CO oxidation on CuO/CeO2 catalyst prepared by solvothermal synthesis: influence of catalyst activation temperature. J Nanopart Res 17, 471 (2015). https://doi.org/10.1007/s11051-015-3282-0
Received:
Accepted:
Published:
DOI: https://doi.org/10.1007/s11051-015-3282-0