
Abstract
Evolved biological network topologies may resist perturbances to allow for more robust information transport across larger networks in which their network motifs may play a complex role. Although the abundance of individual motifs correlate with some metrics of biological robustness, the extent to which redundant regulatory interactions affect motif connectivity and how this connectivity affects robustness is unknown. To address this problem, we applied machine learning based regression modeling to evaluate how feed-forward loops interlinked by crosstalk altered information transport across a network in terms of packets successfully routed over networks of noisy channels via NS-2 simulation. The sample networks were extracted from the complete transcriptional regulatory networks of two well-studied bacteria: E.coli and Yeast. We developed 233 topological features for the E.coli subnetworks and 842 topological features for the Yeast subnetworks which distinctly account for the opportunities in which two feed-forward loops may exhibit crosstalk. Random forest regression modeling was used to infer significant features from this modest configuration space. The coefficient of determination was used as a primary performance metric to rank features within our regression models. Although only a handful of features were highly ranked, we observed that, in particular, feed-forward loop crosstalk patterns correlated substantially with an improved chance for successful information transmission. Additionally, both E.coli and Yeast subnetworks demonstrate very similar FFL crosstalk patterns that were considered significant in their contribution to information transport robustness in these two organisms. This finding may potentially allude to common design principles in transcriptomic networks from different organisms.
Similar content being viewed by others
Avoid common mistakes on your manuscript.
1 Introduction
Network motifs are recurrent network structures that exhibit higher statistical significance in biological networks than in random ones. In the past, they have been implicated in facilitating information transport in biological networks by resisting noisy perturbation and successfully convey the cellular state [1]. Past studies indicate that feed-forward loop (FFL) network motifs are important, not just in terms of their topological abundance in such biological networks [2], but also in terms of certain behaviors such as response time [3]. The feed-forward loop structure (Fig. 1) is intriguing because it offers two ways of regulating a protein-expressing gene (node C) via two influential paths: a direct route (A to C), or an indirect path beset by a waypoint (A to B to C). This setup may be efficient in terms of communication due to the signaling modality of multiple regulatory paths to protein expression of a regulated gene. We may therefore hypothesize that higher FFL abundances will lead to better information transmission performance. Pursuing this line of research, we demonstrated that such FFL motifs indeed contribute to the information transport robustness in bio-inspired wireless sensor networks that were designed to exhibit the abstract graph topology of transcriptional regulatory networks (TRNs) [4, 5]; such TRNs are a specific class of biological networks where genes are regulated by transcription factor nodes and demonstrate a sparse and scale-free topology that is rich in FFLs. However, one central question remains: Do FFLs contribute signaling/communication benefits individually, synergistically in combination with others, or not at all? To address this question, in this paper, we examined the extent to which feed-forward loops crosslinked by regulatory interactions (edge-connected motifs) contribute to successful information transport across biological networks, modeled as networks of noisy channels across which information packets are routed via NS-2 simulation. The answers to such questions will have a profound impact in many different areas as follows:
-
Synthetic biology: Due to the efficient communication properties of FFLs, specifically in terms of noise filtering and robust signal transport, they make great candidates for the emerging domain of synthetic biology where larger engineered TRN circuits can be built that are resilient to external perturbations [6]. Early efforts in this direction have shown great promise and the importance of connected FFLs as investigated in this paper can motivate the construction of more efficient genetic circuits in the future.
-
Biological network growing algorithms: Another popular area of research includes the transcriptional network growing algorithms primarily based on the preferential attachment model [7]. Currently, only the TRNs of Escherichia coli (E. coli) and Saccharomyces cerevisiae (Yeast) have been validated experimentally; hence such network growing algorithms are essential to allow the community to study the properties of such TRNs for designing robust networks [8], as well as to predict the TRNs of higher-level organisms. We have recently developed such a network growing algorithm by extending the preferential attachment model to produce directed networks that mimic the topological properties of E. coli [9] in terms of their degree distribution and FFL abundance. The connected FFL configurations reported in this paper can motivate more accurate TRN prediction algorithms in the future.
-
Bio-inspired engineered networks: Wireless sensor networks form a special class of engineered systems wherein sensor nodes forward data packets that are routed through adjacent sensors to a sink capable of processing the sensed information. Resemblance between gene regulation systems and wireless sensor networks (herein WSNs) can be described through transcription, where genes process signals from adjacent neighbors in the form of transcription factors that excite/repress other genes by generating mRNA molecules. WSNs operate in a similar manner, where sensor nodes send signals to others in the form of data packets. Recently, we have shown that wireless sensor networks adopting the transcriptional regulatory topologies (of E. coli), designated as bio-inspired WSNs, are more efficient than those adopting Erdos Renyi random graph topologies of the same size in terms of conveying packets to sink nodes [10,11,12,13,14,15,16]. The connected FFL patterns investigated in this paper will motivate the design of smart WSN topologies that exhibit similar FFL abundance and distribution as observed in TRNs and hence will exhibit better efficiency in terms of their average packet receipt rates under node/link failures and channel noise.

(Top Left) A canonical feed-forward loop is one that is free of additional interactions. (Top Right) Embedded feed-forward loops are contained within more complicated topological configurations. (Bottom) Feed-forward loops inter-connected by an edge
Existing network robustness metrics are predominantly “static” [17, 18], in that they do not consider dynamical information transport. Chan et al. [17] provides an in-depth review of existing robustness metrics. Notably absent however are metrics which consider motif-based features including the possibility of crosslinked feed-forward loops. Here we are concerned with the successful transmission of information packets routed across a biological network, modeled using the discrete event network simulator NS-2. These simulations account for the dynamics of information flow among the nodes in a network under controlled conditions such as channel noise and congestion-based information loss. To this effect, we define informational “robustness” as the ratio between the total number of packets received at perfectly absorbing “sink” nodes to the total number of packets emitted from potentially many source nodes. We will refer to this metric colloquially as the packet receipt rate (PRR), which accounts for network behavior resulting from graded perturbations, and is more comprehensively detailed in our earlier reports [4, 19]. We employ discrete event simulations and machine learning techniques to develop a model trained using feature data to predict robust network topologies for information transport. We use these analyses to rank-order the differing configurations of linked feed-forward loops, seeking to answer the following questions: Does abundance positively correlate with information-transport robustness? If so, which features are primary contributors to such robustness?
Additionally, considering the TRN topologies of two well known organisms, i.e., E. coli and Yeast, we seek to investigate the existence of common features that may significantly impact the information transport robustness of both organisms. Such features can have profound impacts on efficient network design principles as both E. coli and Yeast are efficient information transport networks with different topologies in terms of size and other
2 Proposed methodology
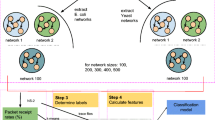
Our basic methodology is illustrated in Fig. 2. First, subnetworks are extracted (Section 2.1) from the known transcriptional regulatory networks of the Escherichia coli (E. coli) and Saccharomyces cerevisiae (Yeast), and passed to the network simulator platform NS-2 (Section 2.2) to generate packet receipt rates; these rates are considered as a measure of robustness for the individual subnetworks. Next, FFL crosstalk based feature values are determined using Python, from which we remove all the duplicate feature vectors and retain a unique feature vector with minimal PRR among all feature vectors. Features are further scaled to the interval [0,1], which reduces the processing time of our regression models; although, scaling is not necessary for Random Forest regression that was used here. These data are processed (Section 2.4) into a format illustrated by Step 1 of Fig. 2, upon which a random forest regression based machine learning technique is applied for ranking the features. The coefficient of determination is calculated to identify the optimal number of estimators (Section 2.5). Before feature ranking is actually done, we perform feature selection which is a process to reduce the feature set (from the original 233 features set for E.coli and 842 features set for Yeast). Finally, features are ranked using feature importance–a method used to determine feature significance in regression trees. Section 2.6 shows the parameters used for creating the random forests based regression models and subsequent accuracy measurement.

a Network extraction and NS-2 simulation methodology. b A feed-forward loop transcriptional network motif
2.1 Network datasets
Transcriptional regulatory networks of several simple organisms have been experimentally and exhaustively interrogated and validated. Among these are the transcriptional regulatory network of the bacteria Escherichia coli (E. coli) and S. cerevisiae (Yeast). These two TRNs are best studied in the literature with high fidelity in their network topologies and readily available network datasets and hence were used in this work. Model transcriptional networks of varying size were first extracted for E. coli using the GeneNetWeaver tool [20], which provides directed subnetworks of user-defined size from the full E. coli TRN, comprising of 1565 nodes and 3758 edges. GeneNetWeaver uses computational tools to extract subgraphs from the original TRNs that approximately preserve the degree and motif distributions, as well as the modularity (in terms of clustering coefficient) in the extracted networks when compared to the original TRN. The S. cerevisiae sub-networks were similarly derived from its original TRN, that comprises of 4441 nodes and 12873 links.
In order to create a large enough sample space for our subsequent machine learning models, we extracted directed transcriptional subnetworks of sizes 300, 400, and 500 nodes (including genes and transcription factors) from the E. Coli bacterium; we extracted 1000 separate networks for each size resulting in a total of 3000 directed networks to consider. Similarly, we extracted directed transcriptional subnetworks of sizes 300, 400, and 500 nodes from Yeast to compare the feature importances contributing to information transport robustness across multiple organisms; in this case, we extracted 1200 separate subnetworks for each network size giving us a total of 3600 directed networks to consider. Regulatory information was retained in these extracted networks while auto-regulatory loops were discarded. Table 1 shows the details of the network counts considered here. Additionally, we discarded the subnetworks that were disconnected. This step pruned the datasets down to 957, 932, and 941 networks respectively for the 300, 400, and 500 network sizes of E.coli. Subnetworks extracted from the Yeast did not have any disconnected components and hence all 1200 networks were retained for each network size. These datasets are then used to explore network dynamics in two ways: a) simulate the information transport robustness in these networks using NS-2 (Section 2.2) and b) determine structural features of importance that contribute to such robustness. Feature vectors were generated by extracting FFL-crosstalk based topological features from the pruned networks, and all duplicate feature vectors were removed. The resulting number of unique feature vectors are reported in Table 1; more details on how the feature vector was constructed is shown in Section 2.3.
2.2 NS-2 simulation setup
The problem of information flow across a biological network can be mapped onto the problem of packet transport over a wireless sensor network as we have shown earlier [8, 10,11,12]; here each extracted directed subnetwork from the previous step is considered to be a wireless sensor network where the nodes (genes or transcription factors) are mapped to individual sensor nodes. In the NS-2 model, each node relays finite-sized packets of information along its outgoing edges to neighboring nodes. Packets are relayed in this manner using a flooding protocol until they reach (perfectly absorbing) sink nodes, which do not retransmit. Genes are considered to be sink nodes as they do not have any outgoing edges while transcription factor nodes are considered as source nodes in the network that generate new packets or relay previously received packets from other nodes; the regulatory interactions between transcription factors and genes conceptualize communication channels in the sensor network model which determine the destination nodes of transmitted packets. Biology is inherently noisy, and we account for this by using noisy channel models with varying noise amplitude wherein 10%, 20%, 35%, 50%, 60%, 75% or 90% of packets will be, on average, lost during transmission across any individual inter-node route (i.e., a single hop between two nodes). Packet receipt rate in the network is measured as the percentage of the number of packets received at sink nodes to the number of packets sent by all source nodes. Networks with higher packet receipt rate are considered to be more robust. Packet receipt rates of the networks range in between 0 (least robust) and 100 (most robust). The detailed NS-2 simulation set-up with discussions on how each wireless network parameter may abstractly map onto corresponding TRN parameters was reported in [19].
2.3 Feature identification
We next used topology-based network characteristics to understand which network qualities and features contribute primarily to information transport robustness and routing over biological networks. While some of these characteristics, such as average shortest path, network density, and betweenness centrality have been considered before, we emphasize on using them to evaluate information transport robustness by considering the network dynamics measured in terms of the PRR. Previously, we identified fifteen different network features and ranked them using unsupervised learning techniques [4, 5]. These features comprise of the standard graph measures mentioned above as well as features derived from properties of individual FFLs. In this paper, we focus on understanding how FFL crosstalk, i.e., FFLs connected by one or more edges, behave within the embedding environment of the network and their contribution to robustness. Two FFLs can be coupled by crosstalk in 262,144 possible ways (see [21] for an explanation), however we selected only unique features which occur in the largest connected component of the TRNs for E.coli and Yeast respectively. To this effect, we identified 233 unique features for E.coli (selected features from this set are shown in Table 3 for E.coli) and 3227 unique features for Yeast (selected features from this set are shown in Table 4 for Yeast) that capture the abundance of the corresponding connected FFL structures in E.coli and Yeast using the following strategy. First, we identified all possible ways in which two FFL motifs could be connected by one or more edges; second, we counted the occurrence of each such pattern in the above mentioned transcriptional networks and recorded only the patterns that returned a non-zero count in the largest connected component of the corresponding TRNs. Such features are termed as the unique features; note that the set of unique features were overlapping but of substantially different cardinality for the TRNs of the two bacteria considered here.
We used machine learning techniques to identify significant features among a list of several features, and employed different machine-learning strategies by leveraging the widely recognized sci-kit module in Python [22]. We do not exhaustively tabulate data on the abundance of all FFL crosstalk patterns for different network sizes considered here as they are not directly related to main focus of this paper; this information is included in the Additional Materials section. Nevertheless we provide a summary of these data in Section 2.4. Testing for correlations between feature abundance and feature importance is described in Section 2.7.
2.4 Data preparation
Data is constructed similar to the procedures previously described in [4]. Each network is represented as a combination of feature values, feature ids and output labels determined by the PRR values from the NS-2 simulation. Each network (from Section 2.1) is represented as a combination of output labels and 233 features for E.coli, and 842 features for Yeast; as mentioned before, each such feature signify unique FFL crosstalk patterns. In the field of machine learning, such a combination is referred to as a feature instance. Results from the NS-2 simulations are used as output labels and the corresponding features are calculated using the networkX [23] module in Python. In previous work [4, 5], we considered the problem of ranking features to be an unsupervised one, and used an analysis of variance (ANOVA) F-value to determine the significance of each feature. In this paper, however, we consider this problem to be a supervised one and retain the output labels, which range between 0 and 100, as floating points. Regression techniques are suitable when the value of output labels is continuous. Furthermore, we introduce feature selection here as an improvement from our earlier work wherein the entire feature set was used to rank features. Before creating the regression model, data is split into training and testing data with an 80:20 ratio. The accuracy of regression models presented in Fig. 3 is based on testing of the model created on the test data of FFL crosstalk based features.

Coefficient of determination (COD) for FFL crosstalk based features regressor model for different network sizes. a CODs from E.coli subnetwork models (b) CODs from Yeast subnetwork models. A higher COD value indicates an improved performance
2.5 Feature down-selection
Since the number of unique features are very high when compared to the number of subnetworks for each network size in the Yeast subnetworks, we employ a feature down-selection technique. To reduce the features, we counted the occurrence of the 3227 unique features in the largest connected component of the entire Yeast TRN and selected only those features which occurred at least 100 times; this step pruned the Yeast unique feature set to 842. Next, we selected only a subset from all the entire set of 233 FFL crosstalk features from E.coli because some of these features may be be correlated with the others (as discussed in Section 2.3) or some of them might display a higher variance; the same step was applied to the entire 842 FFL crosstalk feature set from Yeast to identify the uncorrelated features. To begin with, we first selected the features that occurred in more than one network. The second column of Table 2 shows these feature counts; as a result of this step, we can observe a significant reduction in the number of E.coli features for each network size however, a very negligible reduction of the features from the Yeast TRNs. We believe that this is due to the initial feature selection step that was done based on the occurrence of features in the largest connected component of Yeast. As our aim is to deduce a minimal set of features that are important for information transport across these networks, we eliminated pairs of features that were positively correlated with a Spearman’s correlation coefficient > 0.95. The third column of Table 2 shows these counts upon removing such correlated features. Finally, we considered different feature reduction methods and examined those residing at the intersection. Randomized PCA was considered but ignored since it does not exploit the output label data to minimize the feature space. To this effect, a feature reduction step was performed using random forests with regression.
Random forest models [24] are well-suited to solve classification and regression problems. A “random forest” refers to the trees (estimators) used by ensemble machine learning models to predict the outcome of data. Mean squared error (MSE) is used to determine the best number of estimators (number of decision trees) used in the random forests algorithm. A number of estimators (e.g, 100 to 300) incremented by steps of 10, were used here in creating the random forest model. MSE is determined for each estimator and the average of the number of estimators is used as the MSE value for that specific estimators’ number. The variation in MSE noted before and after feature reduction, is shown in Fig. 4 for a singular case of a 300 node network with 90% channel loss model of E.coli. Before reduction, MSE is lowest for 290 estimators, while it is lowest for 200 estimators after feature reduction. The estimator for which MSE is the least was selected for calculating feature importance, as shown in Fig. 4.

Mean squared error (MSE) for different number of Random Forest estimators for networks of size 300 and high channel loss (90%). A lower MSE here indicates a better performance
Our analyses reveal that feature importance depends heavily on the network size and channel loss model over time. All the features with importance values ≥ 0.03 were selected, and in cases where the resulting features were less than five, we selected the top five features. We use these selected features to model the final regressor for prediction. Figure 5 shows the final counts of selected features for different network sizes and channel loss models from the E.coli subnetworks. Similarly, Fig. 6 shows the feature counts of selected features for different network sizes and channel loss models from the Yeast subnetworks; for all of the sizes and loss models, less than five features were selected based on threshold, and in such cases we selected the top five features to build the final regressor for the prediction.

Selected features for E.coli from a total of 233 features for every model at a given network size and channel loss model, as described in Section 2.5

Selected features for Yeast from a total of 842 features for every model at a given network size and channel loss model. Top five features are selected when number of features selected were reduced to less than five features, as described in Section 2.5
2.6 Regression modeling
Before carrying out the feature reduction step, we conducted random forests regression to determine the coefficient of determination (COD) calculated using all uncorrelated features identified for a given network size. Important features are selected from the set using the feature importance attribute of random forests regression. We then create a random forests regressor to predict outcomes based on the model of the new feature set, and this model is tested using the test dataset.
Regressors performance is measured using the COD, which quantifies how predicted values provided by the model compare against real values. Adequate regressor models typically exhibit a COD near 1, while poorly performing models exhibit values near 0. As evident from Fig. 3a, the COD determined from the reduced feature set (Section 2.5) either improved the model accuracy or showed no substantial difference from the set of all features for E.coli. In a majority of the cases, it is evident that feature reduction did not affect performance in a negative way, suggesting that the set of reduced features plays a much stronger role in information transport in these transcriptional networks than all other features. Additionally, we observe that our models perform well at higher levels of noise or channel loss. It is evident from Fig. 3a that E.coli models perform better at higher loss rates.
Hence, in order to compare the feature significance across organisms, we considered the Yeast models only for higher losses (i.e. 60%, 75% and 90%). Figure 3b shows the coefficient of determination for Yeast subnetworks at higher loss models. We used the top five features to build the final models of Yeast, as explained in Section 2.5. In all the cases of Yeast, it is evident that top five selected feature models’ performances are comparable or have very little loss in accuracy when compared to the models with all uncorrelated features. This suggests that the top five features play a very strong role in information transport in these Yeast transcriptional networks.
Figure 5 shows the number of features selected by our feature-selection process from all 233 features in E.coli. The maximum number of important features was 15 for the network size 300 and channel loss model of 35% and 60%, with 8 as the least number of important features for 400 node networks operating under a channel loss model of 10% and 20%. We find that many scenarios exhibit 11 and 12 important features.
Feature importance (Section 2.5) for E.coli is shown in Fig. 7a. Heat maps were generated for all the networks at channel loss models of 10%, 20%, 35%, 50%, 60%, 75%, and 90%. Figure 7 represents one such case for a network size of 500 and all loss models created with every reduced feature set. We observe that features with IDs 1, 2, 3, 8, 9, 11, 14 and 52 are important for all levels of packet loss. Additionally, features 62 and 81 are important for 75% and 90% packet loss. Topologies of these features have been collected into the Table 3. Here, the abundance of each feature is provided for the largest connected component of the entire E. coli transcriptional network.

a Feature significance in size 500 E.Coli subnetworks for all loss models and reduced feature sets. b Feature significance in size 500 Yeast subnetworks for all loss models and reduced feature sets. The darker the color, the higher is the feature significance. Additionally, numbers are included to indicate feature rank; higher is better
Figure 7b represents the feature importance for Yeast network size of 500 and all loss models. The feature with ID 7, consistently show up to be important in all of the loss models. Additionally, features 16, 59, 291 are important in two of the three loss models while features 1 and 2 are important in loss model 60%. Topologies of Yeast features have been collected into the Table 4. Here, the abundance of each feature is provided for the largest connected component of the entire Yeast. Note that the same FFL crosstalk pattern may have different IDs in the E.coli and Yeast feature set and the reader is referred to Tables 3 and 4 to identify the important features based on the IDs reported here.
2.7 Correlation of feature importance with feature abundance
To test the hypothesis that high feature counts correlate positively with high feature importance, we performed the following task executed at the network level. For each network size, the significant features were identified for all models for different levels of packet loss as follows:
-
1.
Identify the top five features using random forest regression (feature importance as a metric);
-
2.
Calculate the number of times each feature occurs within the top five ranks at different channel loss models and network size;
-
3.
Plot the distribution of these features (Fig. 8).

Feature value distribution of E.Coli subnetworks of size 500. Refer to Table 3 for depictions of features 1, 2, 3, 8, and 14
We found good correlation between abundant feature values with high variance and their corresponding feature importance. From all the models and across the two organisms studied here, E.coli features 1, 2, 3, 8, 14 and Yeast features 1, 2, 7, 29, 109 consistently rank in the top five features in their respective models; hence, these features are strong indicators of information transport robustness in TRNs across different organisms. Figures 8 and 9 show the feature value (abundance) distribution of the top five features of E. coli and Yeast as mentioned before. We can observe that all the features have high abundance with high variance. In E.coli, it is important to note that certain features, such as features with IDs 62, 81, 125, make their impact distinctively in specific network sizes at specific loss scenarios. This can be attributed to the fact that these specific features might be expressed more during the network extraction step (Section 2.1). Figure 8 illustrates a boxplot of the distribution of feature values of the top five features; feature-value distributions for other networks are not shown here (see Section 4).

Feature value distribution of Yeast subnetworks of size 400. Refer to Table 3 for depictions of features 1, 2, 7, 29, and 109
Once we determine the significant features for both E.coli and Yeast, we compared them to understand the topological feature importance towards network robustness across these two organisms. We found that E.coli and Yeast indeed have common significant features which indirectly prove our hypothesis that specific FFL crosstalk patterns are instrumental in imparting information transport robustness in such TRNs. Figure 10 shows the five most significant features that occur both in the E.coli and Yeast transcriptional networks.

Common important FFL crosstalk features in E.coli and Yeast TRNs
3 Discussion & conclusions
In this paper we studied how differing topological configurations of FFL crosstalk affected the information transport robustness in transcriptional subnetworks of two well studied bacteria: E.coli and Yeast. We evaluated information transport in such TRNs using the packet transport and routing events enabled by NS-2 simulations. Random forest based regression models revealed that a handful of FFL crosstalk features, such as feature IDs 1, 2, 3, 8 and 14 (Table 3) of E.coli, and feature IDs 1, 2, 7, 11, 62 (Table 4) of Yeast may have an important role in enabling the robust communication of molecular information across the sub-cellular transcriptional-regulatory machinery of the cell. Interestingly, we observe that the identified similar FFL crosstalk patterns are all sparsely connected, with three or fewer crosstalk edges between the two FFLs; this observation is consistent with our recent report that FFL crosstalk with three of fewer edges are enough to regulate the dynamics of the coupled-FFL circuit and more edges between them may largely be redundant in altering their dynamics [21]. Certain crosstalk configurations appeared differentially important under varying noise levels inherent to the communication channels. Understanding how noise interferes with communicating the cellular state to distal molecular processes is a great challenge, because the cell is a dynamically evolving environment that continually produces and destroys molecular components from which signaling success is not guaranteed.
Extensions of this work involve investigations in larger E. coli and Yeast transcriptional subnetworks, to explore whether or not trends in feature significance scales with increasing network complexity. Previous results [4] reveal that feature significance varies from one organism to another and scales across network size and perturbation conditions; however these studies did not consider FFL crosstalk based features. As we fine-tune our regression models it is also important to focus on moderately sized networks (e.g., 300 and 500 nodes) with lower channel loss models (e.g., 35% and 50%), to better understand why our regression models did not adequately perform.
Finally, the present work will provide a foundation for the biological network community to better understand the functional role of crosstalk between smaller transcriptional network motifs. In addition, the engineering community may benefit from knowledge that certain network topologies provide more robust communication platforms, transforming the difficult problem of information-preserving dynamical routing across terrain and environmental obstacles into one concerned only with short-range topological interactions.
4 Additional material
Datasets are available for research purposes at: https://github.com/bnetlab/FFL-Crosstalk-pattern-Importance.git
In addition, this address provides results for all the channel loss models not presented here. Sensitivity analyses for variation in mean square error, mean absolute error, and explained variance are also provided here.
Change history
27 November 2017
The original version of this article unfortunately contained a mistake in the author group section. Author Preetam Ghosh’s given name was incorrectly spelled as “Preeetam”.
References
Mayo M, Abdelzaher AF, Perkins E, Ghosh P (2014) Top-level dynamics and the regulated gene response of feed-forward loop transcriptional motifs. Phys Rev E 90(3):032706
Milo R, Shen-Orr S, Itzkovitz S, Kashtan N, Chklovskii D, Alon U (2002) Network motifs: simple building blocks of complex networks. Science 298(5594):824–827
Mangan S, Alon U (2003) Structure and function of the feed-forward loop network motif. Proc Nat Acad Sci 100(21):11980–11985
Kamapantula BK, Mayo M, Perkins E, Ghosh P (2014) Dynamical impacts from structural redundancy of transcriptional motifs in gene-regulatory networks. In: Proceedings of the 8th international conference on bioinspired information and communications technologies (BICT ’14). ICST (Institute for Computer Sciences, Social-Informatics and Telecommunications Engineering), ICST, Brussels, pp 199–206. https://doi.org/10.4108/icst.bict.2014.257928
Kamapantula BK, Mayo M, Perkins E, Abdelzaher AF, Ghosh P (2014) Feature ranking in transcriptional networks: packet receipt as a dynamical metric. In: Proceedings of the 8th international conference on bioinspired information and communications technologies (BICT ’14). . ICST (Institute for Computer Sciences, Social-Informatics and Telecommunications Engineering), ICST, Brussels, pp 1–8. https://doi.org/10.4108/icst.bict.2014.257930
Guo S, Murray RM Prototyping and implementation of a novel feedforward loop in a cell-free transcription-translation system and cells. https://doi.org/10.1101/123190
Barabasi AL, Albert R (1999) Emergence of scaling in random networks. Science 286:509–512
Ghosh S, Ghosh P, Basu K, Das SK (2005) GaMa: an evolutionary algorithmic approach for the design of mesh-based radio access networks. In: Proceedings of the IEEE conference on local computer networks 30th anniversary (LCN ’05). IEEE Computer Society, Washington, pp 374–381. https://doi.org/10.1109/LCN.2005.72
Mayo M, Abdelzaher AF, Perkins EJ, Ghosh P (2012) Motif Participation by Genes in E. coli transcriptional networks. Front Physiol 3:357. https://doi.org/10.3389/fphys.2012.00357
Kamapantula BK, Abdelzaher A, Ghosh P, Mayo M, Perkins E, Das SK (2012) Performance of wireless sensor topologies inspired by E. coli genetic networks. In: 2012 IEEE international conference on pervasive computing and communications workshops (PERCOM Workshops). IEEE, pp 302–307. https://doi.org/10.1109/PerComW.2012.6197500
Ghosh P, Mayo M, Chaitankar V, Habib T, Perkins E, Das SK (2011) Principles of genomi crobustness inspire fault-tolerant wsn topologies: a network science based case study. In: 2011 IEEE international conference on pervasive computing and communications workshops (PERCOM Workshops). IEEE, pp 160–165. https://doi.org/10.1109/PERCOMW.2011.5766861
Kamapantula BK, Abdelzaher A, Ghosh P et al (2014) Leveraging the robustness of genetic networks: a case study on bio-inspired wireless sensor network topologies. J Ambient Intell Human Comput 5:323. https://doi.org/10.1007/s12652-013-0180-0
Nazi A, Raj M, Di Francesco M, Ghosh P, Das SK (2016) Efficient communications in wireless sensor networks based on biological robustness. In: Proceedings of the 2016 international conference on distributed computing in sensor systems (DCOSS), pp 161–168
Nazi A, Raj M, Di Francesco M, Ghosh P, Das SK (2015) Exploiting gene regulatory networks for robust wireless sensor networking. In: Proceedings of the 2015 IEEE global communications conference (GLOBECOM), pp 1–7. https://doi.org/10.1109/GLOCOM.2015.7416957
Nazi A, Raj M, Di Francesco M, Ghosh P, Das SK (2013) Robust deployment of wireless sensor networks using gene regulatory networks. In: Proceedings of the 2013 international conference on distributed computing and networking, pp 192–207
Nazi A, Raj M, Di Francesco M, Ghosh P, Das SK (2014) Deployment of robust wireless sensor networks using gene regulatory networks: an isomorphism-based approach. Pervas Mob Comput 13:246–257
Chan H, Akoglu L, Tong H (2014) Make it or break it: manipulating robustness in large networks. In: Proceedings of the 2014 SIAM data mining conference. SIAM, pp 325–333
de la Peña JA, Gutman I, Rada J (2007) Estimating the Estrada index. Linear Algebra Appl 427:70–76
Kamapantula BK, Abdelzaher AF, Mayo M, Perkins E, Das SK, Ghosh P (2017) Quantifying robustness in biological networks using NS-2. Model Methodol Tools Molecular Nano-scale Commun 9:273–290
Schaffter T, Marbach D, Floreano D (2011) GeneNetWeaver: in silico benchmark generation and performance profiling of network inference methods. Bioinformatics 27:2263–2270
Rowland MA, Abdelzaher AF, Ghosh P, Mayo ML (2017) Crosstalk and the dynamical modularity of feed-forward loops in transcriptional regulatory networks. Biophys J 112:1539–1550
Pedregosa F, Varoquaux G, Gramfort A, Michel V, Thirion B, Grisel O, Blondel M, Prettenhofer P, Weiss R, Dubourg V et al (2011) Scikit-learn: machine learning in Python. J Mach Learn Res 12:2825–2830
Schult DA, Swart PJ (2008) Exploring network structure, dynamics, and function using NetworkX. Proc 7th Python Sci Conf (SciPy 2008(2008):11–16
Breiman L (2001) Random forests. Mach Learn 45:5–32
Acknowledgments
Funding was provided by the US Army’s Environmental Quality and Installations 6.1 Basic Research program. Opinions, interpretations, conclusions, and recommendations are those of the author(s) and are not necessarily endorsed by the US Army.
Author information
Authors and Affiliations
Corresponding author
Additional information
The original version of this article was revised: There was a mistake in the author group section. Author Preetam Ghosh’s given name was incorrectly spelled as “Preeetam”.
Rights and permissions
About this article

Cite this article
Syed, K., Abdelzaher, A., Mayo, M. et al. Similar Feed-forward Loop Crosstalk Patterns may Impact Robust Information Transport Across E. coli and S. Cerevisiae Transcriptional Networks. Mobile Netw Appl 25, 1970–1982 (2020). https://doi.org/10.1007/s11036-017-0944-4
Published:
Issue Date:
DOI: https://doi.org/10.1007/s11036-017-0944-4