
Abstract
Background
Candida auris is a multidrug-resistant pathogen that causes nosocomial outbreaks and high mortality. We conducted this study to investigate the molecular mechanisms of antifungal resistance in our clinical isolate of C. auris with a high level of resistance to three main classes of antifungals.
Material and Methods
A clinical C. auris isolate was identified by MALDI-TOF MS and antifungal susceptibilities were determined by the Sensititre YeastOne YO10 panel. After sequencing the whole genome of the microorganism with Oxford Nanopore NGS Technologies, a phylogenetic tree was drawn as a cladogram to detect where the C. auris clade to this study’s assembly belongs.
Results
The C. auris isolate in this study (MaCa01) was determined to be a part of the clade I (South Asian). The resistance-related genes indicated that MaCa01 would most likely be highly resistant to fluconazole (CDR1, TAC1b, and ERG11), none or little resistant to amphotericin B (AmpB) and echinocandins, and sensitive to flucytosine. The mutations found in the above-mentioned genes in the Türkiye C. auris isolate reveals an antifungal resistance pattern. This molecular resistance pattern was found consistent with the interpretation of MIC values of the antifungals according to CDC tentative breakpoints.
Conclusion
We detected the well-known antifungal resistance mutations, responsible for azole resistance in C. auris. Despite no ERG2, ERG6, and FKS mutation identified, the isolate was found to be resistant to AmpB and caspofungin based on the CDC tentative breakpoints which could be related to unidentified mutations.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
The emerging nosocomial fungal pathogen C. auris causes life-threatening outbreaks, predominantly in intensive care units (ICU). Due to the misidentification of the organism and the multidrug-resistant (MDR) phenotype, treatment failures, and mortality rates are high. C. auris is resistant to multiple antifungal drugs commonly used in the treatment of invasive Candida infections [1].
After its first identification in 2009, C. auris has rapidly spread all around the world. The various four C. auris clades have been determined in different geographic regions by phylogenetic studies: South Asia (clade I), East Asia (clade II), South Africa (clade III), and South America (clade IV) and potential fifth clade Iranian [2, 3]. Despite the limited epidemiological and clinical data, the Centers for Disease Control and Prevention (CDC) has defined tentative antifungal breakpoints based on susceptibility data of hundreds of clinical C. auris isolates [4]. Using recent tentative MIC breakpoints by the CDC, various reports in the USA indicate that the resistance to fluconazole is over 80%, to amphotericin B (AmpB) 30%-50%, and to echinocandins 5% [3,4,5]. Just as C. auris isolates intrinsically reduce susceptibility to antifungal drugs, they can rapidly acquire resistance. Nearly 90% of isolates are estimated to be resistant to at least one antifungal, 30 to 40% resistant to two antifungals and approximately 4% resistant to the three antifungal drug classes [3, 4, 6]. Because of this resistance pattern, C. auris infections have been associated with treatment failure and high mortality rates. Although the resistance levels vary considerably between clades, clade I C. auris isolates have a higher antifungal resistance [7].
The molecular mechanisms of antifungal resistance of C. auris have been defined in previous studies. According to these studies, reduced susceptibility to fluconazole may link to efflux pump overexpression, point mutations, or ERG 11 overexpression, while resistance to echinocandins may be associated with a mutation in FKS1 [8]. The mutation in ERG6 was linked to resistance to polyenes in C. auris by limited data, while it was associated with a 5-flucytosine resistance in the FUR1 [8,9,10].
In this study, we investigated the genetic antifungal resistance mechanisms of one of our clinical C. auris isolates. Although several studies have revealed the genetic profiles of C. auris from different countries, this is the first report from Türkiye that studied molecular mechanisms of antifungal resistance in C. auris.
Material and Methods
The isolate, MaCa01, grew in the blood culture of a 75-year-old woman. She was hospitalized in ICU with COVID-19 and was under broad-spectrum antibiotics. She developed C. auris candidemia on her 21st day of ICU stay. Unfortunately, she passed away the day after likely due to not being under antifungal therapy.
The blood culture was incubated in the BACT/ALERT® 3D system (bioMérieux, France) until growth was detected. The yeast cells were observed in a Gram stain prepared from the bottle with a positive signal, and the bottled fluid aspirate was inoculated onto Sabouraud dextrose agar. After 24–48 h of incubation at 37 °C, yeast colonies were identified by matrix-assisted laser desorption ionization-time of flight mass spectrometry (MALDI-TOF MS) (VITEK MS, v3.0, bioMérieux, France). Antifungal susceptibilities of the isolate were determined by the Sensititre YeastOne YO10 panel (Trek Diagnostic Systems, UK) according to the manufacturer’s instructions.
gDNA Isolation
A C. auris culture plate belonging to a patient with candidemia was obtained and preserved at + 4 °C. The culture was collected from the plate and gDNA extraction processes were started within the first week after the culture plate was obtained.
The collected cells from the plate culture were dissolved in a 2 ml microcentrifuge tube in 1 ml Tris–EDTA(10–100 mM) by means of pulse vortexing 10–15 times. The 2 ml tube containing the fungal cells and Tris–EDTA mixture were centrifuged for 5 min at 20,000 × g and + 4 °C constant temperature. The supernatant was removed, and the extraction process was continued with the pellet by following the ZymoBIOMICS DNA Miniprep Kit (Zymo Research, Cat. No. D4300) DNA isolation procedure. Afterward, the actual DNA concentrations were measured with Qubit 2.0, and the DNA purity levels were determined with NanodropOne (Thermo Fisher, Cat. No. ND-ONE-W). The reference values to confirm the purity of the extracted gDNA were obtained from ONT (260/280 ~ 1.80 and 260/230 ~ 2.0–2.2). The samples that couldn’t reach the purity requirements were purified using Agencourt AMPure XP beads (Beckman Coulter, Cat. No. A63881).
Whole Genome Shotgun Sequencing
To perform the whole genome sequencing of C. auris with approximately 12.5 million base-pairs length genome and 7 main chromosomes excluding other chromosomal copies and a mitochondrial plasmid, the study was planned to include ONT-Ligation Sequencing Kit (ONT, Cat. No. SQK-LSK109) for the sequencing experiment. The prepared sequencing library contained MaCa01’s genomic DNA with adapters, sequencing buffers, and loading beads, and it was loaded on the ONT MinION Flowcell (v. 9.4.1, Cat. No. FLO-MIN106D).
Bioinformatics and Phylogenetic Analyses
During the sequencing, the data were obtained as.fast5 signal files using MinKNOW (v. 22.03.5) GUI program. The adapter and barcode removal and first quality filtering were performed with ONT-guppy (v. 6.0.6) CLI program, and the.fastq formatted sequencing files were made ready for downstream analyses after this process for each sample. The quality score of the completed NGS assay was determined with FastQC (v. 0.11.9) and the average score was found to be Q18 (phred score). By using the flye (v. 2.8) de novo assembler pipeline for Nanopore reads de novo assembler, the contig fasta sequences were obtained [11]. These sequences were mapped to NCBI nt reference database (as of 12/07/2022).
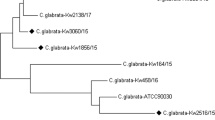
The blast (v. 2.12) alignment results were used to determine reference clades and strains of C. auris which were later used to make the consensus sequences of each sample. Finally, a phylogenetic tree was drawn to determine the relationship between the sample and predefined Clades (I, II, III, and IV) of C. auris (Fig. 1). Unlike using multiple alignments and plotting the tree using these calculated distances, which are done with shorter genomes, ANI (average nucleotide identity) algorithm was used for distance calculation with the implementation of tANI_matrix, CLI program [12]. Identity and coverage cutoff values were determined to be 0.85 and magicblast as the ANI task option was fed to the CLI program as its arguments. After the ANI matrix creation, R (v. 4.1.6) (stringr, reshape2, Matrix, MASS, ape, and phangorn packages) based buildtree.R function of tANI was used to create the phylogenetic tree and visualized with iTOL [13].

The figure above shows the four clades, the Iran variant, and sample MaCa01(blue-marked organism) in an unrooted phylogenetic cladogram. The blue-marked organism represents MaCa01 while the others were obtained from previous studies. The numbers represent the distances between each branch separation. As reference points, 8 strains from Clade I, 2 from Clade II, 3 from Clade III, 2 from Clade IV, and the Iran strain were included in the study
Eukaryotic annotation of features was carried out with the dfast (v. 1.2.17) annotation pipeline, which included TIGR, PFAM, KOG, NCBI CDD (Conserved Domains Database and Resources), and SMART databases, along with Aragorn (v. 1.2.38), for tRNA and tmRNA, Barrnap (v. 0.8) and RNAmmer (v. 1.2), for rRNA, and Prodigal (v. 2.6.3), GhostX (v. 1.3.6) and ORFfinder (v. 0.4.3), for general CDS prediction, were used in the process. As for the ortho-search database, 41 feature annotated NCBI [Candida] auris strains (of which the accessions are available in Supplementary Table 1) were decided to be included. While using the abovementioned CLI programs, eukaryotic databases and translation tables 12 and 3 (alternative yeast and yeast mitochondria) were chosen to specify the annotation type and method.
The antifungal resistance-related genes were compared with the UniProt protein database and MARDy for previously detected antifungal resistance-related mutations with tblastn CLI program (v. 2.12) [14]. The genes were translated according to the alternative yeast translation table and the resulting amino acid sequences’ 3D structures were modeled using SWISS-MODEL, then visualized with ChimeraX (v.1.6rc) [15, 16].
The detected mutations in the assembly were compared with the references (UniProt-C. auris), and the novel mutations found in ERG2 and ERG11 were sequenced using the Sanger technology for confirmation. The forward and reverse PCR primers (ERG2 Forward 5’-GCTGATGCCAAAACGCTCAT-3’, ERG2 Reverse 5’-ACTGCTTCACTTGGCCTCTC-3’/ERG11 Forward 5’-ATTTGATGCCTCCTTCGCCA-3’, ERG11 Reverse 5’-GTGTGCTGACCTCCCATCAA-3’) were designed for each of the mutation locations. After the PCR process was completed, the samples were sent to be sequenced. The Sanger sequencing was carried out by Eurofins Scientific, Germany.
Results
The average quality of the sequencing assay was found to be Q18 (phred score) with the FastQC cli (command line interface) program. The GC percentage of the genome was 43% and N50 was found to be 3898 base pairs. In total, there were 162,457 sequences obtained from the assay, the max length of the reads being 41,395 base pairs-long. The assembly of the raw reads of MaCa01 (NCBI accession number: SRR19393399) obtained with the flye cli program resulted in 12 contigs, out of 12, 1 represented the mitochondrial circular plasmid (99.80% identity with NCBI NC_053321), with an average coverage of 1326, and 1 hit with NCBI E. coli plasmid CP077071.1, while the rest of the contigs represented complete genome of chromosomes 1, 2, 3, 4, 5, 6 and 7, along with the partial sequences of copies of 3rd and 4th chromosomes with an average coverage of 56x. NCBI blast alignment with an average of 99.89% identity to the NCBI reference sequence C. auris (RefSeq [Candida] auris Cand_auris_B11221_V1) was observed.
A phylogenetic tree was constructed with a cladogram to detect where this study’s assembly belongs among all the C. auris clades (Fig. 1). With 16 other strains retrieved from NCBI included in the ANI-based phylogenetic tree, the C. auris sample in this study (MaCa01) was determined to be a part of the clade I, which is also known as South Asian Clade which includes India, Pakistan, Iran, Saudi Arabia, Oman, Kenya, Spain, United Kingdom and Canada [17].
Various genes, such as ERG11, FKS1, CDR1, TAC1b, and FUR1, were previously focused on and found to cause the development of resistance to azole and similar types of antifungals (Table 1). According to the gene mutations listed in Table 1 in resistance-related genes, MaCa01 was demonstrated most likely to be highly resistant to fluconazole, none or little resistant to AmpB and echinocandins, and sensitive to flucytosine.
Our assembly showed two deletion mutations causing frameshifts at the homopolymeric low-complexity regions for ERG2 and ERG11 genes. The novel mutations were tried to be validated using Sanger sequencing (Supplementary Tables 2 and 3). For ERG2, even though the high coverage nanopore sequences were assembled to create the whole genome, the Sanger sequencing result indicates that due to the homopolymeric low-complexity region, the nanopore reads couldn’t accurately predict the repetition of 7 thymine bases, resulting in one-base deletion (Supplementary Table 2). A similar result was obtained with ERG11 with even higher coverage values, instead of 5 guanine bases, we observed only 4 in the MaCa01 assembly (Supplementary Table 3).
The mutation of the 132nd amino acid in ERG11 to phenylalanine (from tyrosine, Y132F both being bulky and hydrophobic amino acids) was shown to be important in the MARDy database for azole resistance (Table 1: ERG11) [14].
A homologous gene to CDR1, responsible for antifungal resistance in C. albicans, was also identified in C. auris in 2019 by Rybak et al. [21]. Due to a mutation in this gene from glutamic acid to aspartic acid, the gene function is not expected to be altered greatly, as they are both acidic amino acids. Therefore, this gene could play a role in the azole resistance of MaCa01 (Table 1: CDR1).
TAC1b was shown to play an important role in azole resistance by Carolus et al. in 2021. In the study, increased azole resistance of C. auris was observed after deletion mutation of a codon at the 191st location (results in removal of phenylalanine) of the TAC1b transcription factor gene. However, MaCa01 didn’t show the mutations detailed in Table 1. Hence, any impact of the mutations in this gene on the azole resistance could not be definite (Table 1: TAC1b).
Additionally, MIC values of MaCa01 and CDC tentative clinical breakpoints for C. auris were summarized in Table 2.
Discussion
C. auris, the first fungus that has been listed among urgent antimicrobial resistance threats by CDC in 2019, is an opportunistic fungal pathogen with reduced antifungal susceptibility [22]. The most distinguishing features of C. auris from other Candida species are resistance to three major antifungal classes, the azoles, polyenes, and echinocandins, and the need for contact isolation rules to prevent transmission. The tentative breakpoints of C. auris released by CDC, clearly show that C. auris has high MICs for all classes of antifungal drugs [4]. Although the mechanism of this level of high resistance is still poorly understood, two main mechanisms are emphasized: molecular and biofilm-associated resistance [8, 23].
The vast majority of C. auris isolates are resistant to fluconazole, the most prescribed antifungal agent. The fluconazole MIC value of our isolate was very high (≥ 256 µg/mL), which is quite higher than CDC tentative breakpoints declared and may be interpreted as decreased susceptibility. There are no defined tentative breakpoints for other azole antifungals. However, the CDC has stated that fluconazole susceptibility may be used as a guide for posaconazole, voriconazole, and itraconazole [4]. Although the molecular mechanism of azoles is relatively little known, some gene-encoding mutations in C. auris have been repeatedly identified [19]. Previous studies have shown that decreased azole susceptibility is linked to CDR1 efflux pump overexpression and TAC1b transcription factor mutation [8, 19, 20]. Moreover, the single point mutation (SNP-single point polymorphism) causing Tyrosine to mutate into Phenylalanine in the ERG11 gene was shown to be related to resistance against azole antifungals [8, 18]. The aforementioned mutation was detected in MaCa01, as well (Table 1).
Limited data about the mutation in the CDR1 shows that the ATP-binding cassette (ABC)-type efflux pump-encoding gene CDR1 contributes to the azole resistance [21]. We found a novel mutation, E709D, in MaCa01. This mutation may be linked to decreased azole susceptibility in C. auris isolates. However, further studies are needed to confirm this result. Similarly, we found various novel mutations encoding TAC1b in MaCa01. Since TAC1b positively regulates the expression of ABC transporter CDR1, in the case of mutation in TAC1b, CDR1 expression significantly increases [19, 20]. As a result, the azoles-resistance mechanism may be again triggered.
Approximately 30–50% of C. auris isolates are resistant to AmpB, according to CDC tentative breakpoints [4]. The AmpB MIC value of the MaCa01 isolate was found as 4 µg/mL. Based on the CDC's tentative breakpoints, this result suggests MaCa01 may be resistant to AmpB. Although the molecular mechanism of this mutation remains unclear, the alternations in the ergosterol pathway are considered to be a potential [18, 24] as ERG2, ERG3, ERG5, ERG6, or ERG11 gene mutations were previously shown to be associated with AmpB resistance in various Candida species [25]. Similarly, Rybak et al. have identified ERG6 gene mutation, as a novel mutation in an AmpB-resistant C. auris isolate [9]. On the other hand, Rhodes et al. have not found any mutations in these genes in 27 C. auris isolates displaying reduced susceptibility to AmpB [10]. Though we couldn’t identify a mutation in ERG2 and ERG6 in our study, the ERG3 and ERG5 genes, which remain unidentified in Candida auris (UniProt) and are related to AmpB resistance, might have an impact on its low susceptibility according to the results of our MIC assays. Since few data are contributing to AmpB resistance in the literature, further research is needed to determine responsible mutations.
Applying CDC tentative antifungal breakpoints, echinocandin resistance in C. auris is quite low, approximately 5% [4]. This resistance has been associated with the mutation of the FKS1 gene, which encodes 1,3-β-D-glucan synthase complex, the key component of the fungal cell wall synthesis. Previous studies have discovered three mutations linked to echinocandin resistance in C. auris at codon 639 (S639F, S639P, and S639Y) within hotspot-1 of FKS1 [8, 18, 26]. Our isolate showed a high MIC value for caspofungin while MIC values for anidulafungin and micafungin were low. We have not observed any mutation in FKS1 in our strain. The patient did not have a history of previous echinocandin exposure; therefore, the high caspofungin MIC value is puzzling. In literature, the adaptive stress responses that cause cell wall chitin elevation are also responsible for echinocandins resistance in C. auris [27]. In a study, the elevated cell wall chitin presence in Candida species triggered a decrease in caspofungin activity [28]. Similarly, Fayed et al. showed that caspofungin-treated C. auris exhibited elevated MIC50 and chitin content [29]. We could not evaluate it in our study and the reason for the high MIC value against caspofungin remains unclear in our strain.
Finally, the MIC value of flucytosine was determined as very low, 0.12 µg/mL, although there are no defined tentative breakpoints for flucytosine. F211I amino acid substitution in the FUR1 gene has been found associated with flucytosine-resistant C. auris isolates [8, 10]. This mutation has been reported just in one isolate so far, but we did not detect such a mutation in our strain [10].
In conclusion, we detected the well-known antifungal resistance mutations, which could be responsible for azole resistance in C. auris, in our sample. We also demonstrated that CDC’s tentative breakpoints for C. auris are consistent with the susceptibility results of azoles, flucytosine, and except caspofungin of echinocandins for our isolate. Since limited data exists regarding the molecular mechanisms of the antifungal resistance of C. auris, further research both in vitro and in vivo is warranted.
References
Di Pilato V, Codda G, Ball L, Giacobbe DR, Willison E, Mikulska M, et al. Molecular epidemiological investigation of a nosocomial cluster of C. auris: evidence of recent emergence in Italy and ease of transmission during the COVID-19 pandemic. J Fungi (Basel). 2021;7(2):140. https://doi.org/10.3390/jof7020140.
Chow NA, de Groot T, Badali H, Abastabar M, Chiller TM, Meis JF. Potential fifth clade of Candida auris, Iran, 2018. Emerg Infect Dis. 2019;25(9):1780–1. https://doi.org/10.3201/eid2509.190686.
Lockhart SR, Etienne KA, Vallabhaneni S, Farooqi J, Chowdhary A, Govender NP, et al. Simultaneous emergence of multidrug-resistant Candida auris on 3 continents confirmed by whole-genome sequencing and epidemiological analyses. Clin Infect Dis. 2017;64(2):134–40. https://doi.org/10.1093/cid/ciw691.
Centers for Disease Control and Prevention (CDC): C. auris: antifungal susceptibility testing and interpretation. https://www.cdc.gov/fungal/candida-auris/c-auris-antifungal.html (2020). Accessed 29 May 2020.
Chowdhary A, Prakash A, Sharma C, Kordalewska M, Kumar A, Sarma S, et al. A multicentre study of antifungal susceptibility patterns among 350 Candida auris isolates (2009–17) in India: role of the ERG11 and FKS1 genes in azole and echinocandin resistance. J Antimicrob Chemother. 2018;73(4):891–9. https://doi.org/10.1093/jac/dkx480.
Magobo RE, Corcoran C, Seetharam S, Govender NP. Candida auris-associated candidemia, South Africa. Emerg Infect Dis. 2014;20(7):1250–1. https://doi.org/10.3201/eid2007.131765.
Chow NA, Munoz JF, Gade L, Berkow EL, Li X, Welsh RM, et al. Tracing the evolutionary history and global expansion of Candida auris using population genomic analyses. MBio. 2020. https://doi.org/10.1128/mBio.03364-19.
Chaabane F, Graf A, Jequier L, Coste AT. Review on antifungal resistance mechanisms in the emerging pathogen Candida auris. Front Microbiol. 2019;10:2788. https://doi.org/10.3389/fmicb.2019.02788.
Rybak JM, Barker KS, Munoz JF, Parker JE, Ahmad S, Mokaddas E, et al. In vivo emergence of high-level resistance during treatment reveals the first identified mechanism of amphotericin B resistance in Candida auris. Clin Microbiol Infect. 2022;28(6):838–43. https://doi.org/10.1016/j.cmi.2021.11.024.
Rhodes J, Abdolrasouli A, Farrer RA, Cuomo CA, Aanensen DM, Armstrong-James D, et al. Genomic epidemiology of the UK outbreak of the emerging human fungal pathogen Candida auris. Emerg Microbes Infect. 2018;7(1):43. https://doi.org/10.1038/s41426-018-0045-x.
Kolmogorov M, Yuan J, Lin Y, Pevzner PA. Assembly of long, error-prone reads using repeat graphs. Nat Biotechnol. 2019;37(5):540–6. https://doi.org/10.1038/s41587-019-0072-8.
Gosselin SFM, Feng Y, Gogarten JP. Expanding the utility of sequence comparisons using data from whole genomes. bioRxiv. 2020. https://doi.org/10.1101/2020.01.15.908137.
Letunic I, Bork P. Interactive Tree Of Life (iTOL) v5: an online tool for phylogenetic tree display and annotation. Nucleic Acids Res. 2021;49(W1):W293–6. https://doi.org/10.1093/nar/gkab301.
Nash A, Sewell T, Farrer RA, Abdolrasouli A, Shelton JMG, Fisher MC, et al. MARDy: mycology antifungal resistance database. Bioinformatics. 2018;34(18):3233–4. https://doi.org/10.1093/bioinformatics/bty321.
Waterhouse A, Bertoni M, Bienert S, Studer G, Tauriello G, Gumienny R, et al. SWISS-MODEL: homology modelling of protein structures and complexes. Nucleic Acids Res. 2018;46(W1):W296–303. https://doi.org/10.1093/nar/gky427.
Pettersen EF, Goddard TD, Huang CC, Meng EC, Couch GS, Croll TI, et al. UCSF ChimeraX: structure visualization for researchers, educators, and developers. Protein Sci. 2021;30(1):70–82. https://doi.org/10.1002/pro.3943.
Rhodes J, Fisher MC. Global epidemiology of emerging Candida auris. Curr Opin Microbiol. 2019;52:84–9. https://doi.org/10.1016/j.mib.2019.05.008.
Frias-De-Leon MG, Hernandez-Castro R, Vite-Garin T, Arenas R, Bonifaz A, Castanon-Olivares L, et al. Antifungal resistance in Candida auris: molecular determinants. Antibiotics (Basel). 2020;9(9):568. https://doi.org/10.3390/antibiotics9090568.
Rybak JM, Munoz JF, Barker KS, Parker JE, Esquivel BD, Berkow EL, et al. Mutations in TAC1B: a novel genetic determinant of clinical fluconazole resistance in Candida auris. MBio. 2020. https://doi.org/10.1128/mBio.00365-20.
Carolus H, Pierson S, Munoz JF, Subotic A, Cruz RB, Cuomo CA, et al. Genome-wide analysis of experimentally evolved Candida auris reveals multiple novel mechanisms of multidrug resistance. MBio. 2021. https://doi.org/10.1128/mBio.03333-20.
Rybak JM, Doorley LA, Nishimoto AT, Barker KS, Palmer GE, Rogers PD. Abrogation of triazole resistance upon deletion of CDR1 in a clinical isolate of Candida auris. Antimicrob Agents Chemother. 2019. https://doi.org/10.1128/AAC.00057-19.
Centers for Disease Control and Prevention (CDC): Antibiotic resistance threats in the United States. https://www.cdc.gov/drugresistance/pdf/threats-report/2019-ar-threats-report-508.pdf (2019). Accessed 26 March 2019.
Kean R, Delaney C, Sherry L, Borman A, Johnson EM, Richardson MD, et al. Transcriptome assembly and profiling of Candida auris reveals novel insights into biofilm-mediated resistance. mSphere. 2018. https://doi.org/10.1128/mSphere.00334-18.
Lockhart SR. Candida auris and multidrug resistance: defining the new normal. Fungal Genet Biol. 2019;131:103243. https://doi.org/10.1016/j.fgb.2019.103243.
Arendrup MC, Patterson TF. Multidrug-resistant Candida: epidemiology, molecular mechanisms, and treatment. J Infect Dis. 2017;216(suppl_3):S445–51. https://doi.org/10.1093/infdis/jix131.
Asadzadeh M, Mokaddas E, Ahmad S, Abdullah AA, de Groot T, Meis JF, et al. Molecular characterisation of Candida auris isolates from immunocompromised patients in a tertiary-care hospital in Kuwait reveals a novel mutation in FKS1 conferring reduced susceptibility to echinocandins. Mycoses. 2022;65(3):331–43. https://doi.org/10.1111/myc.13419.
Beyda ND, Lewis RE, Garey KW. Echinocandin resistance in Candida species: mechanisms of reduced susceptibility and therapeutic approaches. Ann Pharmacother. 2012;46(7–8):1086–96. https://doi.org/10.1345/aph.1R020.
Walker LA, Gow NA, Munro CA. Elevated chitin content reduces the susceptibility of Candida species to caspofungin. Antimicrob Agents Chemother. 2013;57(1):146–54. https://doi.org/10.1128/AAC.01486-12.
Fayed B, Jayakumar MN, Soliman SSM. Caspofungin-resistance in Candida auris is cell wall-dependent phenotype and potential prevention by zinc oxide nanoparticles. Med Mycol. 2021;59(12):1243–56. https://doi.org/10.1093/mmy/myab059.
Funding
This study was supported by Gilead Sciences.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
The authors declared no potential conflicts of interest concerning this article's authorship and/or publication.
Ethical Approval
Marmara University School of Medicine Institutional Ethical Review Board issued approval 09.2021.578. The collected data did not include any patient or employee identifying information.
Additional information
Handling Editor: Ferry Hagen.
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Below is the link to the electronic supplementary material.
Rights and permissions
Springer Nature or its licensor (e.g. a society or other partner) holds exclusive rights to this article under a publishing agreement with the author(s) or other rightsholder(s); author self-archiving of the accepted manuscript version of this article is solely governed by the terms of such publishing agreement and applicable law.
About this article

Cite this article
Erturk Sengel, B., Ekren, B.Y., Sayin, E. et al. Identification of Molecular and Genetic Resistance Mechanisms in a Candida auris Isolate in a Tertiary Care Center in Türkiye. Mycopathologia 188, 929–936 (2023). https://doi.org/10.1007/s11046-023-00787-1
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s11046-023-00787-1