
Abstract
Background
Protease S (PrtS) from Photorhabdus laumondii belongs to the group of protealysin-like proteases (PLPs), which are understudied factors thought to play a role in the interaction of bacteria with other organisms. Since P. laumondii is an insect pathogen and a nematode symbiont, the analysis of the biological functions of PLPs using the PrtS model provides novel data on diverse types of interactions between bacteria and hosts.
Methods and results
Recombinant PrtS was produced in Escherichia coli. Efficient inhibition of PrtS activity by photorin, a recently discovered emfourin-like protein inhibitor from P. laumondii, was demonstrated. The Galleria mellonella was utilized to examine the insect toxicity of PrtS and the impact of PrtS on hemolymph proteins in vitro. The insect toxicity of PrtS is reduced compared to protease homologues from non-pathogenic bacteria and is likely not essential for the infection process. However, using proteomic analysis, potential PrtS targets have been identified in the hemolymph.
Conclusions
The spectrum of identified proteins indicates that the function of PrtS is to modulate the insect immune response. Further studies of PLPs’ biological role in the PrtS and P. laumondii model must clarify the details of PrtS interaction with the insect immune system during bacterial infection.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Protease S (PrtS) is a secretory zinc metalloprotease of the M4 family found in Photorhabdus bacteria [1]. This enzyme belongs to the group of protealysin-like proteases (PLPs) widespread in bacteria but also occurring in fungi and archaea [2, 3]. The biological functions of PLPs are currently being discussed. However, the available data indicates the involvement of PLPs in the bacterial interaction with animals and plants and possibly in the pathogenesis [1, 3,4,5,6,7,8,9,10,11,12,13,14]. Moreover, PLPs are thought to play a role in interbacterial competition [15, 16]. The study of PLP biological functions can clarify interactions between bacteria and other organisms.
Photorhabdus laumondii are both symbionts of Heterorhabditis nematodes and insect pathogens, providing an opportunity to investigate PLP functions via PrtS in various ecological contexts. Photorhabdus models are being actively used to study the mechanisms of bacteria-host interactions during infection. A wide range of Photorhabdus protein toxins with potent insecticidal activity, as well as a number of low molecular weight metabolites that affect host insect immunity, are known [17]. Yet, there is limited data regarding the PrtS’ role. The injection of substantial quantities of PrtS induces insect death [13]; nevertheless, neither the dose-effect relationship nor the mechanism of action has been defined. PrtS is recognized for inducing melanization in insects, which is attributed to the overstimulation and disturbance of the insect’s innate immune response [13]. The hemolymph of infected insects displays PrtS activity and in vitro exposure to PrtS reduces the antibacterial activity of hemolymph by half, likely resulting from the hydrolysis of antimicrobial peptides [1]. The transcription of the prtS gene was observed during infection [18] but decreased in a phenotypic variant of P. laumondii that exhibits delayed pathogenicity [19]. The evidence suggests that PrtS plays a role in bacterial pathogenesis in insects.
In the present study, to come closer to understanding this role, the recombinant PrtS was produced by expressing the prtS gene in Escherichia coli and purified. Using the purified enzyme, we investigated the interaction between PrtS and photorin, its endogenous inhibitor that was recently discovered [20], studied how the PrtS amount affects insect toxicity and melanization, and identified potential PrtS substrates in the larval hemolymph of the greater wax moth Galleria mellonella.
Materials and methods
Chemical and reagents
In this work we used tris-(hydroxymethyl)aminomethane (Tris) (Amresco, USA), ammonium bicarbonate, 1,10-phenanthroline, piperazine-N, N′-bis(2-ethanesulfonic acid) (PIPES), N-(2-hydroxyethyl) piperazine-N′-(2-ethanesulfonic acid) (HEPES), N-(2-hydroxyethyl)piperazine-N′-(4-butanesulfonic acid) (HEPBS), dithiothreitol (DTT), 2-iodoacetamide, 2,5-dihydroxybenzoic acid, trypsin, phenylthiourea, 3-[(3-cholamidopropyl)dimethylammonio]-1-propanesulfonate(CHAPS), NP-40 detergent (Sigma-Aldrich, Germany), dimethyl sulfoxide (DMSO) (MP Biomedicals, France), ampicillin, imidazole (PanReac, Germany), isopropyl-β-D-thiogalactopyranoside (IPTG) (Thermo Fisher Scientific, USA), sodium dodecyl sulfate (SDS) (Helicon, Russia), acrylamide (Dia-M, Russia), N,N’-methylenebisacrylamide (Serva, Germany), and phosphate-buffered saline (PBS, Eco-Service, Russia).
The following reagents were used for genetic construction: Tersus DNA polymerase (Evrogen, Russia), Klenow fragment, T4 DNA ligase (SibEnzyme, Russia), and restriction endonucleases XhoI, XbaI (New England Biolabs, USA), FauNDI, and BglII (SibEnzyme, Russia).
Lennox LB broth (10 g/l peptone (Difco, USA), 5 g/l yeast extract (Difco, USA), and 5 g/l NaCl (PanReac, Germany)) was used to culture bacteria.
Protealysin (Pln), emfourin (M4in), and photorin (Phin) were overproduced in E. coli and purified as described previously [15, 20, 21] Thermolysin (Tln) was obtained from Serva (Germany).
2-Aminobenzoyl-L-arginyl-L-seryl-L-valyl-L-isoleucyl-L-(ε-2,4-dinitrophenyl)lysine (Abz-RSVIK(dnp)) from Peptide 2.0 (Chantilly, USA) was used as a substrate for protease activity determination.
All other reagents were of reagent grade and were purchased from Helicon (Russia).
Protein analysis
Protein concentration was assayed after Bradford with modifications [22] using IgG as a standard.
The concentrations of purified proteins were determined spectrophotometrically. Published extinction coefficients ε280nm = 52,370 M− 1 cm− 1, ε280nm = 58,200 M− 1 cm− 1, ε280nm = 10,810 M− 1 cm− 1 and 11,460 M− 1 cm− 1 were used for Pln, Tln, M4in, and Phin, respectively [20]. The PrtSt extinction coefficient (ε280nm = 48,360 M− 1 cm− 1) was calculated using the ProtParam tool (http://www.expasy.org/tools/protparam.html).
Proteins were analysed by SDS-PAGE in 12.5% polyacrylamide gel according to Laemmli [23]. Proteins were visualized by Coomassie R-250 Brilliant Blue (Sigma-Aldrich, Germany) staining. Pierce Unstained Protein MW Marker (ThermoFisher Scientific, USA) was used as molecular weight standards.
Transformation of E. coli cells with plasmid DNA was carried out as described by Sambrook et al. [24].
Cloning and construction of expression vectors
Plasmid pETDuet-PrtS was constructed to express a gene encoding PrtS from P. laumondii subsp. laumondii TT01 with a His6-tag at the C-terminus. The first step of pETDuet-PrtS construction included PCR amplification of the protease S gene from P. laumondii subsp. laumondii TT01 using pTT01 [20] as a template and PrtS_F (TGCAAATACAAAACAATAACTACAGAG) and PrtS_XhoI (GAATCTCGAGTTAATGGTGATGGTGATGGTGCTCTTAGTTTTATCTTTATTTTTGTCCT) as primers. The resulting fragment was digested with XhoI. The pET23c vector (Novagen, USA) was treated with FauNDI, Klenow fragment, and XhoI. After ligation, the resulting construct pET23-PrtS was used as the PCR template with primers T7-uni (GTAATACGACTCACTATAGGG) and T7-rev (TATGCTAGTTATTGCTCAGCGG). The amplification product was ligated into the XbaI and XhoI sites of pETDuet-1 (Novagen, USA).
pETDuet-PrtSt plasmid encodes a PrtS with a non-conserved C-terminal region replaced by the AHHHHHH sequence. The resulting protein variant was designated as PrtSt (PrtS truncated). This construction included PCR amplification of a protease S gene fragment from pETDuet-PrtS using primers PrtSt_H6 (CAGACTCGAGTTAATGGTGATGGTGATGGTGCGCAACGCCCACTTCTGTCC) and PrtSt_BglII (ATGCCTGGGAAAGAGCAG). The region corresponding to Ala and His6-tag is boldfaced and the XhoI site is underlined. The resulting fragment was ligated into the XhoI and BglII sites of pETDuet-PrtS.
All cloned fragments were confirmed by sequencing. The obtained genetic constructs were sequenced and all oligonucleotides used were synthesized at Evrogen (Russia).
Protease S purification
In an attempt to produce PrtS in E. coli using the pETDuet-PrtS plasmid, the target protein could not be purified by immobilized metal affinity chromatography (IMAC), possibly due to the proteolytic degradation of the C-terminal region with the His6-tag. It should be noted that such C-terminal regions are generally non-conserved in protealysin-like proteases (PLPs) but they are present with some variations in most PLPs of the Photorhabdus bacteria. The role of this particular region remains unknown in Photorhabdus. However, we speculate that its insignificance for the catalytic function stems from its localization outside of the catalytic domain, its distance from the active site, and its likely unstructured nature (Fig. S1). Furthermore, our collected data suggests that this region is removed from PrtS when it is produced in E. coli. Accordingly, we engineered PrtSt (PrtS truncated), a PrtS with a C-terminal region similar to recombinant protealysin (Pln) from Serratia proteamaculans [2].
E. coli T7 Express lysY/Iq (New England Biolabs, USA) cells transformed with pETDuet-PrtSt were grown with agitation in 250 ml of Lennox LB containing 100 μg/ml ampicillin at 37 °C for 2 h; 1 mM IPTG was added to the culture and incubation continued at 16 °C for 30 h. Cells were collected by centrifugation (3000 g, 4 °C, 10 min), resuspended in 15 ml of 50 mM Tris-HCl with 2 mM 1,10-phenanthroline, pH 7.4, and ultrasonicated at 4 °C 3 times for 4 min with 1 s impulse / 2 s pause. The cell lysate was centrifuged (9,000 g, 4 °C, 10 min) and the supernatant was loaded onto a 5 ml Nickel XPure Agarose Resin (UBPBio, USA) column pre-equilibrated with 50 mM Tris-HCl, pH 7.4. Elution was performed with 0-300 mM imidazole gradient (15 ml) in the same buffer. After SDS-PAGE analysis, the protease-containing fractions were pooled and concentrated by ultrafiltration on an Amicon Ultra-15 30,000 NMWL Centrifugal Filter Unit (Millipore, USA). The resulting sample was loaded onto a Superdex 75 Increase 10/300 GL column (GE Healthcare, Sweden) equilibrated with 50 mM Tris-HCl, 150 mM NaCl, 2 mM 1,10-phenanthroline, pH 7.4, and eluted with the same buffer at a flow rate of 0.4 ml/min. The fractions with the highest content of the protease were combined and dialyzed against 100 volumes of 50 mM ammonium bicarbonate using Slide-A-Lyzer G2 dialysis Cassette 3,500 MWCO (Thermo Fisher Scientific, USA) for 3 h at 4 °C. After buffer replacement, dialysis was continued overnight at 4 °C. The dialyzed solution was centrifuged (9,000 g, 15 min) and the supernatant was lyophilized.
N-terminal sequencing
For N-terminal sequencing by Edman degradation, proteins were separated by SDS-PAGE and transferred onto a PVDF membrane (Bio-Rad, USA) using the Trans-Blot Turbo system (Bio-Rad, USA) in 50 mM Borate buffer with 10% (v/v) ethanol, pH 9.0. The membrane was sent to the Protein Core Facility of Innsbruck Medical University for sequencing.
Protease activity and inhibition assays
The previously described substrate Abz-RSVIK(Dnp) [25] and analysis method [15, 20] were used for protease activity and inhibition assays. Mass spectrometry data showed that PrtSt, similar to Pln, hydrolyses the substrate at the Ser-Val bond (Fig. S2). Fluorescence was recorded on a CLARIOstar Plus microplate reader (BMG, Germany) at the excitation and emission wavelengths of 320 and 420 nm, respectively, as described earlier by Berdyshev et al. [26]. PrtSt and Tln concentrations were 50 pM in all reactions. The relationship between the rate of Abz-RSVIK(Dnp) hydrolysis by PrtSt and substrate concentration was studied for the substrate concentration range from 3.75 to 120 μM. In the inhibition assay, Abz-RSVIK(Dnp) concentrations were 30 or 90 μM. M4in concentrations were 0.1–3.5 nM for PrtSt (Fig. S3, PrtS + M4in) and 1–20 μM for Tln (Fig. S3, Tln + M4in). Phin concentrations were 0.1–3.5 nM for PrtSt (Fig. S3, PrtS + Phin).
Determination of slow-binding inhibition kinetic parameters (v0, vs, ka), kinetic constants (k+ 3, k-3), and equilibrium constant (Ki) was performed as described previously [15, 20]. The date on the rate of Abz-RSVIK(Dnp) hydrolysis by PrtSt at different substrate concentrations was fitted to the substrate inhibition Eq. (1) to calculate KM and KiS [27]:
where v is the reaction velocity, [S] is the substrate concentration, Vmax is the maximum reaction velocity, KM is the Michaelis-Menten constant, KiS is the substrate inhibition constant.
Turnover number kcat was calculated from Eq. (2):
where kcat is the turnover number, Et is the concentration of enzyme catalytic sites (for PrtSt, it equals the enzyme concentration).
The cleavage site of Abz-RSVIK(Dnp) by PrtSt was identified in 10 mM NH4HCO3 as described by Karaseva et al. [25]. The volume of the reaction mixture was 100 μl with 30 μM Abz-RSVIK(Dnp) and 0.5 nM PrtSt. The mixture was incubated for 0 and 90 min at 37 °C and immediately frozen at − 20 °C and lyophilized. Control samples were produced in a similar way but without the enzyme or the substrate. The dried samples (≈ 3 nmole of peptide) were dissolved in 300 μl of mixed water-acetonitrile (1:1) with 1 mM 1,10-phenanthroline and then diluted 1:10 in water-acetonitrile (1:1) with 0.1% formic acid. Mass spectrometric analysis of the obtained peptide solutions was carried out on a Q-exactive HF-X mass spectrometer (Thermo Scientific, USA) in the positive ionization mode using an electrospray ion source (ESI). The sample solution was injected into ESI at a rate of 5 μl/min. The following settings were used for mass spectrometric analysis: spray voltage, 4 kV; capillary temperature, 290 °C; and sheath gas flow rate, 8 l/min. Panoramic scanning was performed in the mass range from 200 to 2000 m/z at a resolution of 70,000. The resulting mass spectrum of each sample was obtained by averaging 100 scans.
The proteolytic activity was determined by Azocasein hydrolysis as described previously [1]. Three independent measurements were carried out for each experimental point in all reactions.
Effect of chelating agents and zinc ions on PrtSt activity
Two ml of ZnSO4 (0.5 or 1 mM), 1,10-phenanthroline (0.5 or 1 mM) or EDTA (10 or 50 mM) in 50 mM Tris-HCl, pH 7.4, were supplemented with 2 μl of 1 μM PrtSt in the same buffer and incubated for 10 min. Ten μl of the resulting solution was added to 110 μl of Abz-RSVIK(Dnp) in the same buffer containing the tested compound in the same concentration, and fluorescence changes were measured as described above. The Abz-RSVIK(Dnp) concentration in the reaction mixture was 30 μM. The obtained activity values were checked against the enzyme activity measured similarly but without tested compounds.
Temperature stability of PrtSt
Temperature stability was determined by incubation of 1 nM PrtSt in 50 mM Tris-HCl, pH 7.4, at 40–70°С for 2–150 min, after which samples were placed on ice. Protease activity was determined by hydrolysis of Abz-RSVIK(Dnp) as described above. Its concentration in the reaction mixture was 30 μM.
pH optimum of PrtSt activity
The pH optimum was identified by measuring PrtSt activity in buffers containing 50 mM PIPES, 50 mM HEPES, and 50 mM HEPBS; pH was adjusted to 5.5–9.5 by 1 M HCl or 1М NaOH. Protease activity was determined by Abz-RSVIK(Dnp) hydrolysis as described above. The Abz-RSVIK(Dnp) concentration in the reaction mixture was 30 μM.
Insect toxicity
Insect toxicity of Pln, Tln, and PrtSt was tested on last instar larvae of G. mellonella weighing 250–350 mg (Eco Baits, Russia). Protease solutions with concentrations of 4, 6, 8, 10, 14, 16, 18, 20, and 24 μM were made in sterile PBS. A volume of 10 μl was injected into the left one of the first pair of proleg using a BD Micro-Fine Plus 1 ml syringe (USA) with a 30G×8 needle and an infusion pump LSP04-1 A (Longer Pump, China). The control larvae were injected with 10 μl of PBS. Postinjection larvae were placed in Petri dishes and incubated at 30°С. The living and melanized animals were counted 24 h later. Three independent experiments with 15 larvae in each experimental and control group were conducted.
The LD50 and ED50 values were calculated as described by Agarwal et al. [28]. Data analysis was performed using the GraphPad Prism version 9.5 (GraphPad Software, USA).
Proteomic analysis
The purified PrtSt components were analyzed by SDS-PAGE. The protein bands of interest were excised from the gel and treated with DTT, 2-iodoacetamide, and then trypsin using the standard method [29]. Tryptic peptides were extracted from the gel as described previously [30]. An LC-MS/MS analysis was performed using a nano-flow ultra-high pressure system nanoElute (Bruker Daltonics, Germany) coupled with a mass spectrometer timsTOF (Bruker Daltonics) using a CaptiveSpray (Bruker Daltonics) ion source. The obtained mass spectra were processed by PEAKS Studio X Pro [31].
The effect of PrtSt on hemolymph proteins was studied after incubation of G. mellonella larvae on ice for 15 min. The hemolymph of 50 larvae was collected into 1.5 ml pre-cooled tubes and 2 μg/ml of phenylthiourea was added to prevent melanization. The hemocytes were removed by centrifugation at 200 g, 4 °C for 5 min and the supernatant was centrifuged at 18,600 g, 4 °C for 10 min. The cell-free hemolymph was diluted with sterile PBS to 60 mg/ml protein. The obtained solution (150 μl) was supplemented with an equal volume of purified PrtSt in PBS (0.6 mg/ml). The mixture was incubated at 30°С for 15 min and the reaction was stopped by 0.3 μl 1 M 1,10-phenanthroline, frozen, and stored at -70°С until analysis. In the control, PrtSt and hemolymph solutions were incubated separately, 1,10-phenanthroline was added to 2 mM to protease, and solutions were combined. The PrtSt solution incubated under the same conditions and supplemented with 1,10-phenanthroline was used as an additional control.
Before 2D electrophoresis, the hemolymph of G. mellonella was reprecipitated with a mixture of chloroform/methanol/water and dissolved in a mixture of sample buffer for isoelectric focusing (40 mM Tris-НСl, рН 9.5, containing 8 M urea, 2 M thiourea, and 4% of mixture of CHAPS and NP40), incubated 30 min at the room temperature, and centrifuged at 14,000 g for 15 min. The proteins were labelled using 3Dye 2D DIGE Kit (Lumiprobe, Russia) in accordance with the manufacturer’s instructions.
Differential 2D gel-electrophoresis and tryptic digestion of the proteins were performed as described earlier [32]. Mass spectra were recorded on an Ultraflex II MALDI-ToF-ToF mass spectrometer (Bruker Daltonics) equipped with an Nd laser. The [MH]+ molecular ions were measured in reflector mode; the accuracy of mass peak measurement was 70 ppm.
Data analysis and presentation
Data analysis and construction of charts were performed using the GraphPad Prism version 9.5 (GraphPad Software, USA). SDS-PAGE analysis was performed on a Gel Doc XR + system (Bio-Rad) using Image Lab software. Differential 2D gels were scanned on a Typhoon Trio scanner (Amersham). Identification of protein hemolymph was carried out by peptide fingerprint search by Mascot software (Matrix Science, USA) through the G. mellonella protein database. Protein scores greater than 45 are supposed to be significant (p < 0.05).
Results
Preparing of protease S
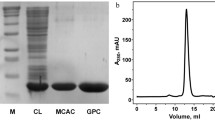
PrtSt (PrtS truncated) accumulated in cells mostly in a soluble activity form and was purified by immobilized metal affinity chromatography (IMAC) and gel permeation chromatography (GPC). Details on protein purification are presented in Fig. 1; Table 1. The purified protease was not electrophoretically homogeneous, which we attribute to autoproteolysis. Along with the major band corresponding to the 35.7 kDa protein (band B36 in Fig. 1A), we also detected a minor constituent (∼ 10%) of approximately 31.6 kDa in the gel (band B32 in Fig. 1A). However, mass spectrometry demonstrated that both the major and minor bands match the protease S (Fig. S4); indicating that the minor constituent is a degradation product.

Purification of protease S (PrtSt) and analysis of the purified protein. A SDS-PAGE analysis. M, molecular weight standards; Lysate, cell lysate of E. coli T7 Express lysY/Iq (pETDuet PrtSt); IMAC, PrtSt sample after immobilized metal affinity chromatography; GPC, PrtSt sample after gel permeation chromatography. The arrows indicate the major (B36) and minor (B32) bands of PrtSt with a mass of ∼ 35.7 kDa and ∼ 31.6 kDa. B Analysis of purified PrtSt by gel permeation chromatography on a Superdex 75 10/300 GL column. The molecular weight of PrtSt was calculated from the calibration curve
The analysis of the obtained PrtSt preparation by GPC demonstrated that the enzyme is in the solution as a monomer (Fig. 1B). The molecular weight of the purified protease determined by SDS-PAGE and GPC corresponds to the mature enzyme (an N-terminal propeptide of about 50 amino acids is removed during PLP maturation). The processing point was identified through N-terminal sequencing of the major and minor components (B36 and B32, respectively, in Fig. 1) of the purified PrtSt. The major N-terminal sequence of B36 is VINES, which corresponds to the previous data for mature PrtS [13]. No such N-terminal sequence was found for B32. Additionally, both samples were found to contain PDNGD and PQPGH (Fig. S4), which likely corresponds to degradation products of the enzyme.
Thus, a preparation of recombinant protease S was acquired with around 95% of the desired enzyme, with the full-length mature protease form comprising approximately 85% of the overall protein.
Enzymatic properties of protease S
The enzymatic activity of PrtSt was evaluated using Abz-RSVIK(Dnp) as a substrate. The relationship between the rate of hydrolysis and the substrate concentration indicated the enzyme inhibition by the substrate (Fig. 2), while no such inhibition was observed for Pln [25, 26]. Abz-RSVIK(Dnp) hydrolysis by PrtSt can be described by the following parameters: KM = 13 ± 3 μM, kcat = 38 ± 4 s-1, and substrate inhibition constant KiS = 310 ± 100 μM (presented as mean ± standard error (SE)).

The dependence of the initial rates of Abz-RSVIK(Dnp) hydrolysis by protease S (PrtSt) on the substrate concentration. PrtSt concentration is 50 pM. Three independent measurements were carried out for each experimental point. Values are represented as mean ± SD
The optimum activity of PrtSt occurs at neutral pH (Fig. 3), which agrees with the published data on protease S from Photorhabdus sp. Az29 (amino acid sequence identity of the enzymes is 81%) and is typical of most metalloproteases of the M4 family [33, 34].

Effect of pH on protease S (PrtSt) activity for Abz-RSVIK(Dnp) hydrolysis. Three independent measurements were carried out for each experimental point. Values are represented as mean ± SD
PrtSt shows no high thermal stability (Fig. 4), which agrees with the published data on protease S from Photorhabdus sp. Az29 [1]. After exposure at 40 and 50°С for 2 h, PrtSt activity is reduced by about a quarter and a half, respectively; while the exposure at 60°С for 30 min reduces it by over 80%.

Protease S (PrtSt) activity after incubation at 40 (●), 50 (▼), 60 (▲), and 70 °C (■). A0 and A, Abz-RSVIK(Dnp) hydrolysis activity by PrtSt before and after incubation, respectively. Three independent measurements were carried out for each experimental point. Values are represented as mean. SD is below 0.1 for all points
The activity of PrtSt is effectively inhibited by 1,10-phenanthroline, whereas EDTA does not inhibit the enzyme completely even at 50 mM (Table 2). The same inhibition pattern was previously demonstrated for protease S from Photorhabdus sp. Az29 [1] as well as some other PLPs [33]. PrtSt activity, as well as other zinc metalloproteases [34], is inhibited by high (more than 0.05 mM) concentrations of Zn2+ (Table 2). Such an effect for PLPs was demonstrated for the first time.
We studied the sensitivity of PrtSt to two protein inhibitors of M4 peptidases, photorin (Phin) from P. laumondii [20] and emfourin (M4in) from S. proteamaculans [15], both of which belong to the recently discovered family I104 (https://www.ebi.ac.uk/merops/cgi-bin/pepsum?id=I104.001;type=I). Phin and M4in act as potent slow-binding competitive inhibitors of PrtSt (Fig. S3). Their inhibition constants (Ki) were determined to be 210 ± 10 and 190 ± 40 pM, respectively. Comparing these values, as well as our determined Ki for the M4in-Tln pair (660 ± 210 pM) (Fig. S3), with the previously determined Ki values for Phin-Tln, Phin-Pln [20], and M4in-Pln [15] interactions makes it clear that inhibitors from the same species most efficiently inhibit PLPs (Fig. 5).

Comparison of inhibition constants (Ki) for photorin (Phin) and emfourin (M4in) effect on protease S (PrtS), protealysin (Pln), and thermolysin (Tln). Constants are represented as mean + SE. Published Ki values for Pln and Tln [16] were used
The specific proteolytic activity of the obtained PrtSt determined by Azocasein hydrolysis was 28,240 ± 1,577 U/mg, which is consistent with the published value (26,518 U/mg) for protease S from Photorhabdus sp. Az29 [1]. At the same time, the activity of PrtSt was significantly higher compared to the Tln (18,940 ± 1,322 U/mg) and Pln (7,170 ± 1,330 U/mg) samples employed in this study.
Insect toxicity and melanization
The study utilized G. mellonella, the greater wax moth, as a model to investigate the insect toxicity of PrtS and its potential as a melanization factor. Similar tests were also conducted on Pln and Tln. Figure 6 presents the dependence of insect melanization and toxicity on the concentration of enzymes, while Table 3 shows the median lethal (LD50) and effective (ED50) doses inducing melanization in half of the animals.

Mortality (red) and melanization (blue) of Galleria mellonella larvae 24 h after injection of protease S (PrtSt), protealysin (Pln), or thermolysin (Tln)
Effect of protease S on hemolymph proteins in G. mellonella
To identify potential protease S substrates, hemolymph samples from G. mellonella larva were treated with the purified PrtS The protease-treated and control specimens were analyzed by two-dimensional difference gel electrophoresis (Fig. 7). Differentially represented protein spots were identified using a MALDI mass-spectrometry after trypsinolysis (Table 4).

Two-dimensional difference gel electrophoresis of hemolymph G. mellonella. Hemolymph proteins after treatment with PrtSt were labelled with Cyanine3 (green). Hemolymph proteins without PrtSt treatment were labelled with Cyanine5 (red). PrtSt incubated without hemolymph was labelled with Cyanine2 (blue). The arrows indicate the identified protein spots, the numbers correspond to Table 4
Discussion
The initial outcome of this study is the production of recombinant protease S. Due to the enzyme’s propensity for degradation, which complicated manipulations with it, we opted to eliminate the protein’s C-terminal segment and conduct purification in the presence of an inhibitor. After analyzing the resultant sample, we found that the protease’s specific activity, thermostability, pH optimum, and inhibition pattern were not impacted by the treatment, thus enabling the enzyme’s utilization for further research.
For the first time, the study examined the impact of recently discovered protein protease inhibitors of the I104 family on PrtS. These inhibitors’ genes in bacteria are colocalized with the genes of protealysin-like proteases (PLPs) [15]. The analysis of the obtained and published data [20] established that I104 inhibitors show significant species-specificity: Phin from P. laumondii most effectively inhibited PrtS activity just as M4in from S. proteamaculans inhibited Pln (Fig. 5). This finding affirms that cognate proteases and inhibitors have undergone evolutionary adaptation, and consequently endogenous proteases represent the natural targets of I104 inhibitors. Therefore, it is crucial to investigate the function of PLPs, especially in the P. laumondii model with regard to the corresponding inhibitors.
For the first time, we established the dependence of the insect toxic effect and melanization on the amount of PrtS (Fig. 6). The insect toxicity of PrtS proved much lower compared to its homologs from the non-entomopathogenic bacteria, Pln and Tln (Table 3). This can be attributed to the low specific activity of PrtS. Nevertheless, the efficiency of hydrolysis of protein (see Results, 3.2. Enzymatic Properties of PrtSt) and Abz-RSVIK(Dnp) peptide (kcat/KM is 2.9 ± 0.4 s-1 μM-1 for PrtSt (this work); 2.0 ± 0.1 s-1 μM-1 for Pln; and 1.5 ± 0.1 s-1 μM-1 for Tln [25]) by PrtS was higher compared to two other enzymes. In contrast to insect toxicity, the melanization ability was indistinguishable for the three enzymes (Table 3). At the same time, it was shown convincingly that both insect toxicity and melanization are defined by the enzyme activity [13]. Thus, it can be inferred that the differences in toxicity observed are attributable to the fine substrate specificity of proteases. Earlier findings on Tln [50] and elastase (LasB) from Pseudomonas aeruginosa [51] suggested a direct relationship between the insect toxicity of M4 proteases and insect melanization. The data obtained here for PrtS argues against this since pronounced melanization is observed in the presence of non-lethal enzyme doses (Fig. 6). Overall, it can be stated that both as insectotoxins and melanization inducers, homologues from non-entomopathogenic bacteria are not inferior to PrtS. Thus, it is not improbable that these PrtS properties are not specific and are not essential for the infection process.
PrtS is a secretory enzyme that can be found in the hemolymph of infected insects [19], suggesting specific targets for this protease in the hemolymph. The proteomic analysis of G. mellonella hemolymph exposed to PrtS was conducted, leading to the identification of potential protein substrates of the enzyme (Table 4). No published experimental findings regarding the function of this proteins in G. mellonella are available, but their orthologs in other organisms are known. Analysis of these data suggests that many of the proteins revealed are associated with the immune response, in particular, in insects. Adapting to the insect immune response is an important factor in the interaction of P. laumondii and other entomopathogenic bacteria with the host. In particular, these bacteria produce a number of low molecular weight compounds and proteins that affect the insect host immunity by acting on host proteins [52,53,54,55]. Thus, effector proteins and toxins such as LopT, TccC3, TccC5, Mcf1 and Photox secreted by Photorhabdus suppress the host insect cellular response through inhibition of phagocytosis by insect haemocytes [56]. In addition, Photorhabdus, like other entomopathogenic bacteria, release secondary metabolites that affect host insect immunity by inhibiting eicosanoid biosynthesis, resulting in suppression of melanization and nodule formation [52, 57]. Apparently, PrtS is in this category and its function is to modulate the insect immune response to bacterial infection.
Conclusion
The data obtained supports the previous proposal that photorin from P. laumondii is a specific inhibitor of PrtS and suggests an evolutionary co-adaptation between I104 inhibitors and protealysin-like proteases. The insect toxicity of PrtS is presumably negligible for the bacterial infection process since its role is to impair the insect’s immune system. Therefore, future investigations concerning the biological function of PLPs on the P. laumondii model should concentrate on the detailed analysis of PrtS interactions (given the involvement of photorin) with the immune system during in vivo infection.
Data availability
No datasets were generated or analysed during the current study.
References
Cabral CM, Cherqui A, Pereira A, Simoes N (2004) Purification and characterization of two distinct metalloproteases secreted by the entomopathogenic bacterium Photorhabdus sp. strain Az29. Applied and environmental microbiology. 70(7):3831–8. https://doi.org/10.1128/AEM.70.7.3831-3838.2004
Demidyuk IV, Gasanov EV, Safina DR, Kostrov SV (2008) Structural organization of precursors of thermolysin-like proteinases. Protein J 27(6):343–354. https://doi.org/10.1007/s10930-008-9143-2
Demidyuk IV, Gromova TY, Kostrov SV (2013) Protealysin. In: Rawlings ND, aGS (eds) Handbook of proteolytic enzymes. Academic, Oxford, pp 507–602
Kyostio SR, Cramer CL, Lacy GH (1991) Erwinia carotovora subsp. carotovora extracellular protease: characterization and nucleotide sequence of the gene. J Bacteriol 173(20):6537–6546. https://doi.org/10.1128/jb.173.20.6537-6546.1991
Feng T, Nyffenegger C, Hojrup P, Vidal-Melgosa S, Yan KP, Fangel JU et al (2014) Characterization of an extensin-modifying metalloprotease: N-terminal processing and substrate cleavage pattern of Pectobacterium carotovorum Prt1. Appl Microbiol Biotechnol 98(24):10077–10089. https://doi.org/10.1007/s00253-014-5877-2
Eshwar AK, Wolfrum N, Stephan R, Fanning S, Lehner A (2018) Interaction of matrix metalloproteinase-9 and Zpx in Cronobacter turicensis LMG 23827(T) mediated infections in the zebrafish model. Cell Microbiol 20(11):e12888. https://doi.org/10.1111/cmi.12888
Bozhokina E, Kever L, Khaitlina S (2020) The Serratia grimesii outer membrane vesicles-associated grimelysin triggers bacterial invasion of eukaryotic cells. Cell Biol Int 44(11):2275–2283. https://doi.org/10.1002/cbin.11435
Khaitlina S, Bozhokina E, Tsaplina O, Efremova T (2020) Bacterial actin-specific endoproteases Grimelysin and Protealysin as virulence factors contributing to the invasive activities of Serratia. Int J Mol Sci 21(11). https://doi.org/10.3390/ijms21114025
Tsaplina OA, Efremova TN, Kever LV, Komissarchik YY, Demidyuk IV, Kostrov SV et al (2009) Probing for actinase activity of protealysin. Biochemistry 74(6):648–654. https://doi.org/10.1134/s0006297909060091
Tsaplina O, Efremova T, Demidyuk I, Khaitlina S (2012) Filamentous actin is a substrate for protealysin, a metalloprotease of invasive Serratia proteamaculans. FEBS J 279(2):264–274. https://doi.org/10.1111/j.1742-4658.2011.08420.x
Tsaplina O, Demidyuk I, Artamonova T, Khodorkovsky M, Khaitlina S (2020) Cleavage of the outer membrane protein OmpX by protealysin regulates Serratia proteamaculans invasion. FEBS Lett 594(19):3095–3107. https://doi.org/10.1002/1873-3468.13897
Bozhokina ES, Tsaplina OA, Efremova TN, Kever LV, Demidyuk IV, Kostrov SV et al (2011) Bacterial invasion of eukaryotic cells can be mediated by actin-hydrolysing metalloproteases grimelysin and protealysin. Cell Biol Int 35(2):111–118. https://doi.org/10.1042/CBI20100314
Held KG, LaRock CN, D’Argenio DA, Berg CA, Collins CM (2007) A metalloprotease secreted by the insect pathogen Photorhabdus luminescens induces melanization. Appl Environ Microbiol 73(23):7622–7628. https://doi.org/10.1128/AEM.01000-07
Warshan D, Espinoza JL, Stuart RK, Richter RA, Kim SY, Shapiro N et al (2017) Feathermoss and epiphytic Nostoc cooperate differently: expanding the spectrum of plant-cyanobacteria symbiosis. ISME J 11(12):2821–2833. https://doi.org/10.1038/ismej.2017.134
Chukhontseva KN, Berdyshev IM, Safina DR, Karaseva MA, Bozin TN, Salnikov VV et al (2021) The protealysin operon encodes emfourin, a prototype of a novel family of protein metalloprotease inhibitors. Int J Biol Macromol 169:583–96.https://doi.org/10.1016/j.ijbiomac.2020.12.170
Tsaplina O, Khaitlina S, Chukhontseva K, Karaseva M, Demidyuk I, Bakhlanova I et al (2022) Protealysin targets the bacterial housekeeping proteins FtsZ and RecA. Int J Mol Sci 23(18). https://doi.org/10.3390/ijms231810787
Clarke DJ (2020) Photorhabdus: a tale of contrasting interactions. Microbiology 166(4):335–348. https://doi.org/10.1099/mic.0.000907
Lanois A, Pages S, Bourot S, Canoy AS, Givaudan A, Gaudriault S (2011) Transcriptional analysis of a Photorhabdus sp. variant reveals transcriptional control of phenotypic variation and multifactorial pathogenicity in insects. Appl Environ Microbiol 77(3):1009–1020. https://doi.org/10.1128/AEM.01696-10
Marokhazi J, Lengyel K, Pekar S, Felfoldi G, Patthy A, Graf L et al (2004) Comparison of proteolytic activities produced by entomopathogenic Photorhabdus bacteria: strain- and phase-dependent heterogeneity in composition and activity of four enzymes. Appl Environ Microbiol 70(12):7311–7320. https://doi.org/10.1128/AEM.70.12.7311-7320.2004
Berdyshev IM, Svetlova AO, Chukhontseva KN, Karaseva MA, Varizhuk AM, Filatov VV et al (2023) Production and characterization of Photorin, a novel proteinaceous protease inhibitor from the entomopathogenic Bacteria Photorhabdus Laumondii. Biochemistry 88(9):1356–1367. https://doi.org/10.1134/S0006297923090158
Demidyuk IV, Kalashnikov AE, Gromova TY, Gasanov EV, Safina DR, Zabolotskaya MV et al (2006) Cloning, sequencing, expression, and characterization of protealysin, a novel neutral proteinase from Serratia proteamaculans representing a new group of thermolysin-like proteases with short N-terminal region of precursor. Protein Exp Purif 47(2):551–561. https://doi.org/10.1016/j.pep.2005.12.005
Gasparov VS, Degtiar VG (1994) Protein determination by binding with the dye Coomassie brilliant blue G-250. Biochemistry 59(6):763–777
Laemmli UK (1970) Cleavage of structural proteins during the assembly of the head of bacteriophage T4. Nature 227(5259):680–685. https://doi.org/10.1038/227680a0
Sambrook J, Russell DW, Sambrook J (2006) The condensed protocols from molecular cloning: a laboratory manual. Cold Spring Harbor Laboratory Press, Cold Spring Harbor, N.Y.
Karaseva MA, Chukhontseva KN, Lemeskina IS, Pridatchenko ML, Kostrov SV, Demidyuk IV (2019) An internally quenched fluorescent peptide substrate for Protealysin. Sci Rep 9(1):14352. https://doi.org/10.1038/s41598-019-50764-2
Berdyshev IM, Karaseva MA, Demidyuk AIV (2022) Assay for protealysin-like protease inhibitor activity. Bio-protocol 12(19). https://doi.org/10.21769/BioProtoc.4528
Copeland RA (2023) Enzymes: a practical introduction to structure, mechanism, and Data Analysis. Wiley
Agarwal R, Zakharov S, Hasan SS, Ryan CM, Whitelegge JP, Cramer WA (2014) Structure-function of cyanobacterial outer-membrane protein, Slr1270: homolog of Escherichia coli drug export/colicin import protein, TolC. FEBS Lett 588(21):3793–3801. https://doi.org/10.1016/j.febslet.2014.08.028
Shevchenko A, Tomas H, Havlis J, Olsen JV, Mann M (2006) In-gel digestion for mass spectrometric characterization of proteins and proteomes. Nat Protoc 1(6):2856–2860. https://doi.org/10.1038/nprot.2006.468
Grunina TM, Demidenko AV, Lyaschuk AM, Poponova MS, Galushkina ZM, Soboleva LA et al (2017) Recombinant human erythropoietin with additional processable protein domains: purification of protein synthesized in Escherichia coli Heterologous expression system. Biochemistry 82(11):1285–1294. https://doi.org/10.1134/S0006297917110062
Martens L, Vandekerckhove J, Gevaert K (2005) DBToolkit: processing protein databases for peptide-centric proteomics. Bioinformatics 21(17):3584–3585. https://doi.org/10.1093/bioinformatics/bti588
Pobeguts OV, Ladygina VG, Evsyutina DV, Eremeev AV, Zubov AI, Matyushkina DS et al (2020) Propionate induces virulent properties of Crohn’s disease-associated Escherichia coli. Front Microbiol 11:1460. https://doi.org/10.3389/fmicb.2020.01460
Adekoya OA, Sylte I (2009) The thermolysin family (M4) of enzymes: therapeutic and biotechnological potential. Chem Biol Drug Des 73(1):7–16. https://doi.org/10.1111/j.1747-0285.2008.00757.x
Roche RS, Voordouw G (1978) The structural and functional roles of metal ions in thermolysin. CRC Crit Reviews Biochem 5(1):1–23. https://doi.org/10.3109/10409237809177138
Ji JL, Han SJ, Zhang RJ, Yu JB, Li YB, Yu XP et al (2022) Inter-alpha-trypsin inhibitor heavy chain 4 plays an important role in the development and reproduction of Nilaparvata lugens. Insects 13(3). https://doi.org/10.3390/insects13030303
Jiang H, Vilcinskas A, Kanost MR (2010) Immunity in lepidopteran insects. Adv Exp Med Biol 708:181–204. https://doi.org/10.1007/978-1-4419-8059-5_10
Wen D, Luo H, Li T, Wu C, Zhang J, Wang X et al (2017) Cloning and characterization of an insect apolipoprotein (apolipophorin-II/I) involved in the host immune response of Antheraea Pernyi. Dev Comp Immunol 77:221–228. https://doi.org/10.1016/j.dci.2017.08.010
Kamareddine L, Nakhleh J, Osta MA (2016) Functional interaction between apolipophorins and complement regulate the mosquito immune response to systemic infections. J Innate Immun 8(3):314–326. https://doi.org/10.1159/000443883
Mayans O, Benian GM, Simkovic F, Rigden DJ (2013) Mechanistic and functional diversity in the mechanosensory kinases of the titin-like family. Biochem Soc Trans 41(4):1066–1071. https://doi.org/10.1042/BST20130085
Riddell CE, Lobaton Garces JD, Adams S, Barribeau SM, Twell D, Mallon EB (2014) Differential gene expression and alternative splicing in insect immune specificity. BMC Genomics 15(1):1031. https://doi.org/10.1186/1471-2164-15-1031
Galetto L, Abba S, Rossi M, Vallino M, Pesando M, Arricau-Bouvery N et al (2018) Two Phytoplasmas Elicit different responses in the insect vector Euscelidius Variegatus Kirschbaum. Infect Immun 86(5). https://doi.org/10.1128/IAI.00042-18
Zhang YL, Xue RY, Cao GL, Zhu YX, Pan ZH, Gong CL (2013) Shotgun proteomic analysis of wing discs from the domesticated silkworm (Bombyx mori) during metamorphosis. Amino Acids 45(5):1231–1241. https://doi.org/10.1007/s00726-013-1588-8
Zurovcova M, Benes V, Zurovec M, Kucerova L (2019) Expansion of imaginal disc growth factor gene family in Diptera reflects the evolution of Novel functions. Insects 10(10). https://doi.org/10.3390/insects10100365
Kucerova L, Broz V, Arefin B, Maaroufi HO, Hurychova J, Strnad H et al (2016) The Drosophila chitinase-like protein idgf3 is involved in protection against nematodes and in wound healing. J Innate Immun 8(2):199–210. https://doi.org/10.1159/000442351
Brown MD, Sacks DB (2009) Protein scaffolds in MAP kinase signalling. Cell Signal 21(4):462–469. https://doi.org/10.1016/j.cellsig.2008.11.013
Di Lelio I, Varricchio P, Di Prisco G, Marinelli A, Lasco V, Caccia S et al (2014) Functional analysis of an immune gene of Spodoptera littoralis by RNAi. J Insect Physiol 64:90–. https://doi.org/10.1016/j.jinsphys.2014.03.008
Pascale M, Laurino S, Vogel H, Grimaldi A, Monne M, Riviello L et al (2014) The Lepidopteran endoribonuclease-U domain protein P102 displays dramatically reduced enzymatic activity and forms functional amyloids. Dev Comp Immunol 47(1):129–139. https://doi.org/10.1016/j.dci.2014.07.009
Falabella P, Riviello L, Pascale M, Lelio ID, Tettamanti G, Grimaldi A et al (2012) Functional amyloids in insect immune response. Insect Biochem Mol Biol 42(3):203–211. https://doi.org/10.1016/j.ibmb.2011.11.011
Lim SG, Suk K, Lee WH (2020) LETMD1 regulates phagocytosis and inflammatory responses to Lipopolysaccharide via reactive oxygen species generation and NF-kappaB activation in macrophages. J Immunol 204(5):1299–1309. https://doi.org/10.4049/jimmunol.1900551
Kong L, Lu A, Guan J, Yang B, Li M, Hillyer JF et al (2015) Thermolysin damages animal life through degradation of plasma proteins enhanced by rapid cleavage of serpins and activation of proteases. Arch Insect Biochem Physiol 88(1):64–84. https://doi.org/10.1002/arch.21178
Andrejko M, Mizerska-Dudka M (2012) Effect of Pseudomonas aeruginosa elastase B on level and activity of immune proteins/peptides of Galleria mellonella hemolymph. J Insect Sci 12:88. https://doi.org/10.1673/031.012.8801
Mollah MMI, Kim Y (2020) Virulent secondary metabolites of entomopathogenic bacteria genera, Xenorhabdus and Photorhabdus, inhibit phospholipase A(2) to suppress host insect immunity. BMC microbiology. 20(1):359. https://doi.org/10.1186/s12866-020-02042-9
Antonello AM, Sartori T, Silva MB, Prophiro JS, Pinge-Filho P, Heermann R et al (2019) Anti-trypanosoma activity of bioactive metabolites from Photorhabdus luminescens and Xenorhabdus nematophila. Exp Parasitol 204:107724. https://doi.org/10.1016/j.exppara.2019.107724
Darsouei R, Karimi J, Dunphy GB (2019) Functional characterization of outer membrane proteins (OMPs) in Xenorhabdus nematophila and Photorhabdus luminescens through Insect Immune Defense reactions. Insects 10(10). https://doi.org/10.3390/insects10100352
Sanda NB, Hou Y (2023) The symbiotic Bacteria-Xenorhabdus nematophila All and Photorhabdus luminescens H06 strongly affected the phenoloxidase activation of Nipa Palm Hispid, Octodonta nipae (Coleoptera: Chrysomelidae) Larvae. Pathogens 12(4). https://doi.org/10.3390/pathogens12040506
Eleftherianos I, ffrench-Constant RH, Clarke DJ, Dowling AJ, Reynolds SE (2010) Dissecting the immune response to the entomopathogen Photorhabdus. Trends Microbiol 18(12):552–560. https://doi.org/10.1016/j.tim.2010.09.006
Kim Y, Ji D, Cho S, Park Y (2005) Two groups of entomopathogenic bacteria, Photorhabdus and Xenorhabdus, share an inhibitory action against phospholipase A2 to induce host immunodepression. J Invertebr Pathol 89(3):258–264. https://doi.org/10.1016/j.jip.2005.05.001
Acknowledgements
Mass spectrometric analysis of products of Abz-RSVIK(Dnp) hydrolysis by PrtSt were performed using the equipment of the Advanced Mass Spectrometry Core Facility at the Skolkovo Institute of Science and Technology (Skoltech, Moscow, Russia).
Funding
This study was financially supported by the Russian Science Foundation, grant no. 24-24-00122.
Author information
Authors and Affiliations
Contributions
Conceptualization: Ilya V. Demidyuk; Data curation: Maria A. Karaseva; Formal analysis: Anastasia O. Svetlova, Maria A. Karaseva, Igor M. Berdyshev, Ksenia N. Chukhontseva, Ilya V. Demidyuk; Funding acquisition: Sergey V. Kostrov, Ilya V. Demidyuk; Investigation: Anastasia O. Svetlova, Maria (A) Karaseva, Igor M. Berdyshev, Ksenia N. Chukhontseva, Olga V. Pobeguts, Maria А. Galyamina, Igor P. Smirnov, Nikita (B) Polyakov, Maria G. Zavialova; Methodology: Anastasia O. Svetlova, Maria (A) Karaseva, Igor M. Berdyshev, Ksenia N. Chukhontseva, Olga V. Pobeguts, Maria А. Galyamina, Nikita (B) Polyakov, Maria G. Zavialova, Ilya V. Demidyuk; Project administration: Ilya V. Demidyuk; Supervision: Anastasia O. Svetlova, Sergey V. Kostrov, Ilya V. Demidyuk; Validation: Anastasia O. Svetlova, Maria A. Karaseva; Visualization: Anastasia O. Svetlova, Maria A. Karaseva, Igor M. Berdyshev, Olga V. Pobeguts, Maria А. Galyamina, Igor P. Smirnov, Maria G. Zavialova, Ilya V. Demidyuk; Writing – original draft: Anastasia O. Svetlova, Maria A. Karaseva, Igor M. Berdyshev, Olga V. Pobeguts, Maria А. Galyamina, Igor P. Smirnov, Maria G. Zavialova, Ilya V. Demidyuk; Writing – review & editing: Anastasia O. Svetlova, Maria A. Karaseva, Igor M. Berdyshev, Ksenia N. Chukhontseva, Olga V. Pobeguts, Maria А. Galyamina, Maria G. Zavialova, Sergey V. Kostrov, Ilya V. Demidyuk.
Corresponding author
Ethics declarations
Ethical approval
No approval of research ethics committees was required to accomplish the goals of this study because experimental work was conducted with an unregulated insect species.
Competing interests
The authors declare no competing interests.
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
Springer Nature or its licensor (e.g. a society or other partner) holds exclusive rights to this article under a publishing agreement with the author(s) or other rightsholder(s); author self-archiving of the accepted manuscript version of this article is solely governed by the terms of such publishing agreement and applicable law.
About this article

Cite this article
Svetlova, A.O., Karaseva, M.A., Berdyshev, I.M. et al. Protease S of entomopathogenic bacterium Photorhabdus laumondii: expression, purification and effect on greater wax moth Galleria mellonella. Mol Biol Rep 51, 713 (2024). https://doi.org/10.1007/s11033-024-09654-8
Received:
Accepted:
Published:
DOI: https://doi.org/10.1007/s11033-024-09654-8