
Abstract
Iron oxide nanoparticles exhibit highly size-dependent physicochemical properties that are important in applications such as catalysis and environmental remediation. In order for these size-dependent properties to be effectively harnessed for industrial applications scalable and cost-effective techniques for size-controlled synthesis or size separation must be developed. The synthesis of monodisperse iron oxide nanoparticles can be a prohibitively expensive process on a large scale. An alternative involves the use of inexpensive synthesis procedures followed by a size-selective processing technique. While there are many techniques available to fractionate nanoparticles, many of the techniques are unable to efficiently fractionate iron oxide nanoparticles in a scalable and inexpensive manner. A scalable apparatus capable of fractionating large quantities of iron oxide nanoparticles into distinct fractions of different sizes and size distributions has been developed. Polydisperse iron oxide nanoparticles (2–20 nm) coated with oleic acid used in this study were synthesized using a simple and inexpensive version of the popular coprecipitation technique. This apparatus uses hexane as a CO2 gas-expanded liquid to controllably precipitate nanoparticles inside a 1L high-pressure reactor. This paper demonstrates the operation of this new apparatus and for the first time shows the successful fractionation results on a system of metal oxide nanoparticles, with initial nanoparticle concentrations in the gram-scale. The analysis of the obtained fractions was performed using transmission electron microscopy and dynamic light scattering. The use of this simple apparatus provides a pathway to separate large quantities of iron oxide nanoparticles based upon their size for use in various industrial applications.
Graphical Abstract

Similar content being viewed by others


Explore related subjects
Discover the latest articles, news and stories from top researchers in related subjects.Avoid common mistakes on your manuscript.
Introduction
Metal oxide nanoparticles are finding wide-scale applicability in various industrial fields including catalysis (Park et al. 2010; Zhao et al. 2013), biomedicine (Bulte and Kraitchman 2004; Jo et al. 2010; Thorek et al. 2006), enhanced oil recovery (Baran et al. 2003), and environmental remediation (Orbell et al. 1997; Rajeshwar and de Tacconi 2009; Zhang 2003). Iron oxide nanoparticles are especially important in this sector of nanoscale science and engineering because of their controllable magnetic nature, relative safety, and affordability. Nanoparticles within the size range of 1–100 nm have extremely size-dependent physicochemical properties which arise due to their dimensions being intermediate between the atomic and molecular size regimes (Rao et al. 2002). It is important that we harness these size-dependent properties in order to optimize the use of nanoparticles in the applications mentioned above. There are two primary ways to go about controlling the size and therefore the size-dependent properties of nanoparticles. The first way is to directly synthesize monodisperse nanoparticles of desired size using various specialized-synthesis methods such that these nanoparticles can be used in the targeted applications. However, many of these specialized synthesis techniques suffer from certain disadvantages such as requiring high temperatures (Hyeon et al. 2001; Xu et al. 2010; Yu et al. 2004) and expensive reagents (Park et al. 2005; Yu et al. 2006) during the process, making large-scale synthesis of these particles expensive. Another way to affect the nanoparticle size, and therefore their size-dependent properties is to use a more straightforward and inexpensive synthesis technique to generate polydisperse nanoparticles and then process them using various techniques like magnetic separation (Yavuz et al. 2006), centrifugation (Novak et al. 2001), and electrophoresis (Surugau and Urban 2009) to obtain monodisperse particles. Most of these post-synthesis processing methods suffer from certain drawbacks such as requiring expensive equipment, having low throughputs and large amounts of solvent (Bishop et al. 2009; Fletcher 1991; Saunders and Roberts 2009).
Our laboratory has previously developed a method to size-selectively separate nanoparticles using gas-expanded liquids (GXLs) (Anand et al. 2007; McLeod et al. 2005a; Saunders and Roberts 2009). This process involves the controlled precipitation of nanoparticles coated with aliphatic stabilizing ligands therefore initially dispersed in an organic solvent, under applied CO2 pressure. This technique to size-selectively fractionate nanoparticles using GXLs is a modification of the popular liquid solvent–antisolvent (LSAS) precipitation process (Murray et al. 2000). The LSAS process typically generates large quantities of solvent waste and requires several tedious processing steps such as centrifugation in order to recover the particles. In the GXL fractionation process, compressed CO2 serves as the antisolvent for the aliphatic ligands that stabilize the nanoparticles in an organic solvent dispersion. These ligands (which are solvated in the absence of any CO2) tend to progressively self-associate and collapse upon the addition of CO2 (White and Kitchens 2010). This weakened solvation of the ligand tails causes a fraction of the nanoparticles to precipitate out of solution and the extent this precipitation is dictated by the amount of CO2 dissolved in the solvent. As such the larger nanoparticles precipitate out of solution at lower applied CO2 pressures (lower amounts of CO2 dissolved) due to the larger particles having higher van der Waals forces of attraction. Hence, by controlling the amount of CO2 pressure applied to the system one can controllably precipitate and separate nanoparticles based upon their size from an organic solvent. One of the main advantages of this GXL size-selective fractionation technique is the fact that the antisolvent (CO2) can be subsequently separated from the liquid solvent simply by depressurization, and thereby enabling recovery and reuse of these components. Due, in part, to the decreased solvent viscosities and increased diffusivities of species in a GXL, the nanoparticles can also be inclined to precipitate from the GXL dispersion without the need for centrifugation. This makes the GXL process less energy intensive and time intensive (Saunders and Roberts 2012). Other advantages to the use of CO2 include its low cost, relative inertness, and ready availability (Jessop and Subramaniam 2007; Vengsarkar and Roberts 2013).
We have previously demonstrated that nanoparticle dispersions of quantum dots (e.g., CdSe/ZnS) and noble elements (e.g., gold and silver) can be effectively precipitated using GXLs with CO2 as the gaseous antisolvent (Anand et al. 2007; Duggan and Roberts 2014; McLeod et al. 2005b; Saunders and Roberts 2009). Studies which modified the solvent–ligand combinations were successful in reducing and tuning the pressures required to fractionate gold nanoparticles using the GXL technique (Saunders and Roberts 2011; Vengsarkar and Roberts 2013). However, gold nanoparticles have limited use in contemporary industrial applications excluding certain specialized biomedical and catalytic roles (Haruta and Daté 2001; Mikami et al. 2013; Sharma et al. 2006). Iron oxide nanoparticles on the other hand have a wider scope in current and future applications. These include use in areas such as magnetic resonance imaging (MRI) contrast agents (Andreas et al. 2012; Babes et al. 1999; Hong et al. 2008; Hu et al. 2011; Kwak 2005; Qiao et al. 2009), drug delivery agents (Jain et al. 2005; Morales et al. 2008, 2005), catalysts (Hosseinian et al. 2011; Khedr et al. 2009; Park et al. 2010; Torres Galvis et al. 2012), and for the formation of Pickering emulsions (Binks and Whitby 2005; Zhou et al. 2012). The first GXL fractionation apparatus designed was a proof-of-concept spiral tube apparatus (Anand et al. 2007) designed to fractionate approximately 700 μL of dilute (<1 gram total metal or metal oxide present) nanoparticle sample. The next scaled up version of this apparatus was a cascaded-vessel apparatus (Saunders and Roberts 2009) which effectively fractionated dilute 20 mL samples of nanoparticles. This apparatus provided a good avenue to carry out detailed theoretical studies (Saunders and Roberts 2011; Vengsarkar and Roberts 2013) on the GXL fractionation process, due to the ability to fine tune the process parameters and to visually observe the process. However, one of the drawbacks of the cascaded vessel apparatus was that the use of concentrated nanoparticle samples can lead to the formation of Langmuir–Blodgett films (Saunders and Roberts 2011) and lower the effectiveness of the size-selective fractionation process. Also due to the small sample volume used in the apparatus, it is not possible to increase the total amount of nanoparticles being processed due to solubility limitations of the nanoparticles in the solvent. These issues with the cascaded-vessel apparatus make it critical to design a scalable system, capable of fractionating samples on the gram-scale, and targeted for use with iron oxide nanoparticles which are required in large quantities for any industrial application.
The purpose of this study is to demonstrate the utility of the GXL fractionation process to size-selectively separate inexpensively synthesized iron oxide nanoparticles from an organic dispersion at relatively high concentrations. This is achieved through the design of a scalable high-pressure fractionation apparatus capable of handling a high loading of nanoparticles (>1 gram solids in solution) and large sample volumes. This study also provides fundamental insight into the factors that affect the size-selective fractionation of iron oxide nanoparticles using GXLs.
Experimental section
Materials
Iron (III) chloride hexahydrate (FeCl3·6H2O, 99.9 %), iron (II) chloride tetrahydrate (FeCl2·4H2O, 99.9 %), and n-hexane (HPLC grade, 95 %), were obtained from Alfa Aesar. Oleic acid (99 %), was obtained from Sigma Aldrich, and ammonium hydroxide (5 M) was obtained from BDH. Deionized ultra-filtered (DIUF) water was obtained from Fisher. Carbon dioxide (SFC/SFE grade) was obtained from Airgas. All chemicals were used as received without further purification.
Nanoparticle synthesis
Iron oxide nanoparticles coated with oleic acid were synthesized based on a method described previously by Jain et al. (2005). Initially, aqueous solutions of 0.2 M Fe(III) and 0.2 M Fe(II) were prepared using DIUF water. The Fe(III) solution (120 mL, 0.2 M) was mixed with the Fe(II) solution (60 mL, 0.2 M) in a three-necked flask with magnetic stirring. Under constant stirring and under a nitrogen atmosphere, 24 mL of 5 M ammonium hydroxide was added to this mixture to generate a black precipitate of iron oxide nanoparticles. These reagent concentrations theoretically generate 2.78 gm of iron oxide (i.e., ~2.01 gm of iron). The temperature of this system was then increased to 80 °C and maintained for 30 min to evaporate the ammonia out of the solution. While the temperature is being increased, 2.24 mL of oleic acid was added to the mixture. After the end of the heating cycle, the system was allowed to cool down to room temperature, followed by magnetic immobilization and washing with DIUF water to remove any excess reagents and impurities. The particles were then dried using nitrogen and dispersed in 200 mL of n-hexane via sonication (Fischer Scientific FS20). This 200 mL dispersion of iron oxide nanoparticles (concentration ~10 mg/mL of iron) was used for each of the GXL fractionation experiments.
Nanoparticle characterization
The as-synthesized nanoparticles were characterized using transmission electron microscopy (TEM) to obtain their size and size distribution. Carbon-coated TEM grids of each of the collected nanoparticle fractions were then prepared via dropcasting and micrographs were acquired on a Zeiss EM 10 transmission electron microscope and sized using the ImageJ software package (more than a 1000 nanoparticles for each sample). It should be noted that the ImageJ software uses image analysis to determine the area of a nanoparticle, and this area is then used to calculate particle diameter with the underlying assumption that the particle is perfectly spherical. Since, the particles synthesized in this study are not perfectly spherical it is possible to get a slightly skewed size and size distribution using the TEM analysis. Hence, dynamic light scattering (DLS) analysis was also performed on these particles, using a Malvern Zetasizer Nano ZS, to confirm the trends obtained through TEM analysis. Fourier transform infrared spectroscopy (FTIR) was also carried out on the sample using a Nicolet Avatar 360 to investigate the nature of chemical interaction between the ligand (oleic acid) and the nanoparticle core. The solid samples were pelletized using a pellet press along with KBr for the FTIR analysis. A baseline for pure KBr and atmospheric correction was used for each of the samples studied.
Size-selective separation process
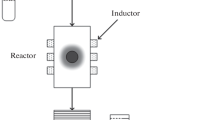
Figure 1 depicts the bench-scale apparatus used to size-selectively separate iron oxide nanoparticles using a gas-expanded liquid system. The vessel used for the bench scale size-selective separation of iron oxide nanoparticles is a modified 1L Parr-reactor Model 4571 (Parr Instruments Company, Moline, Illinois, USA). The Parr reactor is fitted with a dip tube and is also equipped with a pressure transducer and thermocouple. A removable glass liner is used inside the Parr reactor for easy cleaning and to prevent contamination of the iron oxide nanoparticle sample. The reactor system is connected to a high-pressure syringe pump (ISCO 260D) which is used to control the amount of CO2 added to the system. To initiate an experiment, 200 mL of the synthesized iron oxide nanoparticle dispersion in hexane is added to the Parr reactor. The Parr reactor is then sealed and pressure tested using CO2 to ensure that no leaks are present in the system. The reactor system is then purged of air and pressurized to the first experimental pressure of 13.8 bar (200 psi) by injecting CO2 through valve V1 using the syringe pump. The system is then maintained at this pressure for a period of 4 h (enough time for the system to reach equilibrium) using the syringe pump. This is done so as to ensure the complete dissolution of CO2 into the hexane and the complete precipitation (if any) of the iron oxide nanoparticles. After this period of 4 h, the valve V2 is slowly opened, while maintaining the pressure at 13.8 bar using the syringe pump. The pressure difference across valve V2 causes the nanoparticle dispersion inside the vessel to be transferred via the dip tube into an external collection vessel where approximately 2 mL of the nanoparticle dispersion is retrieved. This sample is then transferred to a scintillation vial and the collection vessel is cleaned using pure hexane. The system pressure is then increased to the next desired pressure value (27.6 bar/400 psi), the system allowed to reach equilibrium, and the sample collection process is carried out. After this technique is repeated for each of the desired pressures, the system is then slowly depressurized to atmospheric pressure by opening valve V3. It should be noted that while only 2 mL nanoparticle dispersion samples are drawn from the system at each of the investigated pressures in this particular experiment, it is feasible to recover essentially all of the dispersed nanoparticles from the system at any given pressure by removing all of the remaining liquid dispersion from the vessel (thereby leaving only the precipitated nanoparticles in the vessel). Each of the 2 mL liquid nanoparticle dispersion samples that were retrieved from the Parr reactor at the different system pressure values were then analyzed using TEM and DLS in the same way as the originally synthesized nanoparticle dispersion.

Bench-scale apparatus to size-selectively separate iron oxide nanoparticles using a gas-expanded liquid system
Results and discussion
Oleic acid-coated iron oxide nanoparticles were initially synthesized using the coprecipitation method since it uses inexpensive reagents and lower reaction temperatures compared to other iron oxide nanoparticle synthesis methods (Chen et al. 2009; Hyeon et al. 2001; Park et al. 2005). The surface functionalization of the synthesized particles was examined using FTIR spectroscopy and the results obtained are shown in Fig. 2 along with the spectrum for pure oleic acid. The two sharp peaks present in both spectra, in the range of 2800–3000 cm−1, can be attributed to the –CH2 stretches from the alkyl chain in oleic acid. The spectrum for the iron oxide nanoparticles shows a characteristic Fe–O stretch at 588 cm−1 and the spectrum for pure oleic acid, on the other hand, shows a prominent C=O stretch at 1710 cm−1. In the spectrum of the iron oxide nanoparticles coated with oleic acid, a reduction in intensity of this C=O stretching peak is observed coupled with its splitting into two distinct peaks of lower wavenumber (1623 and 1529 cm−1), thereby signifying a C–O interaction. The presence of this interaction is consistent with previous observations that have been reported in the literature (Lee and Harris 2006; Vengsarkar and Roberts 2014; Wang et al. 2010; Zhang et al. 2006) that indicate the presence of a ligand-core conjugation via the C–O bonds (Zhang et al. 2006).

FTIR spectrum of oleic acid-coated iron oxide nanoparticles synthesized using a coprecipitation technique
The size analysis obtained using TEM and DLS data indicated that the nanoparticles synthesized using the coprecipitation method were relatively polydisperse in nature. It should also be noted that some of the generated particles were not perfectly spherical in nature. which is typical for particles produced using the coprecipitation technique (Babes et al. 1999; Maity and Agrawal 2007; Wang et al. 2010). The core diameter of the iron oxide nanoparticles as determined by TEM analysis was found to be 6.4 ± 3.7 nm. The average volume-weighted hydrodynamic diameter of these same particles was determined to be 21.1 nm using DLS.
DLS measures the hydrodynamic diameter of the dispersed nanoparticles consisting of the metal iron oxide core along with the solvated ligand shell. Thus, DLS size analysis often yields a higher average diameter value than that obtained via TEM analysis which only provides an indication of the diameter of the metal core (Lim et al. 2013).
Size-selective separation of the iron oxide nanoparticle dispersion was performed in the high-pressure Parr-reactor apparatus at a series of predetermined pressures where the fractionated samples were recovered in a collection vessel, as detailed in the Experimental section. The TEM images of the nanoparticle samples obtained at each of the pressure values are shown in Fig. 3a. The reduction in nanoparticle size upon increasing system pressure is clearly visible from these images. From the size analysis of these TEM images, it is observed that there is no significant deviation of size or size distribution of the samples drawn at 13.8 bar (200 psi) and 27.6 bar (400 psi) compared to that of the original sample (6.3 ± 3.7 nm). However, for samples recovered at even higher system pressures, a progressive decrease in average nanoparticle size and standard deviation is observed with increasing pressure. The sample collected at 34.5 bar (500 psi) has an average nanoparticle diameter that is approximately 1 nm smaller than the original nanoparticle dispersion. Further reduction in nanoparticle diameter was obtained at each of the subsequent pressures examined. The lowest nanoparticle size obtained amongst the samples studied is 2.6 ± 1.3 nm for the nanoparticle dispersion obtained at the system pressure of 48.3 bar (700 psi). The nanoparticle dispersion obtained at a highest experimental system pressure of 50 bar (725 psi) was extremely dilute and almost colorless, indicating that very few particles remained dispersed in solution at this stage. The diameter of the nanoparticles present in this particular sample could not be accurately determined given the resolution/magnification of the available TEM equipment and the small number of particles present in the system at this pressure.

a TEM images of the original iron oxide nanoparticle dispersion and the nanoparticle samples retrieved at the experimental pressures. b Trends in average nanoparticle core size and standard deviation (using TEM) under different experimental pressure conditions
The reduction in the size and size distribution of nanoparticles present in the retrieved dispersions is shown in Figs. 3b and 4. It can be deduced from the observed reduction in average nanoparticle size and size distribution that the iron oxide nanoparticle precipitation essentially begins at a system pressure between 27.6 bar and 34.5 bar. At sequentially higher pressure values a steady reduction in nanoparticle size and size distribution is observed (Fig. 3b). This trend is similar to that observed previously in dodecanethiol-coated metal nanoparticle systems (Saunders and Roberts 2012, 2009; White and Kitchens 2010). Our hypothesis is that the increasing CO2 pressure in the reactor system sequentially diminishes the solvent strength of the CO2 + hexane mixture, thereby reducing its ability to sufficiently solvate the oleic acid-coated iron oxide nanoparticles such that they all remain dispersed in solution. Progressively smaller nanoparticles will precipitate from solution upon worsening solvent conditions (i.e., the largest particles will precipitate first as CO2 pressure is applied to the solution). As such, increasing the applied CO2 pressure in the system will cause a fraction of the largest particles in the dispersion to precipitate while leaving the rest of the particles thermodynamically stabilized in solution. A further increase in applied CO2 pressure will then cause the next largest fraction to precipitate where the average nanoparticle size will depend upon the pressure employed (Anand et al. 2007, 2005; Saunders and Roberts 2009).

Size distribution of the iron oxide nanoparticle samples recovered from the GXL apparatus under various pressure conditions obtained using TEM analysis
The mechanism that underpins the precipitation of the oleic acid-coated iron oxide nanoparticles is expected to be same as that attributed to the precipitation of dodecanethiol capped metal nanoparticles from a GXL solution (Von White et al. 2011; White and Kitchens 2010). Oleic acid and dodecanethiol have similar structural configurations once they are conjugated to the nanoparticle core since they both have unbranched aliphatic chains in their chemical structure. The difference in the carbon number and the double bond present in oleic acid could provide for a more densely packed protective layer (Lu et al. 2008) on the surface thus changing the precipitation pressures in the oleic acid-iron oxide nanoparticle system when compared to the dodecanethiol-gold nanoparticle system. As mentioned previously, the largest nanoparticles precipitate first upon worsening solvent conditions (i.e., with increasing applied CO2 pressure) due to larger van der Waals forces (Anand et al. 2007; Saunders and Roberts 2009), leaving the smaller particles dispersed in the solution. These smaller unprecipitated particles are the samples which we recover from the reactor system in the flash vessel. The way the current apparatus is designed causes the nanoparticle size distribution to shift towards the left (lower nanoparticle size) upon increasing system pressure due to what is effectively a ‘sieving effect’ on the original nanoparticle dispersion (i.e., larger particles get held back and smaller particles pass through). In all, these TEM results confirm that progressive increases in applied CO2 pressure causes the larger sized nanoparticles in dispersion to successively precipitate out of solution, leaving the smaller nanoparticles dispersed in the solution and recoverable.
The results from the TEM analysis of the recovered nanoparticle samples are further supported by DLS analysis of the same samples, as shown in Fig. 5 and Table 1. From the size histograms obtained using DLS, the size distributions of the samples obtained at 13.8 and 27.6 bar are very similar to that obtained from the original sample (average size—16.2 nm). However, at and above system pressures of 34.5 bar, a shift towards lower nanoparticle sizes is observed. The DLS size distribution of the recovered samples also decreases as the system pressure is increased above 27.6 bar. A clear trend in average size reduction is observed at 41.4 bar (11.5 nm), 44.8 bar (8.0 nm), and 48.3 bar (4.7 nm). At 50 bar, the highest experimental pressure, DLS analysis was able to provide a size distribution for the extremely small particles (1.7 nm) present in the colorless dilute solution.

Size distribution of the iron oxide nanoparticle samples recovered from the GXL apparatus under various pressure conditions obtained using DLS analysis
By comparing these DLS results to the TEM results (Table 1), the reduction in the average nanoparticle size upon increasing CO2 pressure is evident. The diameters of the nanoparticles obtained from DLS analysis are larger than the diameters obtained from TEM analysis due to the fact that DLS analysis accounts for the presence of the hydration layer and the presence of the stabilizing ligand in the measured particle. This difference in average particle size obtained from these two techniques is similar to that observed in the literature (Lim et al. 2013). For example, Lim et al. observed that while TEM analysis indicated an average iron oxide particle size of 7.2 nm, DLS resulted in a measurement that was 9.7 nm larger for that same sample. It should be noted that the oleic acid layer, which is approximately 2 nm in length (Zhang et al. 2006), affects the hydrodynamic diameter measurement obtained from DLS while this oleic acid layer is not observable in TEM due to the electron-transparent nature of oleic acid. While keeping in mind the inherent differences between these characterization techniques, similar trends in size reduction are obtained through TEM and DLS characterization of the samples. Hence, the current apparatus is very effective at separating large volumes (200 mL) of concentrated oleic acid-coated iron oxide nanoparticle solutions (~10 mg iron/mL solvent) based upon their size through the use of a gas-expanded liquid. A comparison of the current fractionation device and previous apparatuses is shown in Table 2. This bench-scale apparatus is also easily scalable since, in structure, it is very similar to a standard continuous stirred tank reactor.
The ability of the current apparatus to collect multiple samples in a single run/loading allows one to directly determine the pressures at which precipitation occurs. By performing multiple fractionation experiments on a single nanoparticle sample, one can use the obtained experimental data to develop an empirical model to better understand the size and size distribution of the nanoparticles that would be recovered under a given set of GXL fractionation conditions. It should be noted that detailed in situ experiments (including UV–Vis spectroscopy) would be required in order to accurately perform a mass balance on this GXL system (at a given operating pressure) so as to predict the mass of the nanoparticles that could be recovered in each fraction. These experiments are more cumbersome for the iron oxide system compared to previous studies involving metal nanoparticles where UV–Vis spectroscopy and the surface plasmon resonance band of the nanoparticles could be used to directly monitor their precipitation as a function of applied CO2 pressure (Saunders and Roberts 2012). Saunders and Roberts demonstrated that these tools could be used to theoretically determine the size of the nanoparticles that would precipitate from solution at a given set of conditions (Saunders and Roberts 2009). For example, the absorption spectrum of metallic gold nanoparticles is characterized by a strong broad absorption band around 520 nm that is absent in the bulk metal spectra (Amendola and Meneghetti 2009; Haiss et al. 2007). However, for iron oxide nanoparticles, no such absorption band is present and a relatively flat spectrum with a plateau at around 480 nm is observed (Kang et al. 1998; Kebede et al. 2013; Klačanová et al. 2013; Walt et al. 2010). This, unfortunately, prevents accurate in situ monitoring of the precipitation of iron oxide nanoparticles in the gas-expanded liquid system. To better understand the GXL fractionation of iron oxide nanoparticles, more studies should be carried in order to develop a fundamental model that would allow one to predict the size of iron oxide nanoparticles that would precipitate at a given applied CO2 pressure.
Conclusion
In summary, through the design of a simple high-pressure reactor system, large quantities of iron oxide nanoparticles have been successfully fractionated based upon their size using a gas-expanded liquid. This GXL system is effective at separating large volumes (>200 mL) of concentrated oleic acid-coated iron oxide nanoparticle solutions (~10 mg iron/mL solvent). By successively pressurizing the GXL system to applied CO2 pressures of up to 50 bar, the nanoparticles of core sizes varying from 6.3 ± 3.7 to 2.6 ± 1.6 nm were successfully fractionated from the original iron oxide nanoparticle dispersion. The monodispersity of the obtained nanoparticle fractions was observed to increase upon increasing applied CO2 pressure. These trends in nanoparticle precipitation as a function of applied CO2 pressure were found to be similar to those observed previously for metal nanoparticle systems. The use of this simple GXL apparatus provides a pathway to separate large quantities of inexpensively produced iron oxide nanoparticles based upon their size for subsequent use in large-scale applications such as catalysis.
References
Amendola V, Meneghetti M (2009) Size Evaluation of Gold Nanoparticles by UV−vis Spectroscopy. J Phys Chem C 113:4277–4285. doi:10.1021/jp8082425
Anand M, McLeod MC, Bell PW, Roberts CB (2005) Tunable solvation effects on the size-selective fractionation of metal nanoparticles in CO2 gas-expanded solvents. J Phys Chem B 109:22852–22859. doi:10.1021/jp0547008
Anand M, Odom LA, Roberts CB (2007) Finely controlled size-selective precipitation and separation of CdSe/ZnS semiconductor nanocrystals using CO2-gas-expanded liquids. Langmuir 23:7338–7343. doi:10.1021/la700325z
Andreas K, Georgieva R, Ladwig M et al (2012) Highly efficient magnetic stem cell labeling with citrate-coated superparamagnetic iron oxide nanoparticles for MRI tracking. Biomaterials 33:4515–4525. doi:10.1016/j.biomaterials.2012.02.064
Babes L, Denizot B, Tanguy G et al (1999) Synthesis of iron oxide nanoparticles used as MRI contrast agents: a parametric study. J Colloid Interface Sci 212:474–482. doi:10.1006/jcis.1998.6053
Baran Jr, Jimmie R., Cabrera OJ (2003) Use of surface-modified nanoparticles for oil recovery
Binks BP, Whitby CP (2005) Nanoparticle silica-stabilised oil-in-water emulsions: improving emulsion stability. Colloids Surf A 253:105–115. doi:10.1016/j.colsurfa.2004.10.116
Bishop KJM, Wilmer CE, Soh S, Grzybowski BA (2009) Nanoscale forces and their uses in self-assembly. Small 5:1600–1630. doi:10.1002/smll.200900358
Bulte JWM, Kraitchman DL (2004) Iron oxide MR contrast agents for molecular and cellular imaging. NMR Biomed 17:484–499. doi:10.1002/nbm.924
Chen C-J, Lai H-Y, Lin C-C et al (2009) Preparation of monodisperse iron oxide nanoparticles via the synthesis and decomposition of iron fatty acid complexes. Nanoscale Res Lett 4:1343–1350. doi:10.1007/s11671-009-9403-x
Duggan JN, Roberts CB (2014) Aggregation and precipitation of gold nanoparticle clusters in carbon dioxide-gas-expanded liquid dimethyl sulfoxide. J Phys Chem C 118:14595–14605. doi:10.1021/jp502151p
Fletcher D (1991) Fine particle high gradient magnetic entrapment. IEEE Trans Magn 27:3655–3677. doi:10.1109/20.102936
Haiss W, Thanh NTK, Aveyard J, Fernig DG (2007) Determination of size and concentration of gold nanoparticles from UV-vis spectra. Anal Chem 79:4215–4221. doi:10.1021/ac0702084
Haruta M, Daté M (2001) Advances in the catalysis of Au nanoparticles. Appl Catal A 222:427–437. doi:10.1016/S0926-860X(01)00847-X
Hong RY, Feng B, Chen LL et al (2008) Synthesis, characterization and MRI application of dextran-coated Fe3O4 magnetic nanoparticles. Biochem Eng J 42:290–300. doi:10.1016/j.bej.2008.07.009
Hosseinian A, Rezaei H, Mahjoub A (2011) Preparation of nanosized iron oxide and their photocatalytic properties for congo red. World Acedemy Sci Eng Technol 52:736–739
Hu F, Jia Q, Li Y, Gao M (2011) Facile synthesis of ultrasmall PEGylated iron oxide nanoparticles for dual-contrast T(1)- and T(2)-weighted magnetic resonance imaging. Nanotechnology 22:245604. doi:10.1088/0957-4484/22/24/245604
Hyeon T, Lee SS, Park J et al (2001) Synthesis of highly crystalline and monodisperse maghemite nanocrystallites without a size-selection process. J Am Chem Soc 123:12798–12801
Jain TK, Morales MA, Sahoo SK et al (2005) Iron oxide nanoparticles for sustained delivery of anticancer agents. Mol Pharm 2:194–205. doi:10.1021/mp0500014
Jessop PG, Subramaniam B (2007) Gas-expanded liquids. Chem Rev 107:2666–2694. doi:10.1021/cr040199o
Jo J, Aoki I, Tabata Y (2010) Design of iron oxide nanoparticles with different sizes and surface charges for simple and efficient labeling of mesenchymal stem cells. J Control Release 142:465–473. doi:10.1016/j.jconrel.2009.11.014
Kang Y, Lee D, Stroeve P (1998) FTIR and UV-vis spectroscopy studies of Langmuir-Blodgett films of stearic acid/γ-Fe2O3 nanoparticles. Thin Solid Films 329:541–544
Kebede A, Singh AK, Rai PK et al (2013) Controlled synthesis, characterization, and application of iron oxide nanoparticles for oral delivery of insulin. Lasers Med Sci 28:579–587. doi:10.1007/s10103-012-1106-3
Khedr MH, Abdel Halim KS, Soliman NK (2009) Synthesis and photocatalytic activity of nano-sized iron oxides. Mater Lett 63:598–601. doi:10.1016/j.matlet.2008.11.050
Klačanová K, Fodran P, Šimon P (2013) Formation of Fe (0)-nanoparticles via reduction of Fe(II) compounds by amino acids and their subsequent oxidation to iron oxides. J Chem. doi:10.1155/2013/961629
Kwak B (2005) Synthesis of MRI contrast agent by coating superparamagnetic iron oxide with chitosan. IEEE Trans Magn 41:4102–4104. doi:10.1109/TMAG.2005.855338
Lee S-Y, Harris MT (2006) Surface modification of magnetic nanoparticles capped by oleic acids: characterization and colloidal stability in polar solvents. J Colloid Interface Sci 293:401–408. doi:10.1016/j.jcis.2005.06.062
Lim J, Yeap SP, Che HX, Low SC (2013) Characterization of magnetic nanoparticle by dynamic light scattering. Nanoscale Res Lett 8:381. doi:10.1186/1556-276X-8-381
Lu Y, Lu X, Mayers BT et al (2008) Synthesis and characterization of magnetic Co nanoparticles: a comparison study of three different capping surfactants. J Solid State Chem 181:1530–1538. doi:10.1016/j.jssc.2008.02.016
Maity D, Agrawal DC (2007) Synthesis of iron oxide nanoparticles under oxidizing environment and their stabilization in aqueous and non-aqueous media. J Magn Magn Mater 308:46–55. doi:10.1016/j.jmmm.2006.05.001
McLeod MC, Anand M, Kitchens CL, Roberts CB (2005a) Precise and rapid size selection and targeted deposition of nanoparticle populations using CO2 gas expanded liquids. Nano Lett 5:461–465. doi:10.1021/nl047966j
McLeod MC, Kitchens CL, Roberts CB (2005b) CO2-expanded liquid deposition of ligand-stabilized nanoparticles as uniform, wide-area nanoparticle films. Langmuir 21:2414–2418. doi:10.1021/la047576c
Mikami Y, Dhakshinamoorthy A, Alvaro M, García H (2013) Catalytic activity of unsupported gold nanoparticles. Catal Sci Technol 3:58. doi:10.1039/c2cy20068f
Morales MA, Jain TK, Labhasetwar V, Leslie-Pelecky DL (2005) Magnetic studies of iron oxide nanoparticles coated with oleic acid and Pluronic® block copolymer. J Appl Phys 97:10Q905. doi:10.1063/1.1850855
Morales M, Finotelli P, Coaquira J et al (2008) In situ synthesis and magnetic studies of iron oxide nanoparticles in calcium-alginate matrix for biomedical applications. Mater Sci Eng 28:253–257. doi:10.1016/j.msec.2006.12.016
Murray CB, Kagan CR, Bawendi MG, Murray C (2000) Synthesis and characterization of monodisperse nanocrystals and close-packed nanocrystal assemblies. Annu Rev Mater 30:545–610
Novak JP, Nickerson C, Franzen S, Feldheim DL (2001) Purification of molecularly bridged metal nanoparticle arrays by centrifugation and size exclusion chromatography. Anal Chem 73:5758–5761
Orbell JD, Godhino L, Bigger SW et al (1997) Oil spill remediation using magnetic particles: an experiment in environmental technology. J Chem Educ 74:1446. doi:10.1021/ed074p1446
Park J, Lee E, Hwang N-M et al (2005) One-nanometer-scale size-controlled synthesis of monodisperse magnetic iron oxide nanoparticles. Angew Chemie 117:2932–2937. doi:10.1002/ange.200461665
Park J-Y, Lee Y-J, Khanna PK et al (2010) Alumina-supported iron oxide nanoparticles as Fischer-Tropsch catalysts: effect of particle size of iron oxide. J Mol Catal A 323:84–90. doi:10.1016/j.molcata.2010.03.025
Qiao R, Yang C, Gao M (2009) Superparamagnetic iron oxide nanoparticles: from preparations to in vivo MRI applications. J Mater Chem 19:6274. doi:10.1039/b902394a
Rajeshwar K, de Tacconi NR (2009) Solution combustion synthesis of oxide semiconductors for solar energy conversion and environmental remediation. Chem Soc Rev 38:1984–1998. doi:10.1039/b811238j
Rao CNR, Kulkarni GU, Thomas PJ, Edwards PP (2002) Size-dependent chemistry: properties of nanocrystals. Chemistry 8:28–35
Saunders SR, Roberts CB (2009) Size-selective fractionation of nanoparticles at an application scale using CO2 gas-expanded liquids. Nanotechnology 20:475605. doi:10.1088/0957-4484/20/47/475605
Saunders SR, Roberts CB (2011) Tuning the precipitation and fractionation of nanoparticles in gas-expanded liquid mixtures. J Phys Chem C 115:9984–9992. doi:10.1021/jp2019878
Saunders SR, Roberts CB (2012) Nanoparticle separation and deposition processing using gas expanded liquid technology. Curr Opin Chem Eng 1:1–11. doi:10.1016/j.coche.2011.12.004
Sharma P, Brown S, Walter G et al (2006) Nanoparticles for bioimaging. Adv Colloid Interface Sci 123–126:471–485. doi:10.1016/j.cis.2006.05.026
Surugau N, Urban PL (2009) Electrophoretic methods for separation of nanoparticles. J Sep Sci 32:1889–1906. doi:10.1002/jssc.200900071
Thorek DLJ, Chen AK, Czupryna J, Tsourkas A (2006) Superparamagnetic iron oxide nanoparticle probes for molecular imaging. Ann Biomed Eng 34:23–38. doi:10.1007/s10439-005-9002-7
Torres Galvis HM, Bitter JH, Khare CB et al (2012) Supported iron nanoparticles as catalysts for sustainable production of lower olefins. Science 335:835–838. doi:10.1126/science.1215614
Van Der Walt H, Chown L, Harris R et al (2010) Fe3O4 and Fe3O4@ Au nanoparticles: synthesis and functionalisation for biomolecular attachment. World Acedemy Sci Eng Technol 4:1048–1052
Vengsarkar P, Roberts C (2013) Effect of ligand and solvent structure on size-selective nanoparticle dispersability and fractionation in gas expanded liquid (GXL) systems. J Phys Chem C 117:14362–14373
Vengsarkar PS, Roberts CB (2014) Solid-stabilized emulsion formation using stearoyl lactylate coated iron oxide nanoparticles. J Nanopart Res 16:2627. doi:10.1007/s11051-014-2627-4
Von White G, Mohammed FS, Kitchens CL (2011) Small-angle neutron scattering investigation of gold nanoparticle clustering and ligand structure under antisolvent conditions. J Phys Chem C 115:18397–18405. doi:10.1021/jp112020r
Wang CY, Hong JM, Chen G et al (2010) Facile method to synthesize oleic acid-capped magnetite nanoparticles. Chin Chem Lett 21:179–182. doi:10.1016/j.cclet.2009.10.024
Von White G, Kitchens C (2010) Small-angle neutron scattering of silver nanoparticles in gas-expanded hexane. J Phys Chem C 114:16285–16291
Xu Z, Shen C, Tian Y et al (2010) Organic phase synthesis of monodisperse iron oxide nanocrystals using iron chloride as precursor. Nanoscale 2:1027–1032. doi:10.1039/b9nr00400a
Yavuz CT, Mayo JT, Yu WW et al (2006) Low-field magnetic separation of monodisperse Fe3O4 nanocrystals. Science 314:964–967. doi:10.1126/science.1131475
Yu WW, Falkner JC, Yavuz CT, Colvin VL (2004) Synthesis of monodisperse iron oxide nanocrystals by thermal decomposition of iron carboxylate salts. Chem Commun (Camb) 20:2306–2307. doi:10.1039/b409601k
Yu WW, Chang E, Sayes CM et al (2006) Aqueous dispersion of monodisperse magnetic iron oxide nanocrystals through phase transfer. Nanotechnology 17:4483–4487. doi:10.1088/0957-4484/17/17/033
Zhang W (2003) Nanoscale iron particles for environmental remediation: an overview. J Nanopart Res 5:323–332
Zhang L, He R, Gu H (2006) Oleic acid coating on the monodisperse magnetite nanoparticles. Appl Surf Sci 253:2611–2617. doi:10.1016/j.apsusc.2006.05.023
Zhao H, Zhu Q, Gao Y et al (2013) Iron oxide nanoparticles supported on pyrolytic graphene oxide as model catalysts for Fischer Tropsch synthesis. Appl Catal A 456:233–239. doi:10.1016/j.apcata.2013.03.006
Zhou J, Wang L, Qiao X et al (2012) Pickering emulsions stabilized by surface-modified Fe3O4 nanoparticles. J Colloid Interface Sci 367:213–224. doi:10.1016/j.jcis.2011.11.001
Acknowledgements
This research was made possible in part from a grant from the Gulf of Mexico Research Initiative through the Consortium for Molecular Engineering of Dispersant Systems. The authors also thank Dr. Michael Miller and the Auburn University Research and Instrumentation Facility for access to the transmission electron microscope. The authors also greatly appreciate the assistance in the use of characterization equipment by Dr. Allan David and Steven Moore at Auburn University.
Author information
Authors and Affiliations
Corresponding author
Additional information
Pranav S. Vengsarkar and Rui Xu have contributed equally to this work.
Rights and permissions
About this article

Cite this article
Vengsarkar, P.S., Xu, R. & Roberts, C.B. Scalable fractionation of iron oxide nanoparticles using a CO2 gas-expanded liquid system. J Nanopart Res 17, 387 (2015). https://doi.org/10.1007/s11051-015-3196-x
Received:
Accepted:
Published:
DOI: https://doi.org/10.1007/s11051-015-3196-x