
Abstract
A strategy involving the preparation of silver nanoparticles in a biodegradable polymer stemming from an ex situ or an in situ method using a green chemistry process is reported. The influence of the reducing agent concentration and the silver nanoparticles' generation route were investigated on the structure, the morphology, and the properties of the nanocomposite films. Two distinct silver nanoparticle populations in size were highlighted from the ex situ route (diameter around 5 nm for the first one and from 20 to 50 nm for the second one), whereas one population was highlighted from the in situ route (around 10 nm). No modification on the crystalline structure of the starch matrix was observed in presence of silver. Crystalline silver nanoparticles were obtained only from the in situ generation route. The decrease of the water sorption and the improvement of water and oxygen barrier properties were found to be not dependent on the reducing agent concentration but mainly on the crystalline structure of the silver nanoparticles associated to the presence of strong interface between the silver nanoparticles and the starch polymer matrix.
Similar content being viewed by others


Explore related subjects
Discover the latest articles, news and stories from top researchers in related subjects.Avoid common mistakes on your manuscript.
Introduction
Metal nanoparticle-incorporated polymers attract great attention because of widened application scope. Particularly, silver nanocomposite films are promising functional materials in fields such as optical, magnetic, electronic, and antimicrobial properties (Vodnik et al. 2009; Marambio-Jones and Hoek 2010; Singh et al. 2011; An et al. 2015). Many of these properties are found to be controlled by the selection of the polymer as well as the controlled distribution of the uniformly shaped and sized nanoparticles (Valmikanathan et al. 2008). Improved properties are obtained when small dispersed nanodomains are formed. Silver nanoparticles (AgNPs) in various synthetic polymers have been published to give well-dispersed nanoparticles such as polyvinylpyrrolidine, polyethylene glycol, polymethyl methacrylate, polycarbonate, and polyacrylate (Balan et al. 2010; Dhakal et al. 2012; Chen et al. 2012; Udayabhaskar et al. 2012; Sawant et al. 2013). However, in most of these silver nanocomposite films preparations, organic solvents and non-environmentally friendly components are mainly used. In order to minimize or eliminate pollution to the environment, green chemistry processes are required to prepare silver nanoparticles. The uses of nontoxic chemicals, environmentally benign solvents, and renewable materials are some of the key issues that deserve important consideration in a green chemistry strategy.
In the last decade, efforts have been made in the incorporation of AgNPs into biodegradable polymers (Chichova et al. 2014). Polysaccharide polymers, such as chitosan, alginate, and starch, are generally used as host matrices (Brayner et al. 2007; Wei et al. 2009; Božanić et al. 2011; Khan et al. 2013). However, very few papers are reported in the literature concerning the elaboration of silver nanocomposite films from a biodegradable polymer (Yoksan and Chirachanchai 2010; Božanić et al. 2011; Rhim et al. 2013). In the family of renewable-sources and biodegradable polymeric materials, starch is considered as one of the most promising materials in terms of availability and cost. Nevertheless, in the absence of plasticizers, films made from starch are very brittle due to extensive intermolecular forces (Arvanitoyannis 1999; Prakash Maran et al. 2013). So, starch is commonly pre-treated with a plasticizer to overcome film brittleness and to make enabling processing (Mali et al. 2002; Dole et al. 2004; Chen and Evans 2005). However, plasticized starch cannot meet all the requirements of packaging applications. In particular, these materials remain water sensitive and therefore lose their gas barrier properties at high relative humidity (Arvanitoyannis and Biliaderis 1998; Yılmaz et al. 2004; Chen et al. 2005). In this context, the nanocomposites' concept can be a promising option. Thus, a nanocomposite of starch and AgNPs would ideally be suited for antimicrobial food packaging application, especially if generated using a green method (Ranjan et al. 2014).
Different routes to get AgNPs into polymer matrices have been developed over the past few years. Usually, ex situ and in situ methods are the two main routes. The ex situ method consists in dispersing beforehand produced AgNPs within a polymer solution. The resulting mixture is subsequently cast in order to obtain the nanocomposite films (Radheshkumar and Münstedt 2005). The beforehand colloidal AgNPs can be synthesized by physical, biological, or chemical methods. The chemical approach is mostly used and consists in the treatment of silver precursor with a chemical reducing agent (Rafey et al. 2011). To prepare silver nanoparticles (AgNPs) via a green chemistry process, environmental friendly solvent, reducing agent, and stabilizer should be used (Nadagouda et al. 2011; Kahrilas et al. 2014). Water is generally used as the solvent. Aldehyde functions such as in saccharide molecules have been shown of great interest to reduce silver ions (Mehta et al. 2010). Glucose is one of the most widely known green reducing agents due to its chemical reaction rate which allows a compromise between the number of nuclei created and the rate of growth of the AgNPs (Panigrahi et al. 2004; Mehta et al. 2010; Mori et al. 2011). Polyethylene glycol and polysaccharides such as soluble starch and chitosan have been reported as efficient stabilizers of AgNPs (Sharma et al. 2009; Shameli et al. 2012; Fanta et al. 2013). In a nanocomposite approach, the choice of the capping agent is also crucial to allow a good incorporation and final dispersion of AgNPs in the polymer matrix. In the in situ method, a silver precursor is dissolved in a polymer solution. Then, the solution is cast to obtain a precursor film. Afterwards, the AgNPs are formed via a reduction process directly within the film. Depending on the polymer and on the silver precursor, chemical, physical, or thermal treatments can be used to obtain the metal ion reduction (Clémenson et al. 2007, 2008; Ramesh et al. 2009). Whatever the process, the high performance of the nanomaterials depends on the controlled distribution of uniformly shaped and sized nanoparticles. Therefore, the strategy to control nanoparticles growth and agglomeration during nanocomposite preparation appears as a key challenge.
Among all the studies related with AgNPs, very few works have been concerned with the determination of the effect of the in situ and ex situ nanocomposite synthesis methods on the structure and properties of the resulting silver nanocomposite films (Nimrodh Ananth et al. 2011). The originality of this work persists mainly in the preparation of AgNPs in a biodegradable polymer stemming from an ex situ and an in situ methods via a completely green chemistry process. A particular attention is given to have similar parameters in both compared methods. The structure, the morphology, and the properties of the nanocomposite films could be then compared as a function of the method. It was found that the synthesis route had a significant impact on the morphological characteristics and the resulting water and oxygen barrier properties of the nanocomposite films.
Materials and methods
AgNO3 (ACS reagent >99.0 %) was purchased from Aldrich and used as silver precursor. D(+)-Glucose from Merck was used as reducing agent, and soluble starch, supplied from Aldrich, was introduced as nanoparticles' stabilizer. To elaborate nanocomposite films, native potato starch with a weight ratio of amylopectin to amylase equal to 77/23 was purchased from Sigma and glycerol (99 % purity-supplied from Aldrich) was used as plasticizer.
Preparation of the nanocomposite films
Two routes were investigated to elaborate AgNPs starch nanocomposite films. In both processes, the film preparation consisted in the dissolution of potato starch with glycerol (weight ratio 85:15) in distilled water at a concentration of 3 wt%. The starch/glycerol (SG) solution was heated to the gelatinization temperature (85 ± 0.5 °C) and continuously stirred at this temperature for 3 h.
In the first approach called ex situ method, the colloidal silver nanoparticles were firstly prepared. The experimental conditions have already been presented in a previous paper (Cheviron et al. 2014). In a typical experiment, 85 mg of soluble starch was dissolved in 25 mL of distilled water at 85 °C for 30 min. 10 mL of a 6.10−2 M silver nitrate was added into 25 mL of hot aqueous solution of soluble starch under vigorous stirring away from light. Then 15 mL of a glucose solution (0.06 or 0.12 M) was added into the reactive system, which was held at 85 ± 0.5 °C with 700 rpm stirring. A thermostated oil bath and a magnetic stirrer were used to maintain a constant temperature and constant stirring throughout the reaction process. The obtained colloidal dispersion was cooled down at room temperature after 48 h of reaction and characterized. A previous work has shown that silver reduction was achieved after 48 h of reaction. The already prepared colloidal silver nanoparticles' suspension was added to the SG solution and stirred at room temperature. The obtained mixture was poured into polystyrene petri dishes, and water evaporation was carried out at ambient temperature away from light during 4 days. The obtained ex situ nanocomposite films are denoted as Ag–SGexX:Y, with X:Y the molar fractions of AgNO3/glucose of the prepared colloidal AgNPs. The molar fractions used were 1:0, 1:1.5, and 1:3.
In the second approach called in situ method, a silver nitrate solution and a glucose solution were added to the SG solution and stirred at room temperature. The resulting mixture was poured into polystyrene petri dishes, and water evaporation was carried out at ambient temperature away from light during 4 days. After this evaporation, the precursor films were removed from the mold and then directly heated at 85 ± 0.5 °C for 48 h to obtain the nanocomposite films. This temperature and time of annealing were chosen to allow a comparison between the in situ and the ex situ approaches. In the same way, the molar fractions of AgNO3/glucose used were 1:0, 1:1.5, and 1:3 as well. The in situ prepared films are denoted as Ag–SGinX:Y(TT) after the thermal treatment. As previously described, X:Y corresponds to the molar fractions of AgNO3/glucose.
Whatever the process, the silver content in the films was 2 wt% and was expressed with respect to the total matter content including glycerol. Neat matrices were also prepared as control using the same experimental conditions. The neat matrices are denoted as SG 0:0 (without any glucose) and SG 0:3 (with the same glucose amount as the ex situ or the in situ silver/starch nanocomposite films with the AgNO3/glucose ratio 1:3). When neat matrices are also thermal treated (48 h at 85 °C), the denominations are SG 0:0 (TT) and SG 0:3 (TT).
All films were conditioned in dark place at 25 °C and 40 % of relative humidity, between two aluminum sheets before characterization. In all cases, the film thickness was approximately 60 ± 5 µm.
Characterization methods
Ultraviolet–visible (UV–vis) absorption studies were performed on the different materials with a Perkin Elmer Lambda 750 spectrophotometer in the wavelength range of 200–700 nm. The absorption values were normalized taking into account the real thickness of the films.
Transmission electron microscopy (TEM) analysis of AgNPs was carried out with a Philips CM120 electron microscope with an accelerating voltage of 120 kV. Samples were cut with a cryo-ultramicrotome at −90 °C with a Reichert Ultracut S instrument equipped with a diamond knife to obtain ultrathin sections of about 40-nm thicknesses. For each analysis, low electron beam intensity was used and short time of exposure was performed to avoid any evolution of the samples during their exposure to the electron beam. The average diameter and size distribution of the nanoparticles were determined by the ImageJ Software based on the data of an average of 5000 nanoparticles in the nanocomposite films.
X-ray diffractometry (XRD) analyses were carried out on the nanocomposite films using a Cu tube and a Bruker D8 Advance diffractometer. The diffraction patterns were obtained at room temperature in the range of 2θ between 1° and 50° by step of 0.02°. The films were deposited on neutral monosubstrates with a thin transfer adhesive on sides with low scattering response.
Thermogravimetric analyses (TGA) were performed on the nanocomposite films with a TGA Q500 Thermal Analysis system. Data were collected on samples of 3–5 mg during a ramp of temperature from 30 to 600 °C at 10 °C/min under dry helium atmosphere. The first derivate weight/temperature (%/°C) obtained from the mass loss curves allowed determining the degradation temperature at the maximum of the derivate curve (T max).
Dynamic vapor sorption analyzer, DVS Advantage, was used to determine water sorption isotherms of the different samples. The vapor partial pressure was controlled by mixing dry and saturated nitrogen, using electronic mass flow controllers. The experiments were carried out at 25 °C. The initial weight of the sample was approximately 30 mg. The sample was predried in the DVS Advantage by exposure to dry nitrogen until the equilibrated dry mass of the sample was obtained (m 0). A partial pressure of vapor (p) was then established within the apparatus, and the mass of the sample (m t) was followed as a function of time (t). The mass of the sample at equilibrium (m eq) was considered to be reached when changes in mass with time (dm/dt) were lower than 2.10−4 mg min−1 for at least five consecutive minutes. Then, vapor pressure was increased in suitable activity up to 0.9 by step of 0.1. The value of the concentration of water at equilibrium in the material (C) for each water activity (a w) allowed to plot the water sorption isotherm for each sample. C is expressed in \( {\text{cm}}_{\text{STP}}^{ 3} \) per g of material according to the following equation:
The sorption rate was also estimated at each water activity by applying the Fick’s diffusion law. Taking into account the film thickness (L), the water diffusion coefficient (D) was calculated for the short time (m water t/m water eq <0.5) according the following equation:
Here m water t is the mass of water absorbed as a function of the time and m water eq is the mass of water absorbed at equilibrium for a water activity. Experiments were performed for the neat matrices and the nanocomposite films with a molar fraction of AgNO3/glucose of 1:0 and 1:3. The precision on the values of the water concentration at equilibrium and values of the diffusion coefficient was estimated to be better than 5 %.
Water permeability measurements were performed on a Mocon Permatran W 3/33 equipped with an infrared sensor. Nitrogen was used as the carrier gas. The film was placed on an aluminum mask with an open testing area of 5 cm2. The test cell was composed of two chambers separated by the film. Prior to testing, specimens were conditioned in nitrogen inside the unit for at least 12 h to remove traces of atmospheric water. Then, water vapor was introduced in the upstream compartment of the test cell. The water molecules transferred through the film were conducted by the carrier gas to the infrared sensor. The water permeability coefficient (\( P_{{{\text{H}}_{ 2} {\text{O}}}} \)) was calculated considering the following equation:
where L is the thickness of the film, \( J_{{{\text{stH}}_{ 2} {\text{O}}}} \) the water stationary flux, and Δp is the difference of pressure between the upstream and the downstream compartments. \( P_{{{\text{H}}_{ 2} {\text{O}}}} \) can be expressed in barrer (1 barrer = 10−10 cm 3STP cm cm−2 s−1 cmHg−1 = 3.36 × 10−16 mol m m−2 s−1 Pa−1 in SI). Measurements were performed at controlled temperature (T = 25 °C) for a water activity range from 0.45 to 0.8. Measurements were doubled, and the precision on the values of the permeability coefficient was estimated to be better than 5 %.
Oxygen permeability measurements were performed on a Mocon Oxtran 2/21 (Minneapolis, USA) equipped with a Coulox sensor. The film was placed on an aluminum mask with an open testing area of 5 cm2. The test cell was composed of two chambers separated by the film. Nitrogen containing 2 % of hydrogen (N2/H2) was used as the carrier gas and pure oxygen was used as the test gas. The water activity of the two gases was controlled by a humidifier. Prior to testing, specimens were conditioned in N2/H2 inside the unit for at least 24 h on the one hand to remove traces of atmospheric oxygen and on the other hand to be at the water uptake equilibrium condition of the film. Subsequently, oxygen was introduced in the upstream compartment of the test cell. Oxygen transferred through the film was conducted by the carrier N2/H2 gas to the coulometric sensor. The oxygen permeability coefficient \( P_{{{\text{O}}_{ 2} }} \) was calculated considering the following equation:
where L is the thickness of the film, \( J_{{{\text{stO}}_{2} }} \) the oxygen stationary flux, and Δp is the difference of pressure between the upstream and the downstream compartments. \( P_{{{\text{O}}_{ 2} }} \) can be expressed in barrer (1 barrer = 10−10 \( {\text{cm}}_{\text{STP}}^{ 3} \) cm cm−2 s−1 cmHg−1 = 3.36 × 10−16 mol m m−2 s−1 Pa−1). Measurements were performed at controlled temperature (T = 25 °C) and for a water activity range from 0.45 to 0.8. Measurements were doubled, and the precision on the values of the permeability coefficient was estimated to be better than 5 %.
Results and discussion
Morphology of the ex situ nanocomposite films (Ag–SGex)
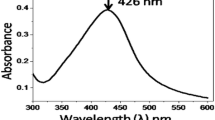
The ex situ nanocomposite films (Ag–SGex), obtained from the dispersion of synthesized colloidal nanoparticles within the matrix by solvent cast process, were handleable and not brittle. Furthermore, the colorless neat matrix film changed to light-orange color when AgNPs were introduced. The UV–visible absorption spectra of the different Ag–SGex and the corresponding neat matrix films are presented in Fig. 1a. Whatever the AgNO3/glucose molar ratio, the absorbance SPR peak confirmed the presence of the AgNPs (Raveendran et al. 2003; Mehta et al. 2010; Mori et al. 2011; Shameli et al. 2012). All UV–visible absorption spectra of nanocomposite films exhibited a symmetrical UV peak centered at λ max = 416 nm. It can be remarked that a SPR peak was observed, although no glucose was introduced in the mixture. Thus, soluble starch used to synthesize silver colloidal suspension had a double role: it can act as stabilizer and reducing agent. As explained by Gao et al., the aldehyde terminal of soluble starch can reduce silver nitrate, while the hydroxyl groups and the glycosidic bonds can stabilize the AgNPs (Gao et al. 2011).

UV–Visible absorption spectra with different AgNO3/Glucose molar ratios (1:3, 1:1.5, and 1:0) of: a corresponding ex situ nanocomposite films (Ag–SGex) and b corresponding in situ nanocomposite films (Ag–SGin)
The sizes and morphology of synthesized AgNPs were imaged using Transmission Electron Microscopy (TEM) analysis. TEM images of the AG–SGex films are shown in Fig. 2a–c. The spherical nanoparticles were homogeneously distributed in the starch matrix. No blank space free from nanoparticles was observed in the whole sample surface examined. However, two populations of nanoparticles coexisted. The first one was composed of large nanoparticles and the second one of very small nanoparticles. A quantitative analysis of the nanoparticle sizes was performed. The obtained mean average nanoparticle size (\( \overline{d} \)) and associated standard deviation (σ) values for the AG–SGex films are given in Table 1. Whatever the amount of glucose during the synthesis of the silver colloidal suspension, the average size of the smallest nanoparticles was similar, around 5 nm. The number of AgNPs bigger than 10 nm in the absence of glucose and in the presence of glucose represented 40 and 15 % of the total number, respectively. Thus, the role of the reducing agent in the nucleation step of the colloidal nanoparticles' synthesis was confirmed. The presence of glucose allowed a greater number of silver nuclei leading to a lower AgNPs size. However, from the ex situ method, some AgNPs bigger than 60 nm were also observed.

Transmission electron micrographs of the nanocomposite films (Ag–SGex and Ag–SGin(TT)) with AgNO3/Glucose molar ratios 1:3, 1:1.5, and 1:0
The XRD patterns of the ex situ nanocomposite films and the associated neat starch/glycerol film are presented in Fig. 3a. The diffraction diagram of the neat matrix showed the typical diffraction peaks of starch tubers, such as B-type crystals (Zobel 1988; Zeppa et al. 2009), at 2θ = 5.6, 15.2, 17.2, 19.8, and 22.5°. After the incorporation of AgNPs into the polymer matrix, no significant difference was observed in the 2θ range from 5 to 50° meaning that either the amount of glucose or the presence of AgNPs had an impact on the matrix crystalline morphology. Lastly, it is noteworthy that the XRD patterns of the Ag–SGex films did not exhibit any characteristic diffraction peak for the silver nanoparticles. Two explanations are possible. Firstly, the low amount of silver and the small size of the metal particles could lead to broadened reflection that could be difficult to distinguish from the background level of the analysis. However, for a similar AgNPs content, silver diffraction peak has been already observed in other polymer matrices (Clémenson et al. 2007, 2008). Secondly, the absence of the silver diffraction peak could also be explained by amorphous nanoparticles. Indeed, few authors studied XRD patterns of AgNPs and most of them applied beforehand a thermal treatment to get the silver diffraction peak observable (Vaidya et al. 2010). Amorphous AgNPs have been clearly evidenced by Liu et al. (Liu et al. 2001). Their nanoparticles were prepared by a pulse sonochemical technique from an aqueous solution of AgNO3 (Salkar et al. 1999) and AgBr in the presence of gelatin (Liu et al. 2001). Amorphous AgNPs have been also obtained by Ghaseminezhad et al. (2012) for a nanoparticle synthesis method similar to the one used in this work. Thus, amorphous nanoparticles were suspected to be obtained from the ex situ route.

XRD patterns of starch neat matrix and different nanocomposite films with different AgNO3/Glucose molar ratios (1:3, 1:1.5, and 1:0): a ex situ nanocomposite films SGex and b in situ nanocomposite films SGin(TT)
Morphology of the in situ nanocomposite films (Ag–SGin(TT))
The in situ silver nanoparticles' generation route consists of dispersing the silver nitrate in a glycerol plasticized potato starch film and reducing it by thermal treatment of the precursor film. The thermal treatment (TT) was set for 48 h at 85 °C in order to get comparable parameters between the in situ and the ex situ route. In this way, the corresponding neat matrices of the in situ nanocomposite films were thermal treated as well to understand the observed evolutions.
After the thermal treatment, the in situ nanocomposite films (Ag–SGin(TT)) became dark orange and brittle. The UV–visible absorption spectra of the different in situ nanocomposite films and the associated neat matrix film after the same thermal treatment were also determined. The UV–visible absorption spectra are presented in Fig. 1b. Whatever the molar ratio of AgNO3/glucose, a saturation effect in the absorbance SPR peak was observed. The dispersion of the AgNPs of the Ag–SGin(TT) films was further analyzed using Transmission Electron Microscopy. Characteristic examples of the obtained images are shown in Fig. 2c–f. The in situ nanocomposite films exhibited a homogeneous dispersion of spherical AgNPs with an average size and standard deviation of only 12 and 5 nm, respectively (Table 1). The presence and the amount of reducing agent did not have an influence on the size of the AgNPs. Contrary to the ex situ generation method, only one population in size with a low size distribution was observed. The number of AgNPs bigger than 20 nm represented less than 2 % of the total number. This low size and low size distribution can be attributed to the presence of extensive hydroxyl groups and glycosidic bonds of starch polymer which can facilitate the interactions of silver ions to starch matrix and also to the high medium viscosity which prevented the nanoparticles' aggregation (Vigneshwaran et al. 2006; Valodkar et al. 2010, 2011; Gao et al. 2011).
The XRD patterns of the in situ nanocomposite films are presented in Fig. 3b. For all films, in addition to the diffraction peaks of the potato starch observed in the 2θ range from 5 to 30°, a broad diffraction peak located at 2θ = 38° was observed. This diffraction peak corresponding to the (111) crystal plan of the face-centered cubic (fcc) silver demonstrates that crystalline silver was formed by the in situ generation route (Johnson et al. 2010). High broadening peaks of the crystalline AgNPs have already been observed by Clémenson et al. and were attributed to the small size of the AgNPs and also to the low amount of these nanoparticles within the polymer matrix (Clémenson et al. 2007, 2008).
Therefore, elaboration of silver starch nanocomposite films exhibited marked differences in size, size distribution, and crystalline structure of AgNPs as a function of the generation route. The properties of the silver starch nanocomposite films obtained from ex situ and in situ methods will now be compared for both AgNO3/glucose molar ratios 1:0 and 1:3.
Thermal properties of the nanocomposite films
The degradation process and the thermal stability of the nanocomposite films were studied by thermal gravimetric analysis (TGA). Three steps were evidenced on the weight loss curve of all films (Fig. 4a, b). The first one between room temperature and 140 °C corresponded to water loss. The second mass loss between 180 and 250 °C was assigned to the evaporation of glycerol in agreement with Yang et al.'s study (Yang et al. 2006). The last step corresponded to the starch and glucose decomposition. No loss of glycerol or glucose occurred at 85 °C, and only water content was evaporated. The values of the temperature of the starch matrix decomposition (T max) were determined at the maximum of the derivate peak. Whatever the silver incorporation process and the amount of glucose, no significant difference was found in the thermal stability of starch. The obtained values of T max were closed to each other, around 319 ± 2 °C. In the literature, the degradation temperature of starch is known around 310–320 °C and is attributed to the decomposition of the strong chemical bonds of glucose and starch (Cyras et al. 2006; Božanić et al. 2007; Magoń and Pyda 2011; Dai et al. 2015). It can then be noted that the stability of the nanocomposite films was slightly higher compared to the associated neat matrix. This result can be attributed to the presence of the AgNPs in the nanocomposite films which can delay the thermal degradation of the starch polymer chains (T max = 315 °C).

Example of the thermal degradation of ex situ and in situ nanocomposite films with the molar ratio AgNO3/glucose 1:3: a Thermogravimetric curves and b Derived thermogravimetric curve
Water sorption properties
In order to evaluate the effect of the thermal treatment and the amount of glucose on the water sorption capacity of the films, water sorption analyses were also performed on the neat matrices with and without glucose and before and after thermal treatment. The sorption isotherms of the different films are presented in Fig. 5a. The experimental data defined a sigmoidal shape corresponding to BET II in the classification of Brunauer–Emmett–Teller (Brunauer et al. 1940; Labes 1965). The BET II model, which is a combination of dual-mode (Langmuir sorption and Henry law) and clustering contribution, is a typical model of water sorption in hydrophilic materials (Hellman et al. 1952; Gouanvé et al. 2006). The first part of the isotherm presented a concave form, which is usually analyzed as the sorption step corresponding to the formation of the primary hydration sphere of the hydroxyl groups which act as Langmuir sites. The second part was linear and corresponded to a Henry sorption mode. The last part of the isotherm presents a convex form, which can be explained by the formation of water clustering (Bessadok et al. 2009). In the range of water activity 0 < a w < 0.6, the amount of water absorbed at equilibrium was lower for neat matrix containing glucose. This could be related to a decrease of the available hydroxyl groups of starch in the presence of glucose molecules. Hydroxyl groups of glucose could form hydrogen bonds with hydroxyl groups of starch, which led to a decreasing number of available sorption sites. (Talja et al. 2007). At higher water activity (a w > 0.6), the water sorbed molecules weaken the glucose starch hydrogen bonds, leading to a same amount of water at equilibrium for the different films. Whatever the amount of glucose, it can be then noticed that the thermal treatment did not have a significant effect on the water sorption capacity of the films. In each case, a single curve was thus obtained showing a superimposition of the sorption isotherms in the whole range of water activity. The effect of the thermal treatment and the amount of glucose on the neat matrices were also investigated on the water sorption kinetic. The evolutions of water diffusion coefficient (D) as a function of the water concentration at equilibrium for the different films are plotted in Fig. 5b. The diffusion coefficients D were not constant in the whole range of water concentration, meaning that D was dependent on the amount of water molecules sorbed by the films. The water diffusion first increased and then decreased. These variations were in agreement with the water sorption mechanism complying with the BET II sorption mode and the trends generally observed for starch-based films (Masclaux et al. 2010). The increase of D for low water content can be explained by the dual-mode sorption contribution. Water molecules predominately were sorbed on hydroxyl groups in which they are partially immobilized. Then, Henry’s type sorption was dominant and the diffusion coefficient increases with the contribution of mobility of the water molecules. For high water content, the decrease of water diffusion coefficient could be attributed to the water clustering phenomenon which can be assigned to the increase of cluster size of the diffusing water molecules which became fewer movables. It can be remarked that the cluster phenomenon occured from water activity of 0.5 and from a water concentration at equilibrium of 120 \( {\text{cm}}_{\text{STP}}^{ 3} \) g−1. Finally, in the presence or absence of glucose, it can be concluded that the thermal treatment did not have significant effect on the water sorption properties in thermodynamic and in kinetic point of view. As a consequence, it was expected that the possible change in water sorption properties of nanocomposite films will mainly depend on the presence and on the structure of the AgNPs.

Water sorption properties expressed for the neat starch/glycerol matrices before and after thermal treatment (TT) as a the water concentration at equilibrium versus the water activity and b the diffusion coefficient versus the water concentration at equilibrium in \( {\text{cm}}_{\text{STP}}^{ 3} \) per g of polymer
The sorption isotherms of the nanocomposite films are presented in Fig. 6a, b. Whatever the molar fraction of AgNO3/glucose, for the Ag–SGex films, in the whole range of water activity, no noteworthy difference of water concentration at equilibrium was observed compared to the associated neat matrix. For the Ag–SGin films, in absence of glucose, the water uptake of the nanocomposite film was lower in the whole range of activity compared to the associated neat matrix. However, in presence of glucose, no significant difference of water uptake was observed in the activity range lower than 0.5. For water activity up to 0.5, a slight decrease of water uptake was only observed for the nanocomposite film compared to the associated neat matrix at higher activity (Fig. 6b). It can be also noticed that the water uptake of both in situ nanocomposite films was not dependent on the presence of glucose. The effect of the presence of AgNPs and the amount of glucose on the water diffusion could also be evidenced in Fig. 6c, d. For each system, in the whole range of water activity, the shape of the curve relative to the nanocomposite films is similar compared to those of the associated neat matrices. D first increased up to a water concentration of 120 \( {\text{cm}}_{\text{STP}}^{ 3} \) g−1 and then decreased. In a quantitative point of view, whatever the molar fraction of AgNO3/glucose in the whole water concentration range, no noteworthy difference was observed for the Ag–SGex films compared to the neat matrix. Concerning the Ag–SGin films, for low concentration of water at equilibrium, typically below 120 \( {\text{cm}}_{\text{STP}}^{ 3} \) g−1, the water coefficient diffusion values were lower compared to the neat matrix. So the silver reduction method had an influence on the water diffusion whatever the glucose amount. Thus, the crystalline structure of the in situ AgNPs could limit in a higher extent the water diffusion in the starch matrix. The increase of water diffusion as a function of the water concentration at equilibrium is generally related to a plasticization phenomenon. This phenomenon could be described by the following relationship:
where D 0 is the limit diffusion coefficient defined at nil concentration in cm2 s−1 and can give information on the microstructure of the materials, γ is the plasticization coefficient in g \( {\text{cm}}_{\text{STP}}^{ 3} \), and C is the water concentration at equilibrium in \( {\text{cm}}_{\text{STP}}^{ 3} \) g−1. The parameters of Eq. 5, D 0 and γ, were deduced from a linear regression of the curve at low water concentration. The obtained values of the parameters D 0 and γ are reported in Table 2. The values were in the same range than the values mentioned by Masclaux et al. for the study of water sorption of starch glycerol films (Masclaux et al. 2010). Taking into account the uncertainty of the determination of the parameters, it can be noticed that the values of γ were identical and were not dependent on the presence of glucose or the way of AgNPs generation. Furthermore, the obtained values of D 0 did not depend on the presence of glucose for the neat matrices. Besides, the values for the Ag–SGex films were nearly identical to those obtained for the neat matrices. The values of D 0 for the Ag–SGin films were also independent on the presence of glucose. However, they were 50 % lower compared to those obtained for the associated neat matrices. The high decrease of D 0 could be firstly explained by the presence of the crystalline AgNPs which are considered as water barriers and an obstacle to water diffusion. And finally, the change of diffusion could also be explained by a strong interaction between the crystalline AgNPs and the potato starch polymer.

Water sorption properties of the different nanocomposite films using different AgNO3/Glucose molar ratios (1:3 and 1:0) and associated neat starch matrix: a, b the water concentration at equilibrium versus the water activity and c, d the diffusion coefficient versus the water concentration at equilibrium
Water permeation
The evolutions of water coefficient permeability \( P_{{{\text{H}}_{ 2} {\text{O}}}} \) as a function of the water activity for the neat matrices are represented with a semi-log plot in Fig. 7a. The value for the neat matrix SG0:0 at a w = 0.75 was in a good agreement with those obtained by Tang et al. (2008). For all systems, the same general trend was observed as a function of the water activity. The water permeability coefficient increases in the range of water activity from 0.4 to 0.8. This variation could be related to the general water sorption mechanism discussed previously. Indeed, the starch matrix polymer was increasingly plasticized by the sorbed water molecules, resulting in the increase of the water permeability. The evolutions of \( P_{{{\text{H}}_{ 2} {\text{O}}}} \) of the neat matrices show that the water permeability did not depend on the presence of glucose in the potato starch films. The relative permeability which is defined as the ratio of the permeability coefficient of the sample on the permeability coefficient of its associated matrix was calculated. To discuss the effect of the silver nanocomposite elaboration route on the water transport properties, the permeability values of the Ag–SGex and Ag–SGin films were divided by the ones of the respective neat matrices (SG0:0 and SG0:3), considering both the amount of glucose. Thus, the relative permeability coefficients could only be dependent on the eventual thermal treatment and on the presence of AgNPs. The evolution of the calculated relative water permeability for the silver starch nanocomposite films is represented in Fig. 7b. The relative permeability values were close to 1 for all Ag–SGex films. Thus, whatever the amount of glucose, the AgNPs from the ex situ route did not influence the water permeability coefficient of starch films. A significant decrease of the relative water permeability coefficient was observed for the Ag–SGin films, and this for the whole tested range of water activity. As example, at a w = 0.5, the relative water permeability coefficients of the Ag–SGin1:0(TT) and Ag–SGin1:3 (TT) films were 0.36 and 0.2, respectively. For the same water activity, the obtained relative permeability of treated neat matrices films was equal to 0.5 for both matrices. Thus, the thermal treatment itself did not explain this high decrease of water permeability of the Ag–SGin films. It can be concluded that the decrease of \( P_{{{\text{H}}_{ 2} {\text{O}}}} \) was also due to the presence of crystalline AgNPs within the starch polymer matrix. However, the tortuosity effect induced by the small amount of silver and the spherical shape of impermeable nanoparticles was not sufficient to explain the high decrease of water permeability. Indeed, the permeability decrease was much higher than the theoretical decrease calculated by Maxwell law (Frisch 1970). Compton et al. have already observed a decrease of permeability higher than the theoretical decrease for the in situ generation of palladium nanoparticles in a polyimide matrix (Compton et al. 2006). Thus, the improvement of barrier properties can also be due to the decrease of the water diffusion rate at the strong filler/matrix interface, as previously evidenced by the water sorption properties analysis. It can also be noticed that a small difference of relative water permeability was observed between the nanocomposite films Ag–SGin1:0(TT) and Ag–SGin1:3(TT). Thus, we could suspect that the interactions between the in situ grown AgNPs and the starch matrix were more effective in the presence of glucose for the water permeation.

a Evolution of the water permeability coefficient as a function of the water activity for the neat matrices and b Evolution of relative permeability of the ex situ and in situ nanocomposite films as a function of the water activity
Oxygen permeation
The evolutions of oxygen permeability coefficient \( (P_{{{\text{O}}_{ 2} }} ) \) as a function of the water activity (a w) for the neat matrices are represented with semi-log plot in Fig. 8a. The obtained value of \( P_{{{\text{O}}_{ 2} }} \) for SG0:0 at a w = 0.75 is in good agreement with those reported in the literature by Gaudin et al. (Gaudin et al. 2000). For all films, the same general trend is observed. \( P_{{{\text{O}}_{ 2} }} \) tended to increase as the water activity increases. Indeed, the plasticization effect due to the water molecules sorbed by the polymer tends to decrease the cohesive density energy of the nanocomposite. Similar behaviors have been reported in the literature for plasticized starch-based films (Gaudin et al. 1999, 2000; Bertuzzi et al. 2007). With the increase of water activity, the amount of water in the films increased and then the water sorbed molecules tended to weaken the intermolecular and intramolecular hydrogen bonding, leading to a drastic decrease of the cohesive energy density of the polymer matrix. Thus, segmental motion of the starch chain increased by mean of plasticization effect resulting in a large increase of oxygen permeability. The comparison between evolutions of \( P_{{{\text{O}}_{ 2} }} \) of the neat matrices SG0:0 and SG0:3 is presented in Fig. 8a and shows that the oxygen permeability did not depend on the presence of glucose within the matrix for the whole range of water activity tested. Then, to discuss more specifically the effect of the thermal treatment and the silver nanocomposite elaboration route on the oxygen transport properties, the oxygen relative permeability values were determined and the obtained values are shown in Fig. 8b. As previously observed for the water permeability, the oxygen permeability coefficients of the Ag–SGex films are identical to the ones obtained for the corresponding matrices. However, for the Ag–SGin films, a significant decrease of the oxygen relative permeability coefficient was observed in the presence or absence of glucose within the film. At a w = 0.5, the relative permeability is around 0.1. As in the case of water permeability, the thermal treatment had an influence on the oxygen permeability of the neat matrices. The obtained relative permeabilities of treated neat matrices at a w = 0.5 are equal to 0.45 and 0.36 for SG0:0(TT) and SG0:3(TT), respectively. Here again, such high decrease of relative permeability for the Ag–SGin films was due to the presence of the crystalline AgNPs within the biodegradable polymer matrix on the one hand and on the other hand to an effect of the nanoparticle/polymer interface. Such effects have been already evidenced in previous work devoted to in situ growth of metal particles within polymer (Compton et al. 2006; Simon and Espuche 2014; Simon et al. 2014).

a Evolution of the oxygen permeability coefficient as a function of the water activity for the neat matrices and b relative oxygen permeability of the ex situ and in situ nanocomposite films as a function of the water activity
Thus, we demonstrated first that the thermal treatment enhanced the oxygen barrier properties and then the silver crystalline nanoparticles improved it further through a strong interface with starch. In this way, the in situ method allows a maximum decrease of 90 % of the oxygen permeability on both effects combined, as shown in Fig. 8b. The in situ elaboration of crystalline silver starch nanocomposite film was finally an efficient method to obtain a barrier biodegradable-based nanocomposite film.
Conclusions
Silver nanoparticles in a starch biodegradable polymer stemming from an ex situ and an in situ methods via a completely green chemistry process were prepared. The obtained nanocomposite films exhibit marked differences in their morphology as a function of the silver incorporation approach. The nanoparticles produced by the ex situ method are spherical and well dispersed within the polymer matrix. However, two distinct nanoparticle populations in size are clearly highlighted. The first population corresponds to the silver nanoparticles characterized by a mean average size contained between 20 and 50 nm which is dependent on the glucose amount in the reaction mixture. The higher amount of glucose leads to lower nanoparticle size. The second population corresponds to very small nanoparticles (around 5 nm) whose size is independent on the amount of glucose in the mixture. The nanoparticles produced by the in situ method are also spherical and well dispersed within the polymer matrix. However, only one population more homogenous and smaller in size is highlighted. It is characterized by a small average size (around 10 nm) which is independent on the amount of glucose. Amorphous silver nanoparticles are obtained using the ex situ method, whereas crystalline silver nanoparticles are obtained using the in situ method. Whatever the silver incorporation approach, no significant difference in starch thermal stability is observed. However, the water sorption, water, and oxygen barrier properties of the nanocomposite films exhibit a strong dependence on the silver nanoparticles' generation method. For the same amount of silver, a decrease of the water uptake and a significant enhancement of the water and oxygen barrier properties are obtained solely for the in situ nanocomposite films with respect to the associated neat matrix. It was then pointed out that the crystalline in situ silver nanoparticles and the cohesive interface between the crystalline silver nanoparticles and the starch matrix improved significantly the water and oxygen barrier properties of the nanocomposite films. The two silver nanoparticles' approaches have a pronounced influence on the nanoparticles' morphology and structure and on the transport properties of the silver starch nanocomposite films.
References
An J, Luo Q, Li M et al (2015) A facile synthesis of high antibacterial polymer nanocomposite containing uniformly dispersed silver nanoparticles. Colloid Polym Sci. doi:10.1007/s00396-015-3589-5
Arvanitoyannis I (1999) Physical properties of polyol-plasticized edible blends made of methyl cellulose and soluble starch. Carbohydr Polym 38:47–58. doi:10.1016/S0144-8617(98)00087-3
Arvanitoyannis I, Biliaderis CG (1998) Physical properties of polyol-plasticized edible films made from sodium caseinate and soluble starch blends. Food Chem 62:333–342. doi:10.1016/S0308-8146(97)00230-6
Balan L, Malval J-P, Schneider R et al (2010) In-situ fabrication of polyacrylate–silver nanocomposite through photoinduced tandem reactions involving eosin dye. Polymer 51:1363–1369. doi:10.1016/j.polymer.2009.05.003
Bertuzzi MA, Castro Vidaurre EF, Armada M, Gottifredi JC (2007) Water vapor permeability of edible starch based films. J Food Eng 80:972–978. doi:10.1016/j.jfoodeng.2006.07.016
Bessadok A, Langevin D, Gouanvé F et al (2009) Study of water sorption on modified Agave fibres. Carbohydr Polym 76:74–85. doi:10.1016/j.carbpol.2008.09.033
Božanić DK, Djoković V, Blanuša J et al (2007) Preparation and properties of nano-sized Ag and Ag2S particles in biopolymer matrix. Eur Phys J E 22:51–59. doi:10.1140/epje/e2007-00008-y
Božanić DK, Djoković V, Dimitrijević-Branković S et al (2011) Inhibition of microbial growth by silver-starch nanocomposite thin films. J Biomater Sci Polym Ed 22:2343–2355. doi:10.1163/092050610X539532
Brayner R, Vaulay M-J, Fiévet F, Coradin T (2007) Alginate-mediated growth of Co, Ni, and CoNi nanoparticles: influence of the biopolymer structure. Chem Mater 19:1190–1198
Brunauer S, Deming LS, Deming WE, Teller E (1940) On a theory of the van der waals adsorption of gases. J Am Chem Soc 62:1723–1732. doi:10.1021/ja01864a025
Chen B, Evans JRG (2005) Thermoplastic starch–clay nanocomposites and their characteristics. Carbohydr Polym 61:455–463. doi:10.1016/j.carbpol.2005.06.020
Chen M, Chen B, Evans JRG (2005) Novel thermoplastic starch–clay nanocomposite foams. Nanotechnology 16:2334–2337. doi:10.1088/0957-4484/16/10/056
Chen C, Li J, Luo G et al (2012) Size-controlled in situ synthesis and photo-responsive properties of silver/poly(methyl methacrylate) nanocomposite films with high silver content. Appl Surf Sci 258:10180–10184. doi:10.1016/j.apsusc.2012.06.102
Cheviron P, Gouanvé F, Espuche E (2014) Green synthesis of colloid silver nanoparticles and resulting biodegradable starch/silver nanocomposites. Carbohydr Polym 108:291–298. doi:10.1016/j.carbpol.2014.02.059
Chichova M, Shkodrova M, Vasileva P et al (2014) Influence of silver nanoparticles on the activity of rat liver mitochondrial ATPase. J Nanoparticle Res. doi:10.1007/s11051-014-2243-3
Clémenson S, David L, Espuche E (2007) Structure and morphology of nanocomposite films prepared from polyvinyl alcohol and silver nitrate: influence of thermal treatment. J Polym Sci Part Polym Chem 45:2657–2672. doi:10.1002/pola.22020
Clémenson S, Léonard D, Sage D et al (2008) Metal nanocomposite films prepared in situ from PVA and silver nitrate. Study of the nanostructuration process and morphology as a function of thein situ routes. J Polym Sci Part Polym Chem 46:2062–2071. doi:10.1002/pola.22541
Compton J, Thompson D, Kranbuehl D et al (2006) Hybrid films of polyimide containing in situ generated silver or palladium nanoparticles: effect of the particle precursor and of the processing conditions on the morphology and the gas permeability. Polymer 47:5303–5313. doi:10.1016/j.polymer.2006.05.048
Cyras VP, Tolosa Zenklusen MC, Vazquez A (2006) Relationship between structure and properties of modified potato starch biodegradable films. J Appl Polym Sci 101:4313–4319. doi:10.1002/app.23924
Dai L, Qiu C, Xiong L, Sun Q (2015) Characterisation of corn starch-based films reinforced with taro starch nanoparticles. Food Chem 174:82–88. doi:10.1016/j.foodchem.2014.11.005
Dhakal TR, Mishra SR, Glenn Z, Rai BK (2012) Synergistic effect of PVP and PEG on the behavior of silver nanoparticle-polymer composites. J Nanosci Nanotechnol 12:6389–6396. doi:10.1166/jnn.2012.6561
Dole P, Joly C, Espuche E et al (2004) Gas transport properties of starch based films. Carbohydr Polym 58:335–343. doi:10.1016/j.carbpol.2004.08.002
Fanta GF, Kenar JA, Felker FC, Byars JA (2013) Preparation of starch-stabilized silver nanoparticles from amylose–sodium palmitate inclusion complexes. Carbohydr Polym 92:260–268. doi:10.1016/j.carbpol.2012.09.016
Frisch HL (1970) In: Crank J, Park GS (eds) Diffusion in polymers. Academic Press, New York, 1968. J Appl Polym Sci 14:1657–1657. doi: 10.1002/app.1970.070140623
Gao X, Wei L, Yan H, Xu B (2011) Green synthesis and characteristic of core-shell structure silver/starch nanoparticles. Mater Lett 65:2963–2965. doi:10.1016/j.matlet.2011.06.020
Gaudin S, Lourdin D, Le Botlan D et al (1999) Plasticisation and mobility in starch-sorbitol films. J Cereal Sci 29:273–284
Gaudin S, Lourdin D, Forssell PM, Colonna P (2000) Antiplasticisation and oxygen permeability of starch–sorbitol films. Carbohydr Polym 43:33–37
Ghaseminezhad SM, Hamedi S, Shojaosadati SA (2012) Green synthesis of silver nanoparticles by a novel method: comparative study of their properties. Carbohydr Polym 89:467–472. doi:10.1016/j.carbpol.2012.03.030
Gouanvé F, Marais S, Bessadok A et al (2006) Study of water sorption in modified flax fibers. J Appl Polym Sci 101:4281–4289. doi:10.1002/app.23661
Hellman NN, Boesch TF, Melvin EH (1952) Starch granule swelling in water vapor sorption. J Am Chem Soc 74:348–350. doi:10.1021/ja01122a016
Johnson L, Thielemans W, Walsh DA (2010) Nanocomposite oxygen reduction electrocatalysts formed using bioderived reducing agents. J Mater Chem 20:1737. doi:10.1039/b922423h
Kahrilas GA, Haggren W, Read RL et al (2014) Investigation of antibacterial activity by silver nanoparticles prepared by microwave-assisted green syntheses with soluble starch, dextrose, and arabinose. ACS Sustain Chem Eng 2:590–598. doi:10.1021/sc400487x
Khan Z, Singh T, Hussain JI et al (2013) Starch-directed green synthesis, characterization and morphology of silver nanoparticles. Colloids Surf B Biointerfaces 102:578–584. doi:10.1016/j.colsurfb.2012.08.057
Labes MM (1965) Physics and chemistry of the organic solid state, volume 2 edition. Wiley, New York
Liu S, Huang W, Chen S et al (2001) Synthesis of X-ray amorphous silver nanoparticles by the pulse sonoelectrochemical method. J Non-Cryst Solids 283:231–236. doi:10.1016/S0022-3093(01)00362-3
Magoń A, Pyda M (2011) Melting, glass transition, and apparent heat capacity of α-d-glucose by thermal analysis. Carbohydr Res 346:2558–2566. doi:10.1016/j.carres.2011.08.022
Mali S, Grossmann MVE, Garcia MA et al (2002) Microstructural characterization of yam starch films. Carbohydr Polym 50:379–386. doi:10.1016/S0144-8617(02)00058-9
Marambio-Jones C, Hoek EMV (2010) A review of the antibacterial effects of silver nanomaterials and potential implications for human health and the environment. J Nanoparticle Res 12:1531–1551. doi:10.1007/s11051-010-9900-y
Masclaux C, Gouanvé F, Espuche E (2010) Experimental and modelling studies of transport in starch nanocomposite films as affected by relative humidity. J Membr Sci 363:221–231. doi:10.1016/j.memsci.2010.07.032
Mehta SK, Chaudhary S, Gradzielski M (2010) Time dependence of nucleation and growth of silver nanoparticles generated by sugar reduction in micellar media. J Colloid Interface Sci 343:447–453. doi:10.1016/j.jcis.2009.11.053
Mori Y, Tagawa T, Fujita M et al (2011) Simple and environmentally friendly preparation and size control of silver nanoparticles using an inhomogeneous system with silver-containing glass powder. J Nanoparticle Res 13:2799–2806. doi:10.1007/s11051-010-0168-z
Nadagouda MN, Speth TF, Varma RS (2011) Microwave-assisted green synthesis of silver nanostructures. Acc Chem Res 44:469–478. doi:10.1021/ar1001457
Nimrodh Ananth A, Umapathy S, Sophia J et al (2011) On the optical and thermal properties of in situ/ex situ reduced Ag NP’s/PVA composites and its role as a simple SPR-based protein sensor. Appl Nanosci 1:87–96. doi:10.1007/s13204-011-0010-7
Panigrahi S, Kundu S, Ghosh S et al (2004) General method of synthesis for metal nanoparticles. J Nanoparticle Res 6:411–414
Prakash Maran J, Sivakumar V, Thirugnanasambandham K, Kandasamy S (2013) Modeling and analysis of film composition on mechanical properties of maize starch based edible films. Int J Biol Macromol 62:565–573. doi:10.1016/j.ijbiomac.2013.09.027
Radheshkumar C, Münstedt H (2005) Morphology and mechanical properties of antimicrobial polyamide/silver composites. Mater Lett 59:1949–1953. doi:10.1016/j.matlet.2005.02.033
Rafey A, Shrivastavaa KBL, Iqbal SA, Khan Z (2011) Growth of Ag-nanoparticles using aspartic acid in aqueous solutions. J Colloid Interface Sci 354:190–195. doi:10.1016/j.jcis.2010.10.046
Ramesh GV, Porel S, Radhakrishnan TP (2009) Polymer thin films embedded with in situ grown metal nanoparticles. Chem Soc Rev 38:2646. doi:10.1039/b815242j
Ranjan S, Dasgupta N, Chakraborty AR et al (2014) Nanoscience and nanotechnologies in food industries: opportunities and research trends. J Nanoparticle Res. doi:10.1007/s11051-014-2464-5
Raveendran P, Fu J, Wallen SL (2003) Completely “green” synthesis and stabilization of metal nanoparticles. J Am Chem Soc 125:13940–13941. doi:10.1021/ja029267j
Rhim JW, Wang LF, Hong SI (2013) Preparation and characterization of agar/silver nanoparticles composite films with antimicrobial activity. Food Hydrocoll 33:327–335. doi:10.1016/j.foodhyd.2013.04.002
Salkar RA, Jeevanandam P, Aruna ST et al (1999) The sonochemical preparation of amorphous silver nanoparticles. J Mater Chem 9:1333–1335. doi:10.1039/A900568D
Sawant SN, Selvaraj V, Prabhawathi V, Doble M (2013) Antibiofilm properties of silver and gold incorporated PU, PCLm, PC and PMMA nanocomposites under two shear conditions. PLoS One 8:e63311. doi:10.1371/journal.pone.0063311
Shameli K, Bin Ahmad M, Jazayeri SD et al (2012) Synthesis and characterization of polyethylene glycol mediated silver nanoparticles by the green method. Int J Mol Sci 13:6639–6650. doi:10.3390/ijms13066639
Sharma VK, Yngard RA, Lin Y (2009) Silver nanoparticles: green synthesis and their antimicrobial activities. Adv Colloid Interface Sci 145:83–96. doi:10.1016/j.cis.2008.09.002
Simon S, Espuche E (2014) Effect of different metal in situ growing routes on the morphology and gas separation properties of polyetherimide/palladium nanocomposite asymmetric membranes. Sep Purif Technol 129:41–49. doi:10.1016/j.seppur.2014.03.023
Simon S, Alcouffe P, Espuche E (2014) Hybrid films of polyetherimide containing in situ grown Ag, Pd, and AgPd alloy nanoparticles: synthesis route, morphology, and gas transport properties. J Polym Sci Part B Polym Phys 52:1211–1220. doi:10.1002/polb.23545
Singh M, Sinha I, Singh AK, Mandal RK (2011) Formation of fractal aggregates during green synthesis of silver nanoparticles. J Nanoparticle Res 13:69–76. doi:10.1007/s11051-010-0001-8
Talja RA, Helén H, Roos YH, Jouppila K (2007) Effect of various polyols and polyol contents on physical and mechanical properties of potato starch-based films. Carbohydr Polym 67:288–295. doi:10.1016/j.carbpol.2006.05.019
Tang X, Alavi S, Herald TJ (2008) Effects of plasticizers on the structure and properties of starch–clay nanocomposite films. Carbohydr Polym 74:552–558. doi:10.1016/j.carbpol.2008.04.022
Udayabhaskar R, Mangalaraja RV, Manikandan D et al (2012) Room temperature synthesis and optical studies on Ag and Au mixed nanocomposite polyvinylpyrrolidone polymer films. Spectrochim Acta A Mol Biomol Spectrosc 99:69–73. doi:10.1016/j.saa.2012.08.066
Vaidya S, Patra A, Ganguli AK (2010) Core–shell nanostructures and nanocomposites of Ag@TiO2: effect of capping agent and shell thickness on the optical properties. J Nanoparticle Res 12:1033–1044. doi:10.1007/s11051-009-9663-5
Valmikanathan OP, Ostroverkhova O, Mulla IS et al (2008) The effect of synthesis procedure on the structure and properties of palladium/polycarbonate nanocomposites. Polymer 49:3413–3418. doi:10.1016/j.polymer.2008.05.036
Valodkar M, Bhadoria A, Pohnerkar J et al (2010) Morphology and antibacterial activity of carbohydrate-stabilized silver nanoparticles. Carbohydr Res 345:1767–1773. doi:10.1016/j.carres.2010.05.005
Valodkar M, Modi S, Pal A, Thakore S (2011) Synthesis and anti-bacterial activity of Cu, Ag and Cu–Ag alloy nanoparticles: a green approach. Mater Res Bull 46:384–389. doi:10.1016/j.materresbull.2010.12.001
Vigneshwaran N, Nachane RP, Balasubramanya RH, Varadarajan PV (2006) A novel one-pot “green” synthesis of stable silver nanoparticles using soluble starch. Carbohydr Res 341:2012–2018. doi:10.1016/j.carres.2006.04.042
Vodnik VV, Vuković JV, Nedeljković JM (2009) Synthesis and characterization of silver—poly(methylmethacrylate) nanocomposites. Colloid Polym Sci 287:847–851. doi:10.1007/s00396-009-2039-7
Wei D, Sun W, Qian W et al (2009) The synthesis of chitosan-based silver nanoparticles and their antibacterial activity. Carbohydr Res 344:2375–2382. doi:10.1016/j.carres.2009.09.001
Yang J-H, Yu J-G, Ma X-F (2006) Preparation and properties of ethylenebisformamide plasticized potato starch (EPTPS). Carbohydr Polym 63:218–223. doi:10.1016/j.carbpol.2005.08.059
Yılmaz G, Jongboom ROJ, Feil H et al (2004) Permeation of volatile compounds through starch films. Biomacromolecules 5:650–656. doi:10.1021/bm034493m
Yoksan R, Chirachanchai S (2010) Silver nanoparticle-loaded chitosan–starch based films: fabrication and evaluation of tensile, barrier and antimicrobial properties. Mater Sci Eng C 30:891–897. doi:10.1016/j.msec.2010.04.004
Zeppa C, Gouanvé F, Espuche E (2009) Effect of a plasticizer on the structure of biodegradable starch/clay nanocomposites: thermal, water-sorption, and oxygen-barrier properties. J Appl Polym Sci 112:2044–2056. doi:10.1002/app.29588
Zobel HF (1988) Molecules to granules: a comprehensive starch review. Starch Stärke 40:44–50. doi:10.1002/star.19880400203
Acknowledgments
The authors gratefully acknowledge Ruben Vera and the “Centre de Diffractométrie Henri Longchambon” of University of Lyon 1 for the reflection XRD experiments, and the “Centre Technologique des Microstructures” of University of Lyon 1 for TEM photomicrographs.
Author information
Authors and Affiliations
Corresponding authors
Rights and permissions
About this article

Cite this article
Cheviron, P., Gouanvé, F. & Espuche, E. Effect of silver nanoparticles' generation routes on the morphology, oxygen, and water transport properties of starch nanocomposite films. J Nanopart Res 17, 364 (2015). https://doi.org/10.1007/s11051-015-3173-4
Received:
Accepted:
Published:
DOI: https://doi.org/10.1007/s11051-015-3173-4