
Abstract
Background
Mutations within the COL12A1 gene have been linked with the onset of congenital Ullrich muscular dystrophy 2 (UCMD2) and Bethlem myopathy. The severity of the symptoms exhibited is dependent on the mutation’s type and whether it is heterozygous or homozygous.
Methods
We used whole-exome sequencing to identify disease-causing variants in a nine-year-old Iranian patient who had weakness, joint contractures, delayed motor development, and other symptoms. We confirmed the pathogenicity of the identified variant using in silico tools and verified its novelty using various databases. We also performed a co-segregation study and confirmed the presence of the variant in the patient’s parents by Sanger sequencing.
Results
Our analysis identified a novel homozygous missense variant in the affected patient in COL12A1 (c.8828 C > T; p.Pro2943Leu). This is the second reported family with UCMD2 caused by a mutation in COL12A1. Our findings confirm that this mutation results in significantly more severe symptoms than Bethlem myopathy.
Conclusion
Our investigation contributes to the expanding body of evidence that links mutations in COL12A1 with UCMD2. Our findings confirm that the homozygous mutation in COL12A1 caused this condition and suggest that genetic testing for this mutation may be useful for diagnosing patients with this disease.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Collagen XII belongs to the interrupted triple helices (FACIT) family of fibrous-associated collagens and is mainly found in tissues abundant in collagen I, such as ligaments, perichondrium, periosteum, dermis, and skeletal muscle [1]. It consists of three α1 chains that are encoded by the COL12A1 gene. Each chain contains two adjacent helical collagen domains (COL1 and COL2), two short carboxy-terminal non-collagenous domains (NC1 and NC2), and an essential amino-terminal non-collagen domain (NC3) [2]. The NC3 domain is composed of 18 fibronectin type III repeats with four von Willebrand factor A domain insertions. It also has a thrombospondin domain at the N-terminus and folds into three arms extending from a trunk formed by two short triple-helical collagen domains, forming trimers [3]. Mutations in several genes, including COL6A1 [4], COL6A2 [5], COL6A3 [6], and COL12A1 [7], lead to a spectrum of diseases ranging from severe Ullrich congenital muscular dystrophy to Bethlehem myopathy. Figure 1 displays a spectrum of variants in COL12A1, the majority of which are missense and splice site mutations, classified as variants of uncertain significance (VUS) and likely benign variants [8]. Patients with collagen VI-associated myopathy exhibit clinical features overlapping both myopathy and Ehlers-Danlos syndrome (EDS), such as distal joint hypermobility and progressive contractures of large joints, which are not typically observed in EDS [9]. In contrast, patients with typical Ullrich symptoms of collagen VI-related myopathy present with neonatal hypotonia, pronounced joint hypermobility, tender skin on the hands and feet, and prominent heel bones [10]. Initially, progressive myopathy appears mostly atrophic but gradually develops a dystrophic appearance upon histological examination, leading to a progressive loss of muscle strength [11]. In this study, we present the identification of a mutation in the COL12A1 gene in a patient exhibiting a notable overlap phenotype characterized by joint hypermobility syndrome and myopathy. This represents the second reported case of a recessive mutation in COL12A1 causing Ullrich congenital muscular dystrophy 2 (UCMD2) (OMIM: 616,470).

A visual representation of the distribution of COL12A1 variants (derived from the ClinVar database) based on their type and pathogenicity
Materials and methods
Patient
In this study, we conducted an examination of a nine-year-old boy who was born to first cousin consanguineous parents. The boy presented with cerebral palsy, weakness, delayed motor development, and joint contractures. These specific criteria were utilized to select the initial study sample. Prior to the commencement of the study, informed written consent was obtained from all participants, including the patient’s parents, siblings, and other subjects. For genetic testing, a volume of five to ten mL of blood was drawn into tubes containing EDTA from each individual. This research was conducted in compliance with the regulations established by the Ethics Committee of Golestan University of Medical Sciences (Ethics Code: IR.GOUMS.REC.1399.382).
DNA extraction
We extracted DNA samples from 1 µl of mononuclear cells extracted from blood using the Kowsar kit (cat# K1135) following the completion of informed consent form. The concentration and purity of DNA were determined using the Nanodrop device after extraction. Sequencing was performed using DNA extracted from whole blood.
Whole exome sequencing
High-quality genomic DNA samples were subjected to random fragmentation using Covaris, resulting in library fragment sizes primarily ranging from 150 to 200 base pairs (bp). Adapters were subsequently ligated to both ends of the resulting fragments. The adapter-ligated template was then purified using Agencourt AMPure SPRI beads, and a fragment with an approximate insert size of 176 bp was isolated. This extracted DNA was then amplified using ligation-mediated PCR (LM-PCR) and purified before undergoing hybridization to Sure Select biotinylated RNA libraries (BAITS) for enrichment. Following a 24-hour incubation, hybridized fragments were bound to streptavidin beads while non-hybridized fragments were removed through washing. The LM-PCR products obtained from this process were analyzed using an Agilent 2100 Bioanalyzer to assess the level of accumulation. Subsequently, each captured library was loaded onto the HiSeq2000 platform, and high-throughput sequencing was performed to obtaini the normal sequencing depth for each sample. Raw image files were processed using Illumina software 1.7 and base-called with default parameters, producing paired-end reads of 90/100 bp in length. Bioinformatics analyses were conducted using the sequence information (referred to as “raw data”) obtained from the Illumina pipeline. Initially, adapter sequences and low-quality reads were removed to obtain “clean data.“ The Burrows-Wheeler Aligner (BWA) was then employed to align the clean reads with the published GRCh37/UCSC hg19 human genome build. Picard was used to add read groups, flag duplicates, and sort the aligned reads, resulting in final BAM files that were used for variant calling with the GATK haplotype caller. This process distinguished between single nucleotide variants (SNVs) and insertions/deletions (indels).
In silico analysis of variants
To validate the pathogenicity of the identified variant, we utilized multiple tools including SIFT [12], CADD [13], Mutation Taster [14], and Polyphen-2 [15], and PANTHER [16]. Moreover, I-Mutant [17] and MUpro [18] web servers were used to evaluate effect of the identified variant on the protein stability.
Protein–protein interaction analysis using STRING
The interaction of COL12A1 with other proteins within the collagen chain terimerization super pathway, were investigated utilizing the online tool STRING [19]. This database uses a combination of gene fusion, co-expression, function and experimental data to predict protein interactors. The results are combined scores of each interactor ranging from 0 to 1, representing highest and lowest confidence in the interaction. To visualize the obtained interaction data, we used the NetworkX python package (https://github.com/networkx).
Conservation analysis
The conservation analysis of the protein sequence of COL12A1 was performed using Clustal Omega [20], and ConSurf [21] web servers. The amino acid sequences of COL12A1 protein of various species, including the western clawed frog (Xenopus tropicalis), red jungle fowl (Gallus gallus), domestic cow (Bos taurus), rhesus macaque (Macaca mulatta), house mouse (Mus musculus), brown rat (Rattus norvegicus), European rabbit (Oryctolagus cuniculus), horse (Equus caballus), domestic cat (Felis catus), human (Homo sapiens), Sumatran orangutan (Pongo abelii), and olive baboon (Papio Anubis), were retrieved from the Uniprot database [22] and submitted to Clustal Omega for multiple sequence alignment. The aim of this analysis was to identify conserved amino acids within the COL12A1 protein sequence across different species. The ConSurf web server is a tool that analyses the evolutionary pattern of amino acids and nucleic acids by predicting the structural and functional regions. The results are conservation scores that range from 1 to 9, with 1 indicating variable regions, 5 indicating mildly conserved regions, and 9 indicating highly conserved regions. Additionally, exposed residues with high scores are predicted as functional residues, while buried residues with high scores are predicted as structural residues. The obtained results provide valuable insights into the functional and structural characteristics of the identified variant.
Sequencing and co-segregation study
To confirm the reported variant in the parents, sanger sequencing was performed. Forward and reverse primers were designed with Oligo 7 software to amplify the variant sites, specifically targeting exon 63. Subsequently, we evaluated the resulting sequence chromatograms using Codon Code Aligner.
Result
Clinical finding
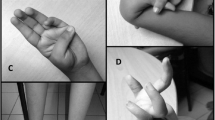
The patient was a six-year-old boy born into a consanguineous family of Persian descent. During the last week of gestation, decreased fetal movement was noted. The patient was delivered at 37 weeks of gestation with a birth weight of 2700 g. Seizures occurred on the third day of life, along with poor feeding and progressive spasticity leading to a prominent delay in motor development. The patient’s motor development continued to be severely delayed, as evidenced by inability to sit, profound weakness, lack of speech, and mild strabismus, as well as poor swelling for solid foods. Other symptoms included pectus excavatum, multiple attacks of pneumonia, proximal contractures, and mild microcephaly (Fig. 2). Unfortunately, the patient passed away at the age of ten years. Neither parent exhibited any symptoms related to the condition.

Nine-year-old patient with Ullrich congenital muscular dystrophy 2 (UCMD2). The patient’s body appears dysmorphic, with physical characteristics like slim and elongated torso (a), absence of teeth (b), narrow shoulders, muscle dystrophy (c) and curved spine. It’s important to note that patients with UCMD2 may experience a range of symptoms beyond those shown in this figure
Genetic testing
After initial diagnosis by a neurologist and orthopaedist, whole exome sequencing (WES) was performed to identify genetic variation. Analysing WES data involved four steps: (1) Removal of synonymous variants, (2) Removal of benign variants (CADD Phred < 20), (3) Removal of minor allele frequency < 0.001, and (4) Retention of homozygous variants. Ultimately, a homozygous mutation in COL12A1: NM_001399.5 exon 63, c.8828 C > T (p.Pro2943Leu), as shown in Fig. 3, related to UCMD2 was identified as the disease-causing variant.

Genomic location of the c.8828 C > T mutation in the COL12A1 gene, from chromosomal to the nucleotide level. (a) The effect of the mutation on protein level, identified as p.Pro3943Leu (b)
In silico analysis of pathogenicity and protein stability
Given that the identified mutation is novel and has not been previously reported, we utilized bioinformatics tools to determine its pathogenicity. The Variant of Uncertain Significance (VUS) was designated in the Varsome database. Table 1 presents the results obtained from SIFT, CADD, Mutation Taster, Polyphen-2, PANTHER, I-Mutant, and MUpro analyses.
Protein-protein interaction
We used the STRING web server and NetworkX python library to demonstrate the impact of COL12A1 in the collagen chain trimerization superpathway. Figure 4 was created after applying the concept of ‘betweenness centrality’ (BC) to the STRING output. In graph theory, BC demonstrates which nodes are more important or the absence of which causes more disruption in the network. In this graph, the size of the nodes indicates their BC, and a color range from dark blue to white indicates the degree (number of edges connected to the node). Thus, darker and larger nodes have a more significant impact on the network. Also, a greater width and darker color indicate a higher interaction score between a pair of proteins.

Prediction of protein–protein interaction based on STRING data, visualized by NetworkX.
Conservation of amino acids
The Clustal Omega tool was utilized to perform multiple sequence alignment of the COL12A1 protein sequence across various species, and the obtained results revealed that amino acid 2943 (Proline), which is the point of variation, was highly conserved in all other species. This finding suggests that the residue plays a crucial evolutionary and functional role in the protein. The results are highlighted in Fig. 5a. Furthermore, the ConSurf tool was used to identify the evolutionary conservation pattern of the COL12A1 protein sequence. This tool predicts each residue’s structural or functional role based on their conservation and solvent accessibility. Residues that are highly conserved and exposed are expected to be functional, while those that are highly conserved and buried are deemed structural. The analysis indicated that amino acid 2943 had a conservation score of 8 (normalized score= -0.696), which was predicted to be functional. Supplementary Material 1 and Fig. 5b contain more details.

Evolutionary conservation analysis of p.Pro2943Leu mutation on COL12A1 using Clustal Omega (a), and Consurf (b)
Co-segregation study
To confirm the segregation of the identified novel variant, sanger sequencing was performed for the proband and his parents. As shown in Fig. 6, the results confirmed that both parents were carriers of the variant.

This figure illustrates the first cousin consanguineous pedigree of an Iranian family with Ullrich congenital muscular dystrophy 2, a rare genetic disorder (a). It also depicts the homozygous and heterozygous mutations identified in affected and unaffected members, respectively (b)
Discussion
In this study, we present a case of a nine-year-old boy from an Iranian family who was suspected of having an inborn error of metabolism and clinically diagnosed with UCMD2. We report the second family affected by UCMD2 and confirm a novel missense mutation, c.8828 C > T, in the COL12A1 gene through genetic sequencing. This mutation has not been previously reported. After the patient’s initial evaluation by a neurologist and orthopaedist, WES was performed to identify any underlying genetic causes. The analysis revealed the c.8828 C > T mutation in the COL12A1 gene, which was also confirmed in the patient’s parents. Since this is a novel mutation and has not been reported, its pathogenicity was assessed using bioinformatics tools. This study confirms that UCMD2 is caused by a homozygous mutation in the COL12A1 gene. Collagen type XII is a structural protein crucial for maintaining the integrity and stability of connective tissue, including skeletal muscle [23]. Mutations in the COL12A1 gene can result in an abnormal or dysfunctional collagen type XII protein, disrupting the normal structure and function of connective tissue [24]. Ullrich-like congenital muscular dystrophy (ULCMD) is clinical a term used to describe a sopectrum of muscular dystrophy disorders that share similarities with Ulrich congenital muscular dystrophy (UCMD) but are caused by mutations in genes other than the collagen VI genes (COL6A1, COL6A2, and COL6A3) [25, 26]. COL12A1 gene mutations have been identified in individuals with UCMD-like symptoms, leading to the inclusion of COL12A1-related UCMD in the spectrum of UCMD disorders. Clinical features of COL12A1-related UCMD resemble those of other forms of UCMD and typically include muscle weakness, joint contractures, and respiratory involvement. The case we present shares similarities with a study by Zou et al., involving two siblings born to consanguineous Turkish parents with clinical features indicative of congenital Ullrich-type muscular dystrophy. The patient in our case also presented with proximal joint contracture and kyphosis in addition to mild facial weakness, a high arched palate, and lack of deep tendon reflexes. Additionally, the patient experienced severe delayed motor development, requiring non-invasive nocturnal ventilation before the age of three and eventually relying on an electric wheelchair for mobility. At nine years old, the patient developed progressive scoliosis that required surgical intervention for spinal stabilization. The patient’s younger brother exhibited similar symptoms at birth, including weakness, significant distal hyperflexia, mild proximal contracture, and an inability to stand or walk [9]. However, it should be noted that unlike the case in the study by Zou et al., the parents of our patient do not exhibit any signs of the disease, even in a mild form. In summary, congenital Ullrich muscular dystrophy is a rare genetic disorder primarily affecting skeletal muscles. The association between the COL6A1, COL6A2, and COL6A3 genes and congenital Ullrich muscular dystrophy is well-established, as pathogenic variants in these genes can lead to the development of UCMD, a severe form of congenital muscular dystrophy characterized by muscle weakness and contractures [27].The study mentioned above represents the only reported correlation between homozygous mutations in COL12A1 and UCMD2. Further research and genetic studies are necessary to gain a better understanding of the specific role of the COL12A1 gene in UCMD2.
Conclusion
Our research presents the second family of UCMD2 associated with a homozygous mutation in the COL12A1 gene, which has only been reported in one other family. The similarities in symptoms between the two cases strengthen the connection between this gene and the disease. Using WES, we identified a novel mutation in COL12A1 (c.8828 C > T). However, further research is required to fully understand the relationship between homozygous mutations in COL12A1 and UCMD2.
Data availability
Data sharing not applicable to this article as no datasets were generated or analysed during the current study.
References
Chiquet M, Birk DE, Bönnemann CG, Koch M, Collagen XII (2014) Protecting bone and muscle integrity by organizing collagen fibrils. Int J Biochem Cell Biol 53:51–54
Snellman A, Keränen M-R, Hägg PO, Lamberg A, Hiltunen JK, Kivirikko KI et al (2000) Type XIII collagen forms homotrimers with three triple helical collagenous domains and its association into disulfide-bonded trimers is enhanced by prolyl 4-hydroxylase. J Biol Chem 275(12):8936–8944
Keesler DA, Slobodianuk TL, Kochelek CE, Skaer CW, Haberichter SL, Flood VH (2021) Fibronectin binding to von Willebrand factor occurs via the A1 domain. Res Pract Thromb Haemostasis 5(5):e12534
Bardakov SN, Deev RV, Magomedova RM, Umakhanova ZR, Allamand V, Gartioux C et al (2021) Intrafamilial phenotypic variability of collagen VI-related myopathy due to a new mutation in the COL6A1 gene. J Neuromuscul Dis 8(2):273–285
Kutluk MG, Kadem N, Bektas O, Randa NC, Tuncer GO, Albayrak P et al (2021) A novel variant of COL6A2 gene causing bethlem myopathy and evaluation of essential hypertension. Ann Indian Acad Neurol 24(2):280
Marakhonov AV, Tabakov VY, Zernov NV, Dadali EL, Sharkova IV, Skoblov MY (2018) Two novel COL6A3 mutations disrupt extracellular matrix formation and lead to myopathy from Ullrich congenital muscular dystrophy and Bethlem myopathy spectrum. Gene 672:165–171
Punetha J, Kesari A, Hoffman EP, Gos M, Kamińska A, Kostera-Pruszczyk A et al (2017) Novel Col12A1 variant expands the clinical picture of congenital myopathies with extracellular matrix defects. Muscle Nerve 55(2):277–281
Baker NL, Mörgelin M, Peat R, Goemans N, North KN, Bateman JF et al (2005) Dominant collagen VI mutations are a common cause of Ullrich congenital muscular dystrophy. Hum Mol Genet 14(2):279–293
Zou Y, Zwolanek D, Izu Y, Gandhy S, Schreiber G, Brockmann K et al (2014) Recessive and dominant mutations in COL12A1 cause a novel EDS/myopathy overlap syndrome in humans and mice. Hum Mol Genet 23(9):2339–2352
Bönnemann CG (2011) The collagen VI-related myopathies: Ullrich congenital muscular dystrophy and Bethlem myopathy. Handb Clin Neurol 101:81–96
Cassandrini D, Trovato R, Rubegni A, Lenzi S, Fiorillo C, Baldacci J et al (2017) Congenital myopathies: clinical phenotypes and new diagnostic tools. Ital J Pediatr 43:1–16
Ng PC, Henikoff S (2003) SIFT: Predicting amino acid changes that affect protein function. Nucleic Acids Res 31(13):3812–3814
Rentzsch P, Witten D, Cooper GM, Shendure J, Kircher M (2019) CADD: predicting the deleteriousness of variants throughout the human genome. Nucleic Acids Res 47(D1):D886–D94
Schwarz JM, Cooper DN, Schuelke M, Seelow D (2014) MutationTaster2: mutation prediction for the deep-sequencing age. Nat Methods 11(4):361–362
Adzhubei I, Jordan DM, Sunyaev SR (2013) Predicting functional effect of human missense mutations using PolyPhen-2. Curr protocols Hum Genet 76(1):7 1–7. 41
Thomas PD, Ebert D, Muruganujan A, Mushayahama T, Albou LP, Mi H (2022) PANTHER: making genome-scale phylogenetics accessible to all. Protein Sci 31(1):8–22
Capriotti E, Fariselli P, Casadio R (2005) I-Mutant2. 0: predicting stability changes upon mutation from the protein sequence or structure. Nucleic Acids Res 33(suppl2):W306–W10
Cheng J, Randall A, Baldi P (2006) Prediction of protein stability changes for single-site mutations using support vector machines. Proteins Struct Funct Bioinform 62(4):1125–1132
Mering Cv, Huynen M, Jaeggi D, Schmidt S, Bork P, Snel B (2003) STRING: a database of predicted functional associations between proteins. Nucleic Acids Res 31(1):258–261
Sievers F, Higgins DG (2014) Clustal Omega, accurate alignment of very large numbers of sequences. Multiple Seq alignment methods. :105–16
Ashkenazy H, Abadi S, Martz E, Chay O, Mayrose I, Pupko T et al (2016) ConSurf 2016: an improved methodology to estimate and visualize evolutionary conservation in macromolecules. Nucleic Acids Res 44(W1):W344–W50
Consortium U (2019) UniProt: a worldwide hub of protein knowledge. Nucleic Acids Res 47(D1):D506–D15
Zhao C, Xiao Y, Ling S, Pei Y, Ren J (2021) Structure of collagen. Methods in molecular biology. (Clifton NJ) 2347:17–25
Delbaere S, Dhooge T, Syx D, Petit F, Goemans N, Destrée A et al (2020) Novel defects in collagen XII and VI expand the mixed myopathy/Ehlers–Danlos syndrome spectrum and lead to variant-specific alterations in the extracellular matrix. Genet Sci 22(1):112–123
Izu Y, Birk DE (2023) Collagen XII mediated cellular and extracellular mechanisms in development, regeneration, and disease. Front Cell Dev Biology 11:1129000
Yonekawa T, Nishino I (2015) Ullrich congenital muscular dystrophy: clinicopathological features, natural history and pathomechanism (s). J Neurol Neurosurg Psychiatry 86(3):280–287
Di Martino A, Cescon M, D’Agostino C, Schilardi F, Sabatelli P, Merlini L et al (2023) Collagen VI in the Musculoskeletal System. Int J Mol Sci 24(6):5095
Funding
The research project has been financially supported by Golestan University of Medical Sciences (Grant Number: 111672).
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
None to declare.
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
Springer Nature or its licensor (e.g. a society or other partner) holds exclusive rights to this article under a publishing agreement with the author(s) or other rightsholder(s); author self-archiving of the accepted manuscript version of this article is solely governed by the terms of such publishing agreement and applicable law.
About this article

Cite this article
Naghipoor, K., Khosravi, T. & Oladnabi, M. Whole exome sequencing identifies a novel variant in the COL12A1 gene in a family with Ullrich congenital muscular dystrophy 2. Mol Biol Rep 50, 7427–7435 (2023). https://doi.org/10.1007/s11033-023-08644-6
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s11033-023-08644-6