
Abstract
Background
Rice crop is damaged extremely by abiotic stress world-wide. The best approach to enhance drought tolerance in rice varieties is to identify and introgress yield QTLs with major effects. The Association mapping approach helps in the identification of genomic regions governing physiological, yield and yield attributes under moisture and heat stress conditions in diverse collections of crop germplasm, based on historic recombination events and linkage disequilibrium across the genome.
Methods and Results
The association mapping panel of 110 rice germplasm lines exhibited significant variation for all the traits in both irrigated and moisture stress conditions. The extent of yield reduction ranged to 83% during rabi, 2018–19, 53% in rabi, 2019–20 and 68% in pooled analysis. The genotypes Badami, Badshabhog, Pankaj, Varalu, Vasundhara, Vivekdhan, Krishna and Minghui63 exhibited drought tolerance with least yield penalty under moisture stress conditions. The genotypes Konark, MTU3626, NLR33671, PR118 and Triguna exhibited minimal reduction in heat stress tolerance traits. Association mapping of germplasm using 37808 SNP markers detected a total of 10 major MTA (Marker-trait association) clusters distributed on chromosomes 1, 3, 4 and 11 through mixed linear model (MLM) governing multiple traits from individual data analysis which are consistent across the years and situations. The pooled data generated a total of five MTA clusters located on chromosome 6. In addition, several novel unique MTAs were also identified. Heat stress analysis generated a total of 23 MTAs distributed on chromosomes 1, 5, 6 and 11. Candidate gene analysis detected a total of 53 and 38 genes under individual and pooled data analysis for various yield and yield attributes under control and moisture stress conditions, respectively and a total of 11 candidate genes in heat stress Conditions.
Conclusion
The major and novel MTAs identified in the present investigation for various drought and heat tolerant traits can be utilized for breeding climate-resilient rice varieties. The candidate genes predicted for key MTAs are of great value to deploy into the rice breeding after functional characterization.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Rice (Oryza sativa L.) is one of the most important staple food crops and a major source of calories for over half of the population in the world. Although rice production doubled after the Green revolution in the 1960s, the projected demand also doubled due to the anticipated 9 billion people by 2050. Moreover, this mammoth challenge has to be achieved with continuously shrinking land and water resources. Of them rice alone consumes 80% of fresh water and specifically rice consumes 3000–4000 L of fresh water to produce 1 kg of rice. These problems are compounded by climate change especially, frequent drought stress and flash floods, gradually rising global temperatures. Rice productivity is often hampered by several abiotic and biotic stresses. Among abiotic stresses, moisture and heat stresses are the most crucial, which needs special attention as around 43% of rice cultivation is under rainfed condition [1]. Global climate change, with frequent episodes of abiotic stress, reduces the productivity of rice as rice is more sensitive to water deficit than other cereals.
In rice, drought can reduce productivity by 53–92% and through heat by 50% respectively, [2]. Rice is more susceptible to water deficit and drought conditions, especially the modern cultivars developed after the Green revolution as they possess shallow root system.
The heat stress is also becoming an increasing threat to rice productivity as the mean surface temperature of the earth is projected to rise a maximum of 4–8 °C by the end of 2100 [3]. The optimum temperature to cultivate rice is 22 to 28 °C and the temperature beyond 35 °C, especially during reproductive stage will adversely affect rice productivity by reducing pollen viability and spikelet fertility [4]. In addition, heat stress reduces the grain filling period and increases chalky grain number. It is predicted that for every 1 °C rise in temperature, especially during anthesis and booting, the rice yield will affect upto 10% [5]. Therefore breeding for heat tolerant varieties by exploring tremendous variability in the rice germplasm besides armed with excellent genomic resources is the need of the hour to address the situation of global warming.
The molecular mechanism regulating drought and heat tolerance is a complex and found to be involved several major and minor genes. Identification of QTLs and candidate genes helps better understanding of mechanism underlying drought and heat tolerance. Several QTLs have been identified for drought and heat tolerance employing linkage or biparental mapping, which can explain the variability between parents only. Further, the success rate of QTLs identified using biparental mapping population is limited in marker-assisted breeding (MAB) due to small phenotypic effect and large intervals of the QTLs. Additionally, inconsistency of the QTLs across environments and genetic backgrounds limits their adoption in MAB.
The advent of Next Generation Sequencing (NGS) technologies offers the plant breeding community a range of modern tools and methods for addressing challenges pertaining to the functional characterization of genes. Recently developed association mapping coupled with NGS sequence—based genotyping led to a new approach called Genome wide association studies (GWAS). Employing Genome wide association studies (GWAS) that combines phenotyping of the germplasm and high diversity SNP panels helps in the detection of large number of haplotypes and genes. This methodology found several advantages over earlier QTL mapping approaches such as exploiting natural variability in the germplasm, reduced time and cost for developing mapping populations, offers greater resolution because of higher recombination events etc. The marker trait associations identified through GWAS facilitates uncovering candidate genes regulating drought and heat tolerance mechanisms and thus can be utilized in the rice breeding to improve cultivars through marker assisted breeding programs. In addition, the candidate genes can also enables better understanding of the key genomic regions and mechanism of drought and heat tolerance.
The projected increase in global temperatures and reduced precipitation will increase the frequency of occurrence and intensity of these stresses, threatening rice production. Despite recognizing the importance of combined stress in rice, the knowledge generated in this area is very limited. Commonly used MABC has not been very effective in developing superior lines for drought tolerance, modern breeding approaches such as MARS and GWAS are powerful tools for pyramiding multiple QTLs for drought tolerance or introgressing multiple complex traits such as heat tolerance in addition to drought tolerance. The availability of a large number of drought QTLs and continued efforts towards identification of heat QTLs would pave the way for stacking large effect drought and heat QTLs for improved combined stress tolerance. Hence, the present investigation was undertaken with the objective of identification of marker—trait association and candidate genes governing drought and heat tolerance employing GWAS.
Materials and methods
Evaluation of the rice genotypes for reproductive moisture stress tolerance
A diverse panel of 110 rice genotypes were grown in field during two seasons viz., Rabi 2018–2019 (Trail 1 December–May) and Rabi 2019–2020 (Trail 2 November–April) at S. V. Agricultural college, ANGRAU, Tirupati. The experiment was laid out in alpha lattice design with three replications in both control (irrigated) and moisture stress conditions. In both years, Trial 1 and Trial 2, seeds of each genotype were planted in nursery beds at staggered dates as per days to flowering for coinciding flowering period with drought stress imposition. Twenty-one days old seedlings were transplanted to the main field. Each genotype was transplanted in three rows of 1 m length with a spacing of 20 × 15 cm. Fertilizer dose of N: P: K in the ratio of 120:60:40 kg/ha was applied. Water stress was imposed by suspending irrigation for stress plot when the genotypes initiated booting and it continued till maturity with life saving irrigation based on the leaf rolling scores and tensiometer readings while the irrigation was maintained normally in control plots. Soil moisture content was measured at regular intervals using gravimetric approach. Measurements on morphological and physiological parameters were recorded after 14 days of stress imposition. Observations were recorded for various yield traits like days to 50% flowering (DFF), days to maturity (DM), plant height (PH), number of productive tillers per plant (NPTP), number of panicles per plant (NPP), panicle length (PL), grains per panicle (GPP), number of filled grains per panicle (NFGP), number of chaffy grains per panicle (NCGP), thousand seed weight (TSW), grain yield per plant (GYP), biological yield per plant (BYP), harvest index (HI), leaf rolling score (LRS), leaf senescence score (LSS), spikelet fertility (SF), SCMR, relative membrane injury (RMI) and drought tolerance index (DTI) at different periods of crop growth till maturity under both stress and normal conditions in both Trail 1 and Trail 2.
Evaluation of the rice genotypes for heat tolerance
All 110 rice genotypes were evaluated for heat stress tolerance using Temperature Induction Response (TIR) technique as proposed by Sudhakar et al. [6]. Sprouted rice seedlings were subjected to different temperature treatments. 50 uniform seedlings were selected from each genotype and shifted to aluminum trays containing blotting paper wetted with water. These trays with seedlings were subjected to sub-lethal temperature with gradual increasing temperature for every half an hour from 38 °C to 50 °C in the environmental chamber (Seed germinator, model-NLSg-200, Nanolab, India). Later these seedlings were exposed to lethal temperature (55 °C) (induced) for 2 h. Another sub set of seedlings were exposed directly to lethal temperature (non-induced). One treatment was maintained at room temperature (control) in Petri dishes. Induced and non-induced rice seedlings were allowed to recover at room temperature for three days. Recovered number of seedlings in induced condition and number of seedlings in control condition were counted. Root and shoot lengths (cm) were measured for seedlings in induced and control conditions. The reduction in growth per cent was estimated.
Statistical analysis
Analysis of variance (ANOVA) was carried out in R software using “agricolae” package to estimate the significant differences among genotypes and blocks. Correlation analysis was performed using “corrplot” package in R software (R core Team, 2013).
Genotyping of rice genotypes
DNA extraction was carried out using the modified Cetyl Tri Methyl Ammonium Bromide (CTAB) method developed by Murray and Thompson [7]. The PCR analysis was carried out using 10 µL reaction mixture consisting of 0.5 μl of 5 pmol primer (both forward and reverse primer), 0.5 μl of 1 mM deoxy ribonucleotides (dNTPs), 1 μl of 10X PCR buffer with Mg2 + (ABM, Canada), 0.1 μl of 5U/μl Taq DNA polymerase (ABM, Canada) and 5.4 μl of sterile distilled water was added to make up the volume to 8 μl along with 2 µl of template DNA. The amplified products of PCR were resolved using 3 percent agarose gel and run was kept for 1 to 3 h in 0.5X TBE at 95 V. The PCR product is visualized in ALPHA IMAGER Gel Documentation Unit (M/s Alpha Innotech, USA).
Molecular marker analysis
A total of 45 SSR markers were used for genotyping of 110 rice accessions out of which 41 tend to be polymorphic and were used for further genotyping (Spp. Table 3).
Population structure analysis
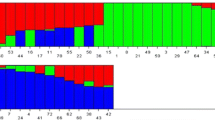
STRUCTURE version 2.3.3 was used to assess the genetic structure of rice genotypes and assign individuals to populations [8]. STRUCTURE HARVESTER ver. 0.6. [9] was used to estimate the most probable K value. The K value, was selected after ten independent runs with a burn-in period of 100,000 steps with 100,000 Monte Carlo Markov chain (MCMC) replicates. The range of genetic clusters was set from K = 1 to 10. The number of subpopulations (K) was determined based on the posterior probability values (LnP(D) and ΔK.
Genome-wide association mapping
Association mapping panel of 110 rice genotypes was subjected to double digest Restriction site associated DNA sequencing (ddRAD) method, which resulted a total of 37,808 SNP markers. SNP data was filtered for minor allele frequency (MAF) ≥ 0.05 and maximum missing sites per SNP was fixed to < 20%. MTAs were identified using Mixed Linear Model (MLM) implemented in GAPIT (genomic association and prediction integrated tool). The extent of kinship among individuals was also estimated with the filtered set of SNP markers using GAPIT in R software [10] (R Development Core Team 2018). Manhattan plots were produced and a threshold value for declaring marker-trait association generated through MLM approach was set at—Log p = 4 (i.e., p-value ≤ 0.0001) and a LOD value of ≥ 3.5 for individual moisture and heat stress data analysis. While, LOD value of ≥ 4 was chosen for pooled data analysis of moisture stress condition. QTLs were named according to the trait name, year, chromosome number, physical map position on the genome and the situation in which QTL was identified. For example, qBY18_6.1C represents QTL for biological yield identified during control condition of rabi, 2018–19 located on chromosome 6 in increasing order of location on the chromosome.
Candidate gene prioritization
The annotated genes for each QTL were prioritized for probable candidate genes using QTG finder software [11]. From the annotated genes, prioritized top three genes were selected and their functions in relation to the trait was examined from the previous literature. For confirming probable candidate gene in each MTA, we considered three criteria viz., literature mining, sequence variation using Ricevarmap and gene expression using RiceXpro.
Results
Evaluation of the rice genotypes for reproductive moisture stress tolerance
Analysis of variance displayed significant variation for all the traits in both control and stress conditions indicating the presence of variation among the genotypes used in the present study. All the genotypes showed the overall mean reduction in many of the yield contributing traits under drought stress compared with non-stress (NS) condition. A mean reduction of 83% observed for grain yield during rabi, 2018–19, while the reduction is 53% in rabi, 2019–20 and 68% in pooled analysis. For grain number, the mean reduction was 62% during rabi, 2018–19, 21% during rabi, 2019–20 and 41% in pooled analysis. For the trait thousand seed weight, the reduction percentage was 29%, 15% and 22% during rabi, 2018–19, rabi, 2019–20 and pooled analysis respectively. Reduction in spikelet fertility was found to be 71% during rabi, 2018–19, 21% in rabi, 2019–20 and 46% in pooled analysis (Spp. Table 2).
The drought resistant genotypes viz., Badami, Badshabhog, Pankaj, Varalu, Vasundhara, Vivekdhan, Krishna and Minghui63 were considered as better performers based on the traits viz., days to 50% flowering, grains yield per plant, spikelet fertility and drought tolerance index during both years under both irrigated and moisture stress conditions.
Evaluation of the rice genotypes for early seedling stage heat stress tolerance
Phenotypic results of heat stress data also revealed the presence of significant variation among treatments indicating the presence of variability which can be exploited for further breeding programs. The mean percent reduction of survival percentage was found to be 6% under sub lethal condition compared to control while, under lethal condition, none of the genotypes were survived. The trait shoot length displayed a percent reduction of 29% while the trait root length showed a reduction of 38% under sub lethal condition compared to control condition (Spp. Table 2).
Under heat stress conditions, the genotypes Konark, MTU3626, NLR33671, PR118 and Triguna have displayed better survival percentage, shoot length, root length along with minimal reduction in shoot and root growth under induced conditions.
Correlation analysis of drought and yield related traits showed a significant positive correlation among almost all the traits with yield under both control and moisture stress conditions. Under irrigated conditions of rabi, 2018–19 the results of correlation analysis revealed a positively significant association of almost all the yield component traits with GYP except NCGP, HI, SCMR and RMI. During rabi, 2018–19 moisture stress condition the traits DFF, DM, NCGP and LSS showed significant negative correlation with grain yield while, the rest of the traits showed significant positive correlation. The results of correlation analysis during rabi, 2019–20 revealed a significant positive correlation of almost all the traits with grain yield except TSW, SF and RMI. Under moisture stress conditions of rabi, 2019–20 majority of the traits showed significant positive correlation with grain yield except for the traits NCGP and TSW having negative significant association. Among the heat stress parameters the trait shoot length alone displayed significant positive correlation with root length and grain yield per plant (Spp. Table 3).
Population structure analysis
Analysis of population structure is critical in association studies as it reduces the effects of Type-I and Type- II errors [8]. Structure analysis was carried out using the genotypic data obtained by screening of 110 rice genotypes with 41 SSR markers. For K = 2 to 10, the highest ΔK of 69.28 was reached at K = 2 with an LnP(D) of − 6671.68 Supp. Figure 1. Thus, the population falls under two groups, POP 1 encompasses a total of 28 genotypes comprising of Aromatic, Japonica and Indica subspecies. POP 2 consists of 82 genotypes comprising of Indica, Japonica, Aus and Aromatic. The genotypes with score > 0.80 were considered as pure while, < 0.80 were considered as admixtures. Among two subpopulations, POP1 consists of 18 pure types and 10 admixed types while, POP2 has 69 pure types and 13 admixed types. The presence of admixtures was perhaps due to the fact that the loci involved in this study revealed only a small part of the genotype/phenotype association of otherwise complex traits or acquisition of a few spontaneous mutations in the genotypes.
Genomic regions governing drought and heat tolerance traits
GWAS analysis detected a total of 115 MTAs for 19 traits related to drought and yield components with a PVE range of 9.73%–27.05% in both control and moisture stress conditions during rabi, 2018–19. All these MTAs were distributed on chromosomes 1, 3, 4, 8, 9, 10 and 11 as presented in Fig. 1 and Spp. Figure 2. Of all MTAs, 82 were found to be novel, which are identified in the present study only and rest are colocalized with previously identified MTAs. The number of significant loci associated with the different agronomic traits under stress conditions ranged from 1 (NTP) to 6 (LSS). Even under control conditions the number ranged from 1 (HI) to 6 (NPTP, GPP).

Manhattan and quantile–quantile (QQ) plots of marker-traits associations under drought stress and control conditions in pooled data analysis. A Grain number per panicle—Control; B Economic yield—Control; C Spikelet fertility – Control; D Grain number per panicle—Drought stress; E Economic yield—Drought stress; F Spikelet fertility—Drought stress
During rabi, 2019–20, a total of 119 MTAs were detected for 19 traits, and of them 90 were novel MTAs with a PVE range of 9.73%- 26.31% under control and moisture stress conditions. All these MTAs are distributed on chromosomes 1, 3, 4, 5, 8, 10, 11 and 12 (Fig. 1 and Spp. Figure 2). The number of significant loci associated with the different agronomic traits under stress conditions ranged from 1 (LRS, LSS, SF and RMI) to 9 (GPP) while, under control conditions the number ranged from 1 (TSW) to 7 (NFGP). Trait wise list of MTAs identified under both control and stress conditions and the common MTAs observed in both the situations during two years were presented in Table 1. Among all the MTAs detected, only 10 MTAs were found to be consistent over both the years and are distributed on chromosomes 1, 3, 4 and 11 (Table 2).
Stable MTAs
In the present study, ten MTAs were found to be governing multiple traits (approx. 10 traits) consistently in two years and are considered as stable MTAs (Table 2). For instance the marker UChr1_20598769 was linked to DFF MTAs (qDFF18_1.2C, qDFF18_1.2S, qDFF19_1.2C, qDFF19_1.2S), DM MTAs (qDM18_1C, qDM18_1S, qDM19_1.1C, qDM19_1.1S), PH MTAs (qPH18_1C, qPH18_1S, qPH19_1C, qPH19_1S), PL MTAs (qPL18_1C, qPL18_1S, qPL19_1S, qPL19_1C), GPP MTAs (qGPP18_1.2C, qGPP18_1.2S, qGPP19_1.3S), GYP MTAs (qGYP18_1.2C, qGYP19_1C), LRS MTAs (qLRS18_1C, qLRS19_1C), LSS MTAs (qLSS18_1.2C, qLSS18_1.2S, qLSS19_1C), SCMR MTAs (qSCMR18_1C, qSCMR18_1S, qSCMR19_1.3C, qSCMR19_1.3S) and DTI MTAs (qDTI18_1S, qDTI19_1S). Usually, major MTAs can be identified across seasons, therefore considered as stable. These stable MTAs of promise can be straight away transferred to elite varieties through marker- assisted breeding (MAB). Further, the stable MTAs can also be targeted for candidate gene identification using various molecular dissection methods.
Environment-specific MTAs
In the present investigation, some MTAs were identified to control specific traits only i.e., either in control or in moisture stress conditions and are considered as environment specific MTAs as presented in Table 2. For instance, SNP UChr1_1064205 was linked to the MTAs qGPP18_1.1C and qGPP19_1.1C; qNFGP18_1.1C and qNFGP19_1.1C were identified only under control conditions. While, the same SNP was linked to MTAs qLSS18_1.1S, qBYP19_1.1S, qSCMR19_1.1S governing LSS, BYP and SCMR under moisture stress conditions only as shown in Table 2. Similarly, UChr1_20598769 was governing GYP (qGYP18_1.2C and qGYP19_1C) and LRS (qLRS18_1C and qLRS19_1C) under control conditions only. The marker UChr4_1739798 was found to govern MTAs expressed only under moisture stress conditions viz., qGPP19_4S, qNCGP18_4S and qLRS18_4S. These environment-specific MTAs are very specific to either control or stress conditions, hence considered as small effect MTAs. Selection of such trait linked markers increases the predictive ability in genomic selection compared to markers selected solely based on neutral linkage disequilibrium.
In addition to these stable and environment specific MTAs, some novel MTAs were identified in a single condition which might be small effect MTAs as presented in Table 3. It is necessary to pool these small effect ones employing marker- assisted recurrent selection and genomic selection.
Pooled data analysis revealed the presence of 48 MTAs or QTLs for 17 traits (Fig. 2 and Spp. Figure 2c). All the 48 MTAs were distributed on chromosomes 1, 2, 3 and 6 as shown in Spp. Table 8 and Spp. Figure 2. Of the 48 MTAs, 24 were identified under irrigated and 24 under moisture stress conditions. Of the identified MTAs, seven were found commonly in both the conditions as depicted in Table 4. Out of the 48 MTAs, 39 were novel identified in the present study only. The PVE for all MTAs ranged from 14.92% to 28.64%.

Manhattan and quantile–quantile (QQ) plots of marker-traits associations under heat stress and control conditions. A Survival percentage – Control; B Shoot length—Control; C Root length—Control; D Survival percentage—Heat stress; E Shoot length—Heat stress; F Root length—Heat stress
Pleiotropic MTAs
Some SNPs showed linkage to more than one trait and considered as pleiotropic MTAs as presented in Tables 2 and 4. The MTA UChr6_6783646 was associated with nine QTLs viz., qDFF_6.2-C, qDFF_6.2-S, qDM_6.2-C, qPL_6.2-S, qBYP_6.1-S, qLRS_6.2-C, qLSS_6.1-C, qLSS_6.1-S, and qSF_6-C related to seven traits. The MTA UChr6_6569011 was associated to qDM_6.1-S, qPH_6.1-C, qPH_6.1-S, qPL_6.1-C, qNFGP_6.1-C, qHI-6-C and qSCMR_6.3-S. The presence of pleiotropic MTAs was supported by the significant correlation among traits which can be witnessed either due to the control of a single gene or due to tightly linked genes.
MTAs controlling heat stress tolerance
In the present study, in all, we have identified a total of 23 MTAs through MLM model at LOD value of 3.5 and above of which all are novel MTAs as presented in Spp. Table 9. All the MTAs identified are distributed over 1, 5, 6 and 11 chromosomes (Fig. 2 and Spp. Figure 2d). The MTAs were having PVE values ranging from 12.30% to 24.48% for traits survival percentage, shoot length, root length, reduction in shoot growth and reduction in root growth.
Candidate genes underlying major MTAs
From individual data analysis a total of 10 major stable MTAs were detected during both seasons, consisting of 155 annotated genes in 100 kb window region as per rice MSU database. Of the detected genes, 14% of genes were coding retrotransposons and hypothetical proteins; 22% of them were expressed proteins and 33% of genes belongs to unique class of gene families having major portion of transcription factors such as ethylene responsive factors, genes involved in hormonal regulation, zinc finger family proteins, MYB family proteins, F box proteins, helix loop helix proteins, MAD box genes, subtilin family proteins etc. In addition, few genes were also involved in hormonal regulation, signal transduction pathways, genes involved in proteolytic machinery, macromolecular proteins, transferases, cyp family genes, pollen methyl esterase inhibitors, methyl transferases, neutral invertases etc.
Under TIR analysis a total of 11 candidate genes were found to be governing the traits SP, SL, RL, RSG and RRG. The identified candidate genes were belonging to transcription family proteins, growth regulators, transferases family proteins etc. The gene regulatory families identified in the current study were presented in Spp. Figure 3.
Discussion
Among abiotic stresses, moisture and heat stress are the most yield depressing factors in rice and demands great attention from the scientific community in the era of climate change and genomics. To address these moisture and heat stresses, a deep understanding at molecular and cellular level intrinsic pathways will offer substantial insights for development of climate resilient varieties. To this end, we uncovered several novel MTAs influencing important moisture and heat tolerance traits employing genome- wide association mapping.
Performance of rice genotypes under drought and heat stress conditions
Imposition of drought stress caused percent reduction in almost all the traits in stress compared to control. This might be due to reduced water availability to plants which reduced the photosynthetic activity in plants causing reduced accumulation of biomass and spikelet fertility. The percent yield reduction during both the seasons ranged from to 53% to 83% which helps in effective screening of drought resistant genotypes from that of susceptible ones. Similar results were also reported by earlier studies [12, 13]. In the current study reduction in grain yield was attributed to poor panicle exertion, reduced number of productive panicles, poor grain filling.
Combined phenotypic data across years revealed few promising rice accessions as valuable genetic resources to increase thousand seed weight (Badami, Pankaj, Vivekdhan), grain yield per plant (Badami, Badshabhog, Varalu, Vasundhara, Vivekdhan, Krishna), spikelet fertility (Badami, Pankaj, Varalu, Vasundhara, Vivekdhan), drought tolerance index (Badshabhog, Vasundhara, Krishna). Certain accessions like Badami, Varalu, Vasundhara and Vivekdhan serve as genetic resources to stack different traits to improve rice plant performance and yielding ability under drought. Thus, the genotypes viz., Badami, Badshabhog, Pankaj, Varalu, Vasundhara, Vivekdhan, Krishna and Minghui63, which showed least yield penalty under moisture could be used as potential donors for development of climate resilient varieties. Of them, the genotypes Badami and Varalu were already being used as donors in rice breeding. Additionally, these genotypes also can be used to identify QTLs for moisture stress tolerance in future map-based cloning research.
Under heat stress conditions, the genotypes Konark, MTU3626, NLR33671, PR118 and Triguna showed minimal reduction in shoot and root growth, and therefore can be used as donors for heat stress tolerance. Notably, the genotypes, Badami, Badshabhog, Pankaj, Varalu, Vasundhara, Vivekdhan, Krishna, Minghui63, Konark, MTU3626, NLR33671, PR118 and Triguna showed impressive performance under both moisture and heat stress conditions. The drought tolerant lines with least reduction can be further recommended for rainfed cultivation also for development of biparental population to uncover novel QTLs. Therefore these genotypes can be used as donors and also for deep understanding of the genes involved in controlling moisture and heat stress.
Identification of MTAs employing GWAS
GWAS analysis of the present study resulted in 234 MTAs under individual and pooled seasons of control and moisture stress and 27 MTAs under heat stress conditions. Among them, 191 and 27 are novel MTAs identified under moisture and heat stress conditions, respectively and 43 MTAs are colocalized with previously identified QTLs. Majority of MTAs are distributed on 1, 3 and 11 chromosomes. It is interesting to note that we have identified both novel and colocalized MTAs using 110 rice genotypes, which is less than previously used genotypes. This suggest that our rice genotypes display substantial variation and can be used as an excellent association mapping panel ideal for GWAS studies to uncover important QTLs. Similar findings were reported by Beena et al. [14] who have used 99 diverse rice panel accessions; while, Melandri et al. [15] used more than 200 accessions in their study.
Of total MTAs, we identified 10 MTAs as stable MTAs found in both the seasons. While, 37 MTAs as environment specific, found in only one season. Usually, the stable MTAs are considered as major MTAs, hence, can be utilized for marker assisted introgression into elite varieties to enhance moisture stress tolerance. Moreover, these stable and novel MTAs are an excellent targets for positional cloning to uncover candidate genes. The environment- specific MTAs with more than 15% PVE can be assembled employing marker assisted gene pyramiding. Further, these MTAs can also be used to breed varieties suitable to particular environment with a specific climatic factor.
Colocalization of MTAs
The SNP UChr1_1064205 association with the traits GPP, NFGP, GYP and BYP were identified earlier Yu et al. [16]. While its association with NPTP, NPP, SCMR, LSS and RMI were novel which are identified in the current study. UChr1_20598769 was found to be related to several of the drought and yield traits whose associations was reported previously by Sasahara et al. [17]. However, a few novel associations were also detected under control conditions. The SNP UChr3_29209246 located on Chr. 3 was found to be novel governing PH, TSW, LSS and RMI. UChr3_29530231 association with NFGP and GPP was reported earlier while, its association with PH, LRS and LSS were novel associations. The SNP UChr4_1739798 was found to be moisture stress specific MTA linked to the traits GPP, NCGP and LRS. Of these associations the earlier two associations were previously reported and the latter association was found to be the novel one. The novel SNP UChr11_3546564 was governing LRS, HI and SF under control conditions having the gene OsRLCK involved in increasing seed weight and plant biomass under abiotic stress conditions [18]. SNP UChr11_3775843 detected on Chr. 11 was found to be another novel major MTA cluster governing several of novel trait associations. UChr11_6034522 was associated to HI, LRS and BYP which were novel associations along with LSS, a previously reported association. Another major SNP cluster UChr11_6656777 was associated to several of the traits. The SNP cluster UChr11_26816152 was also linked to several traits whose associations were novel identified in the current study only.
Pleiotropic MTAs
In the present investigation, a total of 10 MTAs was found to regulate several correlated traits. For instance, UChr1_1064205 was linked to traits NPPP, NPP, GPP, NFGP, GYP, LSS, RMI, BYP and SCMR. Likewise majority of the yield and yield attributing traits along with physiological traits were found to be in pleiotropic gene action imparting drought tolerance mechanism. For such correlated traits with shared QTL, it is likely that either the same causal polymorphism underlies the identified QTL (pleiotropy) or that the genes underlying the QTL are linked.
Candidate gene analysis
Although several genomic regions or MTAs controlling moisture and heat stress tolerance traits have been identified using either linkage mapping or GWAS the underlying candidate genes were left unidentified due to the involvement of laborious and time-consuming strategies of molecular dissection. In the current investigation, we tried to prioritize the candidate genes in a 100 kb window region of each MTA employing publicly available rice genomic resources.
The insilico analysis has displayed the combined action of different gene families like growth regulators, transcription family proteins, kinases, transporters etc. involved in regulation of grain yield through stress signaling as presented in Spp. Table 10. Various growth regulators and proteins like auxin response factor 1 (ARF1), cyclin-like F-box domain containing protein (OsFBox16), basic helix-loop-helix protein (bHLH), plant neutral invertase domain containing protein (OsNIN6) and growth regulator related protein (OsSTA252) are involved in enhancing salt stress tolerance, panicle number, panicle length, tiller number, dry weight, chlorophyll content, grains per panicle, grain yield and reduced plant height [19,20,21]. Insilico analysis also supported the results through their expression pattern observed in ovary, embryo and root tissues. Several kinase family genes were also involved in stress signaling like receptor kinase (OsI-BAK1, OsRLCK), Inositol 1, 3, 4-trisphosphate 5/6-kinase (OsIPK1), tyrosine protein kinase (PTKs), pyruvate kinase (PK), DUF239 and cytochrome P450 (Cyp) were found to enhance root and panicle growth, flowering, spikelet fertility, grain yield, biological yield, tiller number, chlorophyll content, grains per panicle, seed weight, reduced plant height enhances salt and drought stress, detoxification of xenobiotics, biosynthesis of secondary metabolites (flavonoids), antioxidants and phytohormones (Abscisic acid) being expressed in endosperm, leaf blade and anther tissues as evidenced from RiceXPro database [18, 20, 22,23,24,25,26]. In addition several membrane transporters like peptide transporter PTR2 (OsNPF8.5), DNA methyltransferases (MTases) and sulfotransferase domain containing protein (SOT) plays important role in regulating osmotic balance under stress conditions. These genes were involved in plant growth regulation and yield enhancement [27] through enhancement of plant nitrogen metabolism governed by OsGDCi gene [28]. Several other genes like WD repeat-containing protein 5 (OsWDR5), pentatricopeptide repeat containing protein (PPR), pectin methyl esterase inhibitors (PMEI), WD domain, G-beta repeat domain containing protein (OsRBP10), calmodulin binding protein (CaM) and auxin-repressed protein (ARP) were involved in regulating dwarfism, chlorophyll deficiency, flowering and spikelet fertility [29, 30]. Grain development was being influenced by the action of candidate genes like ATL family protein containing RING-H2 finger motif (RRF1), grain setting defect 1 (GSD1), laccase-22 precursor (OsLAC26), serpin domain containing protein (OrysaZ8) and vacuolar-sorting receptor (PV72) [22]. Further, some genes are regulating chloroplast development like photosystem II core complex proteins psbY, chloroplast (PsbY) and succinate dehydrogenase iron-sulfur subunit 2, mitochondrial precursor (OsSDH2-1) through the regulation of electron transport in mitochondria and chloroplast.
In addition to candidate genes under MTA clusters, several other candidate genes were detected under unique MTAs involved in trait regulation as presented in Spp. Table. 10. Thus, a total of 51 candidate genes were found to be governing yield and yield attributes under individual season data analysis of both control and moisture stress conditions.
Under pooled data conditions, of the detected seven MTA clusters four (UChr6_6569011 and UChr6_6569100, UChr6_26976765 and UChr6_26976777) were located in close proximity. The closely positioned clusters were comprising of same prioritized candidate genes. Hence, to avoid repetition these nearby located clusters were discussed as a single cluster in further analysis. Thus a total of five MTA clusters were identified under pooled data analysis.
Majority of candidate genes under pooled data conditions belong to growth regulators, pollen growth development, transferases, osmotic balance regulators, stress signaling family proteins and so on. The candidate genes like cell division inhibitor, ras-related protein (OsRac5), homeobox domain containing protein (HAZ1, HOX1a), GDSL-like lipase/acylhydrolase (GELP8), disease resistance protein (RGA3) sulfiredoxin-1 (OsSrx), LTPL129—Protease inhibitor/seed storage/LTP family protein precursor (ADR11) and phytosulfokine receptor (PSKRs) are involved in regulation of plant growth at seedling stage, stomatal movement and osmotic balance [9, 30,31,32,33]. The insilico analysis inturn supported the gene action of candidate genes through their expression in lemma, leaf, root, embryo and stem tissues. The genes AMP-binding domain containing protein (CL), Fascilin-like arabinogalactan precursor protein (FLA), PPR repeat containing protein (PPR), OsSub52—Putative Subtiliin homologue (OsSUB52) and STRUBBELIG-RECEPTOR FAMILY 3 precursor (SRF) are involved in regulating pollen development and spikelet fertility along with plant height, root length and grain yield [34,35,36,37,38]. Several genes belonging to transferase family proteins like transferase family protein (OsAt20), glutathione S-transferase GSTU6 (OsGSTU17), glycosyl transferase gene (OsGT1) and transporter family protein (OsTMTs) involved in transport of osmolytes and carbohydrates thereby regulates the growth of leaf, leaf sheath, pollen, stem and root tissues [21, 39, 40]. In addition to these F-box domain containing proteins, zinc finger family proteins, MAD box family genes and bhlh transcription factors [41] are also involved in stress signaling mechanisms. The gene OsSADR1 E3 ligase ADR11 acts as a negative regulator under drought conditions by the regulation of ABA-mediated stomatal closure [42]. The gene CSLA3—cellulose synthase-like family A; mannan synthase was involved in storage of carbohydrates, strengthens cell wall, seed hardiness [43].
In addition to these genes several other candidate genes were detected under individual MTAs involved in trait regulation imparting yield under moisture stress conditions as presented in Spp. Table. 11.
The SNP UChr1_20598769 was linked to traits SP, SL and RL. The gene LOC_Os01g37000 encodes protein of unknown function (DUF239) reported to protect the seedlings under abiotic stress conditions [25]. LOC_Os01g36930 encoding ubiquitin-specific processing protease 3 (UBP6) was known to regulate seedling survival percentage and growth under abiotic stress conditions [21]. Another gene LOC_Os01g36940 encoding OsFBOX13—F-box domain containing protein (OsFBox16) was involved in plant growth regulation [19]. The results were further confirmed through the root specific gene expression of as observed from RiceXPro. The SNP UChr11_3775843 was associated to traits SP and RL. LOC_Os11g07440 encodes plant neutral invertase domain containing protein (OsNIN6) involved in regulation of vacuolar invertases (VINs) attributed to roles in cell elongation of seedling hypocotyls in arabidopsis [44] and rapidly expanding tissues in carrot taproot [45]. UChr11_6656777 located on Chr. 11 was governing SP, SL and RL traits. LOC_Os11g11950 encoding disease resistance protein (RPM1) was involved in promotion of root growth [30]. However the gene action was mainly concentrated in leaf tissues as per Rice XPro database indicating the need of further functional analysis of gene. MTA UChr11_26816152 was linked to the traits SP and SL. The gene LOC_Os11g44310 encodes calmodulin binding protein (CaM) which encodes hypothetical conserved gene involved in microsporogenesis [38] and its role in plant survival needs a further analysis. The list of detected candidate genes for heat stress tolerance were presented in Spp. Table. 12.
Conclusion
In the current era of climate change, there is an increased demand for the development of varieties with abiotic stress resistance coupled with high yields in order to meet the ever increasing food demand. This can be accelerated through the combination of physiological profiling of plant traits combined with genetic analysis. The consistent MTAs along with the novel MTAs obtained in this study with high phenotypic variance can be transferred to high yielding varieties and parents of heterotic hybrids through MAB programs for improvement of moisture and heat stress tolerance. Besides, the promising MTAs can also serve as candidates for further fine mapping and map‐based cloning projects in future. Through pyramiding of pleiotropic consistent MTA clusters multiple trait improvement can be attained at the same time reducing the efforts to pyramid multiple minor MTAs. The in silico analysis of candidate genes revealed that several drought-responsive genes were associated with the grain yield under moisture stress conditions. In silico analysis of candidate genes associated with heat stress tolerance were found to be regulating root length and some genes were also having regulatory action in moisture stress and yield enhancement. Some uncharacterized genes were also observed whose function can be characterized and validated with the help of NGS technologies and can be exploited in transgenic rice development. The consistent and accurate MTA obtained in the study has indicated that association mapping can serve as a fascinating option to identify MTAs associated with major yield and its related traits by using genome-wide molecular markers.
Data availability
All the supporting data of the article has been provided in the supplementary files.
References
Pachauri, R.K and Meyer, L.A. 2014. Climate Change: Synthesis Report. Contribution of Working Groups I, II and III to the Fifth Assessment Report of the Intergovernmental Panel on Climate Change. Core Writing Team, Eds.; IPCC: Geneva, Switzerland, 151p.
Fahad S, Bajwa AA, Nazir U, Anjum SA, Farooq A, Zohaib A, Sadia S, Nasim W, Adkins S, Saud S, Ihsan MZ, Alharby H, Wu C, Wang D, Huang J (2017) Crop production under drought and heat stress: plant responses and management options. Frontiers of Plant Science 8:1147–1159
IPCC (2014). “Summary for Policymakers,” in Climate Change 2013: The Physical Science Basis. Contribution of Working Group I to the Fifth Assessment Report of the Intergovernmental Panel on Climate Change. T. F. Stocker, D. Qin, G. K. Plattner, M. Tignor, and Allen (eds). Cambridge University Press, Cambridge
Satake T, Yoshida S (1978) High temperature induced sterility in indica rice at flowering. Japanese J Crop Sci 47(1):6–17
Baker JT, Allen LH Jr, Boote KJ (1992) Response of rice to carbon dioxide and temperature. Agric For Meteorol 60(3–4):153–166
Sudhakar P, Latha P, Babu PR, Sujatha K, Reddy KR (2012) Identification of thermotolerant rice genotypes at seedling stage using tir technique in pursuit of global warming. Indian J Plant Physiol 7(2):185–188
Murray MG, Thompson WF (1980) Rapid isolation of high molecular weight plant DNA. Nucleic Acids Res 8:4321–4325
Pritchard JK, Stephens M, Donnelly P (2000) Inference of population structure using multi locus genotype data. Genetics 155:945–959
Chepyshko H, Lai CP, Huang LM, Liu JH, Shaw JF (2012) Multifunctionality and diversity of GDSL esterase/lipase gene family in rice (Oryza sativa L japonica) genome: new insights from bioinformatics analysis. BMC Genomics 13(309):1–19
Lipka AE, Tian F, Wang Q, Peiffer J, Li M, Bradbury PJ, Gore MA, Buckler ES, Zhang Z (2012) GAPIT: genome association and prediction integrated tool. Bioinformatics 28:2397–2399
Lin F, Fan J, Rhee SY (2019) QTG- Finder: a machine- learning based algorithm to prioritize causal genes of quantitative trait loci in Arabidopsis and rice. G3: Genes/Genomes/Genetics. 2019.
Venuprasad R, Lafitte HR, Atlin GN (2007) Response to direct selection for grain yield under drought stress in rice. Crop Sci 47:285–293
Vikram P, Swamy BP, Dixit S, Ahmed H, Sta-Cruz MT, Singh A, Kumar A (2011) ’qDTY1.1, a major QTL for rice grain yield under reproductive-stage drought stress with a consistent effect in multiple elite genetic backgrounds. BMC Genet 12(89):1–15
Beena R, Kirubakaran S, Nithya N, Manickavelu A, Sah RP, Abida PS, Sreekumar J, Jaslam PM, Rejeth R, Jayalakshmy VG, Roy S, Manju RV, Viji MM, Siddique KHM (2021) Association mapping of drought tolerance and agronomic traits in rice (Oryza sativa L.) landraces. BMC Plant Biol 21(484):1–21
Melandri G, Prashar A, McCouch SR, Linden GV, Jones HG, Kadam N, Jagadish K, Bouwmeester H, Ruyter-Spira C (2020) Association mapping and genetic dissection of drought-induced canopy temperature differences in rice. J Exp Bot 71(4):1614–1627
Yu SB, Li JX, Xu CG, Tan YF, Gao YJ, Li XH, Zhang Q, Maroof MAS (1997) Importance of epistasis as the genetic basis of heterosis in an elite rice hybrid. PNAS 94:9226–9231
Sasahara H, Fukuta Y, Fukuyama T (1999) Mapping of QTLs for vascular bundle system and spike morphology in rice, Oryza sativa L. Breed Sci 49:75–81
Giri J, Vij S, Dansana PK, Tyagi AK (2011) Rice A20/AN1 zinc-finger containing stress-associated proteins (SAP1/11) and a receptor-like cytoplasmic kinase (OsRLCK253) interact via A20 zinc-finger and confer abiotic stress tolerance in transgenic arabidopsis plants. New Phytol 191(3):721–732
Jain M, Nijhawan A, Arora R, Agarwal P, Ray S, Sharma P, Kapoor S, Tyagi AK, Khurana JP (2007) F-box proteins in rice. Genome-wide analysis, classification, temporal and spatial gene expression during panicle and seed development, and regulation by light and abiotic stress. Plant Physiol 143(4):1467–1483
Kumar M, Choi J, An G, Kim SR (2017) Ectopic expression of OsSta2 Enhances salt stress tolerance in rice. Front Plant Sci 8(316):1–14
Moon YK, Hong JP, Cho YC, Yang SJ, An G, Kim TK (2009) Structure and expression of OsUBP6, an ubiquitin-specific protease 6 homolog in rice (Oryza sativa L). Mol Cells 28(5):463–472
Cai Y, Li S, Jiao G, Sheng Z, Wu Y, Shao G, Xie L, Peng C, Xu J, Tang S, Wei X, Hu P (2018) OsPK2 encodes a plastidic pyruvate kinase involved in rice endosperm starch synthesis, compound granule formation and grain filling. Plant Biotechnol J 16(11):1878–1891
Hong Z, Ueguchi-Tanaka M, Shimizu-Sato S, Inukai Y, Fujioka S, Shimada Y, Takatsuto S, Agetsuma M, Yoshida S, Watanabe Y (2002) Loss-of-function of a rice brassinosteroid biosynthetic enzyme, C-6 oxidase, prevents the organized arrangement and polar elongation of cells in the leaves and stem. Plant J 32:495–508
Jiang M, Liu Y, Li R, Li S, Tan Y, Huang J, Shu Q (2020) An inositol 1, 3, 4, 5, 6-pentakisphosphate 2-kinase 1 mutant with a 33-nt deletion showed enhanced tolerance to salt and drought stress in rice. Plants 10(23):1–16
Luhua S, Ciftci-Yilmaz S, Harper J, Cushman J, Mittler R (2008) Enhanced tolerance to oxidative stress in transgenic arabidopsis plants expressing proteins of unknown function. Plant Physiol 148:280–292
Pandian BA, Sathishraj R, Djanaguiraman M, Prasad PVV, Jugulam M (2020) Role of cytochrome P450 enzymes in plant stress response. Antioxidants 9(454):1–15
Jung SE, Bang SW, Kim SH, Seo JS, Yoon HB, Kim YS, Kim JK (2021) Overexpression of OsERF83, a vascular tissue-specific transcription factor gene, confers drought tolerance in rice. Int J Mol Sci 22(14):1–20
Akama K, Akihiro T, Kitagawa M, Takaiwa F (2001) Rice (Oryza sativa) contains a novel isoform of glutamate decarboxylase that lacks an authentic calmodulin-binding domain at the C-terminus. Biochem Biophys Acta 1522(3):143–150
Jiang M, Hu H, Kai J, Traw MB, Yang S, Zhang X (2019) Different knockout genotypes of OsIAA23 in rice using CRISPR/Cas9 generating different phenotypes. Plant Mol Biol 100(5):467–479
Yuan X, Wang Z, Huang J, Xuan H, Gao Z (2019) Phospholipidase Dδ negatively regulates the function of resistance to pseudomonas Syringae Pv. Maculicola 1 (RPM1). Front Plant Sci 9:1–9
Liang W-H, Wang H-H, Li H, Wang J-J, Yang D-D, Hao Y-F, Li J-J, Lou C, Lin Q-T, Hou C-Q (2014) Isolation and characterization of OsMY1, a putative partner of OsRac5 from Oryza sativa L. Mol Biol Rep 41:1829–1836
Liu XP, Liu XY, Zhang J, Xia ZL, Liu X, Huan JQ, Wang DW (2006) Molecular and functional characterization of sulfiredoxin homologs from higher plants. Cell Res 16(3):287–296
Wen BW, Xing MQ, Zhang H, Dai C, Xue HW (2011) Rice Homeobox Transcription Factor HOX1a positively regulates gibberellin responses by directly suppressing EL1. J Integr Plant Biol 53(11):869–878
Gui J, Shen J, Li L (2011) Functional characterization of evolutionarily divergent 4-Coumarate: coenzyme a ligases in rice. Plant Physiol 157(2):574–586
Li J, Yu M, Geng LL, Zhao J (2010) The fasciclin-like arabinogalactan protein gene, FLA3, is involved in microspore development of Arabidopsis. Plant J 64:482–497
Li P, Xue H (2011) Structural characterization and expression pattern analysis of the rice PLT gene family. Acta Biochim Biophys Sin 43(9):688–697
Sharma R, Agarwal P, Ray S, Deveshwar P, Sharma P, Sharma N, Nijhawan A, Jain M, Singh AK, Singh VP, Khurana JP, Tyagi AK, Kapoor S (2012) Expression dynamics of metabolic and regulatory components across stages of panicle and seed development in indica rice. Funct Integr Genomics 12(2):229–248
Zhang L, Liu BF, Liang S, Jones RL, Lu YT (2002) Molecular and biochemical characterization of a calcium/calmodulin-binding protein kinase from rice. Biochem J 368(1):145–157
Bartley LE, Peck ML, Kim SR, Ebert B, Manisseri C, Chiniquy DM, Sykes R, Gao L, Rautengarten C, Vega-Sanchez ME, Benke PI, Canlas PE, Cao P, Brewer S, Lin F, Smith WL, Zhang X, Keasling JD, Jentoff RE, Foster SB, Zhou J, Ziebell A, An G, Scheller HV, Ronald PC (2013) Overexpression of a BAHD acyltransferase, OsAt10, alters rice cell wall hydroxycinnamic acid content and saccharification. Plant Physiol 161(4):1615–1633
Yang X, Sun W, Liu J, Liu Y, Zeng Q (2009) Biochemical and physiological characterization of a tau class glutathione transferase from rice (Oryza sativa). Plant Physiol Biochem 47(11–12):1061–1068
Mukopadhyay P, Tyagi AK (2015) OsTCP19 influences developmental and abiotic stress signaling by modulating ABI4-mediated pathways. Nat Sci Rep 5(1):1–11
Park YC, Chapagain S, Jang CS (2018) A negative regulator in response to salinity in rice: Oryza sativa salt-, ABA- and drought-induced RING finger protein 1 (OsSADR1). Plant Cell Physiol. https://doi.org/10.1093/pcp/pcy009
Liepman AH, Cavalier DM (2012) The CELLULOSE SYNTHASE- LIKE A and CELLULOSE SYNTHASE- LIKE C families: recent advances and future perspectives. Front Plant Sci 3(109):1–7
Sergeeva LI, Keurentjes JJB, Bentsink L, Vonk J, Van-der-Plas LHW, Koornneef M, Vreugdenhil D (2006) Vacuolar invertase regulates elongation of Arabidopsis thaliana roots as revealed by QTL and mutant analysis. Proc Natl Acad Sci 103:2994–2999
Tang GQ, Luscher M, Sturm A (1999) Antisense repression of vacuolar and cell wall invertase in transgenic carrot alters early plant development and sucrose partitioning. Plant Cell 11:177–190
Funding
Madhavilatha Kommana gratefully acknowledge to Bayer for providing fellowship.
Author information
Authors and Affiliations
Contributions
LRV, DMR and MLK conceived the experiment and prepared the manuscript. MLK, LRV, DMR, BVBR and PS conducted the experiment. KA, MVKN, DMW, BRY, KM, MB, EA assisted in field work and LK, SL, NC, VBRL has carried out data analysis.
Corresponding author
Ethics declarations
Conflict of interest
Authors declare that they do not have any conflict of interest.
Ethical approval
This is an observational study. The Research Ethics Committee of the University has confirmed that no ethical approval is required.
Consent to participate
Informed consent was obtained from all individual participants included in the study.
Consent to publication
As there is no personal data of the authors in the article, no ethical approval is required.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Below is the link to the electronic supplementary material.
Rights and permissions
Springer Nature or its licensor (e.g. a society or other partner) holds exclusive rights to this article under a publishing agreement with the author(s) or other rightsholder(s); author self-archiving of the accepted manuscript version of this article is solely governed by the terms of such publishing agreement and applicable law.
About this article

Cite this article
Kommana, M., Reddy, D.M., Amarnath, K. et al. Identification of genomic regions governing moisture and heat stress tolerance employing association mapping in rice (Oryza sativa L.). Mol Biol Rep 50, 1499–1515 (2023). https://doi.org/10.1007/s11033-022-08153-y
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s11033-022-08153-y