
Abstract
Background
Chromosomal microarray analysis is an essential tool for copy number variants detection in patients with unexplained developmental delay/intellectual disability, autism spectrum disorders, and multiple congenital anomalies. The study aims to determine the clinical significance of chromosomal microarray analysis in this patient group. Another crucial aspect is the evaluation of copy number variants detected in terms of the diagnosis of patients.
Methods and results
A Chromosomal microarray analysis was was conducted on a total of 1227 patients and phenotype-associated etiological diagnosis was established in 135 patients. Phenotype-associated copy number variants were detected in 11% of patients. Among these, 77 patients 77 (57%, 77/135) were diagnosed with well-recognized genetic syndromes and phenotype-associated copy number variants were found in 58 patients (42.9%, 58/135). The study was designed to collect data of patients in Kocaeli Derince Training and Research Hospital retrospectively. In our study, we examined 135 cases with clinically significant copy number variability among all patients.
Conclusions
In this study, chromosomal microarray analysis revealed pathogenic de novo copy number variants with new clinical features. Chromosomal microarray analysis in the Turkish population has been reported in the largest patient cohort to date.
Similar content being viewed by others


Explore related subjects
Discover the latest articles, news and stories from top researchers in related subjects.Avoid common mistakes on your manuscript.
Introduction
Chromosomal microarray analysis (CMA) has been the first-tier test for patients presenting with unexplained developmental delay/intellectual disability (DD/ID), autism spectrum disorders (ASD) and multiple congenital anomalies (MCA), since 2010. [1, 6,7,8,9,10,11].
DD/ID are clinically heterogeneous neurodevelopmental disorders seen in 1–3% of children [9]. Thus, genetic testing plays an important role in evaluating patients with DD/ID, ASD, and MCA, however, etiology is still not defined in all patients. Microarray analysis has increased the detection rate of chromosomal imbalances in the human genome, enabling the diagnosis of syndromic phenotypes with previously unknown etiologies. CMA detects microdeletion and microduplication syndromes in this group with a high diagnostic yield [11, 12]. In addition, with the use of high-resolution microarray analyses, it is possible to identify new regions whose copy number changes have not been associated with any phenotype before.
ASD is a complex and genetically heterogeneous disorder, characterized by social communication deficits and interaction as well as restricted, stereotypic behaviors. A recent study estimated that the prevalence is about 1–2% [2, 3]. Previous studies have shown that genetic factors contribute to the diagnosis of ASD [4, 5].
On the other hand, some syndromes been rarely reported despite their potential for diagnosis, such as the Bosch-Boonstra-Schaaf Optic Atrophy Syndrome (BBSOAS; MIM #615,722), 18p monosomy syndrome, 9p duplication syndrome (ORPHA:236), 6q terminal deletion syndrome (ORPHA:75,857), 2q33.1 microdeletion syndrome, and 6q27 terminal deletion syndrome.
The CMA studies plays a significant role in investigating the genetic etiology and identifying new syndromes in patients diagnosed with DD/ID, ASD, and MCA [6,7,8]. The study aimed to identify CNVs and clinical phenotypes by determining the clinical efficiency of CMAs in evaluating DD/ID, ASD, and MCA in Turkish patients.
Materials and methods
In this study, 1227 patients with DD, ASD, and MCA who were consulted by the Department of Medical Genetics at the Health Sciences University Kocaeli Derince Training and Research Hospital were included.
Ethical committee approval for the study was obtained from the Kocaeli Derince Training and Research Hospital Ethical Board (2021–53).
In this study, we presented the CNVs we detected in 135 patients with DD, ASD, and MCA, with or without accompanying dysmorphic features, growth disorder, or epilepsy, and correlated them with the clinical findings. After written informed consents were obtained from the parents of the patients, DNA was isolated from peripheral blood. Microarray analysis was performed using CytoScan Optima, Affimetrix® chips according to the manufacturer’s protocol and the data was analyzed by Chromosome Analysis Suite (ChAS) 3.1 Thermo Fisher Scientific®. Copy number variations (CNVs) with a gene size greater than 100 kb and represented by a minimum of 25 probes were considered in variant calling.
Database of Genomic Variants (DGV, http://projects.tcag.ca/variation), Database of Chromosomal Imbalance and Phenotype in Humans Ensembl Resources (DECIPHER, https://decipher.sanger.ac.uk), PubMed (https://www.ncbi.nlm.nih.gov/pubmed), Online Mendelian Inheritance of Man (OMIM; https://www.ncbi. nlm. nih.gov/omim), ClinVar (https:// www. ncbi. nlm. nih. gov/ clinv ar) and in-house database of Haseki Genetic Diagnosis Center were used to identify the CNVs with three classes: variants of uncertain clinical significance (VUS), likely pathogenic, and pathogenic.
In this study, we examined cases with clinically relevant CNVs, categorized as “pathogenic”, “likely pathogenic”, or “VUS” variants by assessing the diagnostic efficiency of microarray analysis, a frequently used method, in diagnosing patients with DD/ID, ASD, and MCA, according to the American College of Medical Genetics and Genomics guidelines (ACMG) and ClinGen [13].
A retrospective study was conducted on patients with DD/ID, ASD, and MCA within the period between January 1, 2017 and March 30, 2021. A total of 1227 patients with DD/ID, ASD, and MCA were examined using the CMA test. Male individuals underwent conventional karyotyping and Fragile X analysis before introducing microarray analysis. In this study, we analyzed the data from routine microarray analysis performed on patients admitted to the outpatient clinic of the Department of Medical Genetics who were diagnosed with DD/ID, ASD, and MCA.
Results
The results of 1227 patients, 701 male and 526 female, who underwent CMA in the genetics outpatient clinic were evaluated and phenotype-associated CNVs were found in 58 (42.9%, 58/135) patients. Furthermore, 77 out of 135 patients (57%) were diagnosed with well-recognized genetic syndromes via the OMIM database (Table 1). Demographic information and clinical features of the patients are shown in Tables 1, 2, and 3.
Phenotype-associated CNV was detected in 58 patients, where 25 patients were classified as pathogenic CNV (Table 2) and 33 patients had CNV of uncertain clinical significance (Table 3). The diagnosis of well-recognized genetic syndromes was achieved in 77 patients (Table 1). The patients are listed in Tables 1, 2, and 3.
The number and percentage of patients diagnosed with a well-known genetic syndrome are indicated in Table 4. Among the well-known genetic syndrome diagnoses, 22q11.2 microdeletion syndrome was the most common, as it was observed in 4 patients. On the other hand, 3 patients were diagnosed with 16p13.11 microdeletion syndrome, 3 patients were diagnosed with 16p13.11 microduplication syndrome, 3 patients were diagnosed with Williams-Beuren syndrome, and 3 patients were diagnosed with Wolf-Hirschhorn syndrome (Table 4). Table 5 shows the number and percentage of patients diagnosed with multiple CMA findings.
On the other hand, our study identified 16 ASD patients with phenotype-related CNVs including 8 patients with well-recognized genetic CNVs and 8 patients with uncertain CNVs in ASD. The well-known genetic syndromes identified in 8 patients with ASD were; Joubert syndrome (patient 1), Xq28 microduplication syndrome (patient 4), Klinefelter syndrome (patient 17), Coffın-Siris Syndrome 6 (patient 25), 15q13.3 microdeletion syndrome (patient 28), 16p13.11 microduplication syndrome (patient 48), 7q11.23 duplication syndrome (patient 62), and Mosaic trisomy 8 (Warkany syndrome 2) (patient 64) (Table 1).
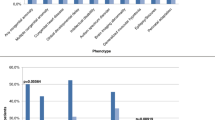
Among the patients diagnosed according to the major clinical findings, 28 had DD/ID, 16 had DD, 9 had epilepsy, 8 had hypotonia, 4 had microcephaly, 3 had corpus callosum hypoplasia, 5 had autism, short stature 4, 12 in cardiovascular anomalies, and 11% of patients with other minor findings (Fig. 1).

Diagnosis rates according to major clinical findings
In this cohort, 111 patients were positive through CMAs but their karyotype analyses results were negative. These findings highlight the importance of CMA analysis in cytogenetic analysis.
Discussion
In 2010, according to the American College of Medical Genetics (ACMG) practice guidelines, CMA testing for CNVs was recommended as a first-line test in the clinical genetic evaluation of unexplained DD/ ID [6]. Since then, CMA has been the first-tier test for patients presenting with DD/ID, ASD, and MCAHowever, another consensus statement was published in 2019 recommended whole exome sequencing as a first-tier test for patients with neurodevelopmental disorders [1].
In the present study, we conducted CMA analysis in 1277 patients with DD/ID, ASD, and MCA. To our knowledge, this study has the highest number of Turkish patients in reports of CMA ever published. The diagnostic rate of chromosomal abnormalities was 11 which is consistent with the results of previous studies 5–20% [7, 14]. Among the 135 patients diagnosed in this study, many patients have additional features that may contribute to the literature.
In previous studies conducted in the Turkish population, CNV rates ranged between 8.5 and 18.55% [15,16,17,18]. Ceylan et al. (2018) reported that of a group of 124 Turkish patients with intellectual disability and global developmental delay, 18.55% had pathogenic and likely pathogenic CNVs detected [15]. A study conducted by Ozyilmaz et al. (2016) reported that 13.6% of a group of 971 Turkish patients with developmental disabilities and congenital anomalies had pathogenic CNVs detected [16]. Özaslan et al. (2021) reported that 8.5% had pathogenic CNVs detected in a group of 47 Turkish patients with ASD [17]. In their study, Türkyılmaz et al.(2021) reported that 17.1% had pathogenic and likely pathogenic CNVs detected in a group of 139 Turkish patients with DD/ID [18]. Pathogenic and likely pathogenic CNVs have been evaluated in several studies. Our study results revealed that 11% of the study cohort showed all clinically relevant CNVs. The differences in CNV rates between the conducted studies, including ours, may be attributed to variations in sample sizes or the effect of VUS variants.
The major clinical findings showed that DD/ID has the highest diagnostic rate (77), followed by DD (16), epilepsy (9), hypotonia (8%), and microcephaly (%4). The diagnostic rates of other findings are shown in Fig. 1. Additional minor findings with very poor diagnostic values, according to radilogical findings, are endocrine disorders, hydrocephaly, behavioral problems, aggressiveness, and minor congenital anomalies such as cleft palate, cryptorchidism, and vesicoureteral reflux. Based on these findings, MCA should be the first step while selecting the test for diagnosing patients with DD/ID.
Patient 1B with phenotype-associated pathogenic CNV, shown in Table 2, presented with intellectual disability, learning disability, mitral valve prolapse (MVP), and bicuspite aortic valve. Microarray analysis revealed a 3,965 kbp deletion at 15q21.3q22.2 including the TCF12 gene. Additionally, craniosynostosis-3 (CRS3)(OMIM #615,314) is caused by heterozygous mutation in the TCF12 (OMIM #600,480) on chromosome 15q21. Pathogenic variants in TCF12 were reported in patients with significant developmental delay or learning disability. Moreover, few reports documented chromosomal deletions including TCF12 in patients with craniosynostosis and intellectual disability [19, 20]. TCF12 heterozygous loss-of-function mutations have been associated with craniosynocytosis. Recently, intragenic deletions and duplications have been reported in 5 cases with TCF-related craniosynostosis and intellectual disability [21, 22]. To date, whole TCF12 deletion has been reported in three patients. Firstly, coronal craniosynostosis and intellectual disability were reported in a patient with a deletion in the 15q21.3q22.2 region, including the TCF12, but a small duplication in the 2q21 region was accompanied as a result of maternal complex chromosomal rearrangement [20]. Second TCF12 deletion was reported in a patient with developmental delay, dysmorphic features, seizures, and atrial septal defect [23]. Two of the four patients with TCF12 deletion had atrial septal defects. Interestingly, MVP and bicuspid aortic valve were reported for the first time in our case. Accordingly, further investigations are required to determine whether MVP and bicuspid aortic valve are related to the TCF12 gene.
Patient 2B with phenotype-associated pathogenic CNV, shown in Table 2, a 12-year-old male, presented with intellectual disability, developmental delay, speech delay, learning disability, gait ataxia, and cerebellar cyst. Microarray analysis revealed a de novo genomic rearrangement including a 5,575 kbp deletion at 12p13.33p13.31 and a 3,057 kbp duplication at 17p13.3. Chromosome 12p deletions included CACNA1C, ERC1, FBXL14, WNT5B, ADIPOR2, CACNA2D4, LRTM2, and DCP1B that have been linked to the developmental verbal dyspraxia (DVD) and childhood apraxia of speech (CAS). Pathogenic variants of the CACNA1C gene are associated with autosomal dominant inheritance, “Neurodevelopmental disorder with hypotonia, language delay, and skeletal defects with or without seizures” (OMIM #620,029). 12p13.33 deletion has been associated to mild intellectual disability, speech delay, and motor skills impairment [24, 25]. Chromosome 17p13.3 duplication syndrome (OMIM #613,215) is characterized by developmental delay, autism, abnormal growth, facial dysmorphism, and structural brain abnormalities [26, 27]. This duplication on the 17p13.3 region, involves PAFAH1B1, YWHAE, and CRK genes. According to Bruno et al., class II duplication encompasses PAFAH1B1 and may also include CRK and YWHAE. Bruno DL et al. suggested class I duplication includes the YWHAE gene, but not specifically PAFAH1B1. However, class II duplication always contained PAFAH1B1, CRK, and YWHAE genes. Class II microduplications are characterized by moderate to mild developmental and psychomotor delay and hypotonia. The patient had similar features with class II microduplication. Patient 5 who had a combined 17p duplication and 12p deletion had a normal constitutional karyotype, and his parents’ karyotypes were also normal. Besides, his parents’ CMAs were normal. However, in the FISH analysis of the parents, it was determined that this complex change occurred as a result of abnormal segregation of the balanced translocation in the father.
As seen in this case, we were able to detect parents with balanced chromosome carriers by applying both chromosome analysis and the FISH method. Accordingly, chromosome analysis and FISH should be performed to determine the balanced chromosome carriers.
Patient 3B with phenotype-associated pathogenic CNV, presented with developmental delay, fallot tetralogy, arachnodactyly, and flexion contractures at the left wrist (Table 2). G-banded karyotype analysis revealed 46,XX,der(15)t(7;15)(p10;q10)pat. Microarray analysis revealed a 57,963 kbp duplication at 7p22.3p11.1. Recently, several cases of 7p duplications have been reported. However, various cardiac anomalies have been reported in 6 of 18 patients with chromosome 7p duplication, this is the first patient that the tetralogy of Fallot reported [28]. Deletions of chromosome 7p may be accompanied by tetralogy of Fallot in 4 patients [29].
Patient 29A with a well-known genetic syndrome, shown in Table 1, a 2-year-old girl who presented with hypotonia. She was unable to sit, walk or talk. Physical examination revealed intellectual disability, developmental delay, microcephaly, scaphocephaly, sparse hair, clinodactyly, and reduced hand subcutaneous fat. Both brain magnetic resonance imaging and echocardiography were within normal limits. A conventional cytogenetic study revealed 46,XX,add(15)(p11.2). Parental chromosomes were normal. Microarray analysis revealed a 38,584 kbp de novo duplication at 9p24.3p13.1. SMARCA2 gene is associated with Nicolaides–Baraitser syndrome (NCBRS; OMIM#601,358) and Coffin-Siris syndrome (CSS; OMIM#135,900) but SMARCA2 mutations causing NCBRS are likely to act through a dominant-negative effect [30].
Patient 43A with a well-known genetic syndrome shown in Table 1, a 19-year-old male patient who presented with intellectual disability and developmental delay. He had learning disability, epilepsy, congenital cataract, scoliosis, and growth hormone deficiency. Additionally, he had a broad nasal root and arachnodactyly. Microarray analysis revealed a 4,350 kbp deletion at 16q23.1q23.3. According to Javadiyan et al. (2017) and Alkhunaizi et al. (2019), MAF gene heterozygote variations are associated with Aymé-Gripp syndrome [31, 32]. It is the first time to report a case with heterozygous deletions of chromosome 16q23 encompassing the MAF gene. The patient’s 48-year-old father and aunt had undergone cataract surgery. The couple is nonconsanguineous, also the mother is healthy.
Also, the karyotype and microarray results of the parents of cases 9B, 10B, 11B, and 14B from Table 2 as well as cases 4C and 29C from Table 3 were found to be normal. These results support the pathogenicity of the variations detected in patients. However, as one of the limitations of our study, we could not perform karyotype and microarray analysis on the mothers and fathers of patients in Tables 2 and 3.
Conclusion
As a result, CMA may be beneficial in identifying microdeletions and microduplications, and has played an important role in the diagnosis and genetic counseling of patients. Here in, we present an analysis of patients with deletions and duplications, as well as novel and previously unreported findings. The study’s findings highlight the importance of CMA analysis in cytogenic examinations since patients exhibited positive results on the CMA test but negative results with karyotype analyses. This article presents multiple cases with new findings. In order to contribute to the literature, some patients are explained in detail above.
Data availability
Available upon a reasonable request.
References
Stankiewicz P, Beaudet AL (2007) Use of array CGH in the evaluation of dysmorphology, malformations, developmental delay, and idiopathic mental retardation. Curr Opin Genet Dev 17(3):182–192
Anagnostou E, Zwaigenbaum L, Szatmari P et al (2014) Autism spectrum disorder: advances in evidence-based practice. CMAJ 186:509–519. https://doi.org/10.1503/cmaj.121756
Khan NZ, Gallo LA, Arghir A et al (2012) Autism and the grand challenges in global mental health. Autism Res 5:56–59. https://doi.org/10.1002/aur.1239
Shen Y, Dies KA, Holm IA et al (2010) Clinical genetic testing for patients with autism spectrum disorders. Pediatrics 125(4):727–735. https://doi.org/10.1542/peds.2009-1684
Waye MMY, Cheng HY (2018) Genetics and epigenetics of autism: a review. Psychiatry Clin Neurosci 72:228–244. https://doi.org/10.1111/pcn.12606
Manning M, Hudgins L (2020) Array-based technology and recommendations for utilization in medical genetics practice for detection of chromosomal abnormalities. Genet Med 22:2126. https://doi.org/10.1038/s41436-020-0847-9
Miller DT, Adam MP, Aradhya S et al (2010) Consensus statement: chromosomal microarray is a first-tier clinical diagnostic test for individuals with developmental disabilities or congenital anomalies. Am J Hum Genet 86:749–764. https://doi.org/10.1016/j.ajhg.2010.04.006
Riggs ER, Wain KE, Riethmaier D et al (2014) Chromosomal microarray impacts clinical management. Clin Genet 85:147–153. https://doi.org/10.1111/cge.12107
Maulik PK, Mascarenhas MN, Mathers CD, Dua T, Saxena S (2011) Prevalence of intellectual disability: a meta-analysis of population-based studies. Res Devel Disabil 32:419–436. https://doi.org/10.1016/j.ridd.2010.12.018
Bartnik M, Nowakowska B, Derwińska K, Wiśniowiecka-Kowalnik B, Kędzior M, Bernaciak J et al (2014) Application of array comparative genomic hybridization in 256 patients with developmental delay or intellectual disability. J Appl Genet 55:125–144. https://doi.org/10.1007/s13353-013-0181-x
Battaglia A, Doccini V, Bernardini L et al (2013) Confirmation of chromosomal microarray as a first-tier clinical diagnostic test for individuals with developmental delay, intellectual disability, autism spectrum disorders and dysmorphic features. Eur J Paediatr Neurol 17:589–599. https://doi.org/10.1016/j.ejpn.2013.04.010
Zarrei M, Burton CL, Engchuan W et al (2019) A large data resource of genomic copy number variation across neurodevelopmental disorders. NPJ Genom Med 7:26. https://doi.org/10.1038/s41525-019-0098-3
Sagoo GS, Butterworth AS, Sanderson S, Shaw-Smith C, Higgins JP, Burton H (2009) Array CGH in patients with learning disability (mental retardation) and congenital anomalies: updated systematic review and meta-analysis of 19 studies and 13,926 subjects. Genet Med 11:139–146. https://doi.org/10.1097/GIM.0b013e318194ee8f
Riggs ER, Andersen EF, Cherry AM et al (2020) Technical standards for the interpretation and reporting of constitutional copy number variants: a joint consensus recommendation of the american college of medical genetics and genomics (ACMG) and the clinical genome resource (ClinGen). Genet Med 22:245–257. https://doi.org/10.1038/s41436-019-0686-8
Ceylan AC, Citli S, Erdem HB et al (2018) Importance and usage of chromosomal microarray analysis in diagnosing intellectual disability, global developmental delay, and autism; and discovering new loci for these disorders. Mol Cytogenet 11:54. https://doi.org/10.1186/s13039-018-0402-4
Ozyilmaz B, Kirbiyik O, Koc A et al (2016) Experiences in microarray-based evaluation of developmental disabilities and congenital anomalies. Clin Genet 92:372–379. https://doi.org/10.1111/cge.12978
Özaslan A, Kayhan G, İşeri E et al (2021) Identification of copy number variants in children and adolescents with autism spectrum disorder: a study from turkey. Mol Biol Rep 48:7371–7378. https://doi.org/10.1007/s11033-021-06745-8
Türkyılmaz A, Geckinli BB, Tekin E et al (2022) Array-based comparatıve genomıc hybrıdızatıon analysıs ın chıldren wıth developmental delay/ıntellectual dısabılıty. Balkan Journal of Medical Genetics 24:15–24
Hiraki Y, Moriuchi M, Okamoto N, Ishikawa N, Sugimoto Y, Equchi K et al (2008) Craniosynostosis in a patient with a de novo 15q15-q22 deletion. Am J Med Genet 146:1462–1465. https://doi.org/10.1002/ajmg.a.32339
Tanno PL, Poreau B, Devillard F et al (2014) Maternal complex chromosomal rearrangement leads to TCF12 microdeletion in a patient presenting with coronal craniosynostosis and intellectual disability. Am J Med Genet 164A:1530–1536. https://doi.org/10.1002/ajmg.a.36467
Piard J, Rozé V, Czorny A et al (2015) TCF12 microdeletion in a 72-year-old woman with intellectual disability. Am J Med Genet 167A:1897–1901. https://doi.org/10.1002/ajmg.a.37083
Goos JAC, Fenwick AL, Swagemakers SMA et al (2016) Identification of intragenic exon deletions and duplication of TCF12 by whole genome or targeted sequencing as a cause of TCF12-related craniosynostosis. Hum Mutat 37:732–736. https://doi.org/10.1002/humu.23010
Yoon JG, Hahn HM, Choi S et al (2020) Molecular diagnosis of craniosynostosis using targeted next-generation sequencing. Neurosurgery 87:294–302. https://doi.org/10.1093/neuros/nyz470
Fanizza I, Bertuzzo S, Beri S et al (2014) Genotype phenotype relationship in a child with 2.3 Mb de novo interstitial 12p13.33-p13.32 deletion. Eur J Med Genet 57:334–338. https://doi.org/10.1016/j.ejmg.2014.04.009
Thevenon J, Callier P, Andrieux J et al (2013) 12p13.33 microdeletion including ELKS/ERC1, a new locus associated with childhood apraxia of speech. Eur J Med Genet 21:82–88. https://doi.org/10.1038/ejhg.2012.116
Chloe SA, Francis D, McGillivray G, Lockhart PL, Leventer RJ (2019) Polymicrogyria associated with 17p13.3p13.2 duplication: case report and review of the literatüre. Eur J Med Genet 63:103774. https://doi.org/10.1016/j.ejmg.2019.103774
Curry CJ, Rosenfeld JA, Grant E et al (2013) The duplication 17p13.3 phenotype: analysis of 21 families delineates developmental, behavioral and brain abnormalities, and rare variant phenotypes. Eur J Med Genet 161:1833–1852. https://doi.org/10.1002/ajmg.a.35996
Chui JV, Weisfeld-Adams JD, Tepperberg J, Mehta L (2011) Clinical and molecular characterization of chromosome 7p22.1 microduplication detected by array CGH. Am J Med Genet 155:2508–2511. https://doi.org/10.1002/ajmg.a.34180
Schmidt B, Cate FU, Weiss M, Koehler U (2012) Cardiac malformation of partial trisomy 7p/monosomy 18p and partial trisomy 18p/monosomy 7p in siblings as a result of reciprocal unbalanced malsegregation–and review of the literatüre. Eur J Pediatr 171:1047–1053. https://doi.org/10.1007/s00431-012-1682-z
Sousa SB, Hennekam RC (2014) Phenotype and genotype in nicolaides-baraitser syndrome. Am J Med Genet 166:302–314. https://doi.org/10.1002/ajmg.c.31409
Javadiyan S, Craig JE, Sharma S et al (2017) Novel missense mutation in the bZIP transcription factor, MAF, associated with congenital cataract, developmental delay, seizures and hearing loss (Aymé-Gripp syndrome). BMC Med Genet 18:52. https://doi.org/10.1186/s12881-017-0414-7
Alkhunaizi E, Koenekoop RK, Saint-Martin C, Russell L (2019) Maternally inherited MAF variant associated with variable expression of aymé-Gripp syndrome. Am J Med Genet 179:2233–2236. https://doi.org/10.1002/ajmg.a.61299
Acknowledgements
Not applicable.
Funding
Not applicable.
Author information
Authors and Affiliations
Contributions
All authors contributed to the study’s conception and design. Material preparation, data collection, and analysis were performed by [Nejmiye Akkus] and [Pelin Ozyavuz Cubuk]. The first draft of the manuscript was written by [Nejmiye Akkus] and all authors commented on previous versions of the manuscript. All authors read and approved the final manuscript.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Ethics approval
This study was performed in line with the principles of the Declaration of Helsinki. Approval was granted by the Kocaeli Derince Training and Research Hospital Ethical Board(08.04.2021–53).
Consent for publication:
Not applicable.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Springer Nature or its licensor (e.g. a society or other partner) holds exclusive rights to this article under a publishing agreement with the author(s) or other rightsholder(s); author self-archiving of the accepted manuscript version of this article is solely governed by the terms of such publishing agreement and applicable law.
About this article

Cite this article
Akkus, N., Cubuk, P.O. Diagnostic yield of the chromosomal microarray analysis in turkish patients with unexplained development delay/ıntellectual disability(ID), autism spectrum disorders and/or multiple congenital anomalies and new clinical findings. Mol Biol Rep 51, 577 (2024). https://doi.org/10.1007/s11033-024-09545-y
Received:
Accepted:
Published:
DOI: https://doi.org/10.1007/s11033-024-09545-y