
Abstract
Background
The Cas9 nuclease is delivered in the form of either Cas9 protein or mRNA along with CRISPR guide RNA (gRNA: dual-crRNA:tracrRNA or chimeric single-guide RNA) or in a plasmid package encoding both Cas9 and the CRISPR gRNA.
Methods and results
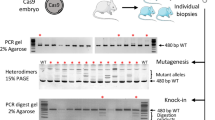
We directly compared the efficiency of producing rat blastocysts with homozygous mutations of the Foxn1 locus by pronuclear injection of Cas9 in the form of protein, mRNA, or plasmid DNA. For highly efficient production of rat blastocysts with homozygous Foxn1 mutations, pronuclear injection of Cas9 protein at 60 ng/µl was likely optimal. While blastocyst harvest in the mRNA groups was higher than those in the protein and plasmid DNA groups, genotype analysis showed that 63.6%, 8.7–20.0%, and 25.0% of the analyzed blastocysts were homozygous mutants in the protein, mRNA, and plasmid DNA groups, respectively. The high efficiency of producing homozygous mutant blastocysts in the 60 ng/µl protein group may be associated with primary genome editing being initiated before the first cleavage. In most cases, homozygous mutations at the target Foxn1 locus are triggered by deletion and repair via nonhomologous end joining or microhomology-mediated end joining. Deletion downstream of the Cas9 break site was more likely than deletion in the upstream direction.
Conclusions
The Cas9 nuclease in protein form, when coinjected with the CRISPR gRNA (ribonucleoprotein) into a rat zygote pronucleus, can access the target genome site and induce double-strand breaks promptly, resulting in the efficient production of homozygous mutants.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Homozygous knockout (KO) rodents, which can be used for investigating gene functions of interest, can be produced by a conventional approach using pluripotent stem cells (PSCs). However, the PSC-based technique is time-consuming and labor-intensive as it requires the construction of a targeting vector, selection of KO PSCs after homologous recombination, generation of germline-competent chimeras by blastocyst injection, and establishment of homozygous KO animals by sib-mating of next generation offspring [1, 2]. The production of homozygous KO rodents has been efficiently facilitated by pronuclear injection of new genome editing tools, such as zinc-finger nucleases [3], transcription activator-like effector nucleases [4,5,6], and the clustered regularly interspaced short palindromic repeat (CRISPR)/CRISPR-associated proteins (Cas) system [7,8,9]. These genome-editing tools can generate targeted mutations by inducing DNA double-strand breaks (DSBs) and error-prone repair by nonhomologous end joining (NHEJ) or microhomology-mediated end joining (MMEJ) [10].
The CRISPR/Cas system is the most cost-effective and reproducible among these new genome-editing tools. The Cas9 endonuclease from Streptococcus pyogenes type II, coinjected with CRISPR RNA (crRNA) and trans-activating crRNA (tracrRNA), can form ribonucleoprotein (RNP) complexes and induce DSBs at the target DNA [11, 12]. The Cas9 endonuclease can be programmed with guide RNA (gRNA), such as a dual-crRNA:tracrRNA or a chimeric single-guide RNA (sgRNA) which fuses crRNA with tracrRNA [12]. The Cas9 endonuclease is delivered either as a recombinant protein [13,14,15,16] or mRNA [9, 17] along with CRISPR gRNA or in an all-in-one plasmid package that encodes both Cas9 and the CRISPR gRNA (mouse: [18, 19]; rat: [20]).
One of the problems in transgenic studies associated with pronuclear injection is the incidence of somatic mosaicism [21, 22], yet little is known about whether the form of CRISPR/Cas9 delivered via pronuclear injection has any effect on the rate of homozygous mutant production. In the present study, we directly compared the efficiency of producing rat blastocysts with homozygous mutations in the Forkhead-Box N1 (Foxn1) locus by pronuclear injection of Cas9 in one of three forms: protein, mRNA, and plasmid DNA. Foxn1, which regulates thymus epithelial lineage specification during organogenesis [23], was used as a model gene.
Materials and methods
Materials
Recombinant Cas9 protein (CP01, PNA Bio, Newbury Park, CA, USA) and Cas9 mRNA (1EA, Sigma–Aldrich, St. Louis, MO, USA) were stored at − 80 °C until pronuclear injection. The gRNA (5′-GAC TGG AGG GCG AAC CCC AA-3′) and all-in-one CRISPR–Cas9 pX330 plasmids for the Foxn1 gene were designed and constructed as described previously [24]. Foxn1-sgRNA, Foxn1-crRNA and tracrRNA (Fasmac, Kanagawa, Japan) were chemically synthesized, purified by polyacrylamide gel electrophoresis, diluted with nuclease-free 0.1 × TE buffer, and stored at − 80 °C until injection.
Animals
Specific pathogen-free Wistar rats (Crlj:WI) were purchased from Charles River Laboratories, Japan Inc. (Kanagawa, Japan). All rats were housed in an environmentally controlled room with a 12-h dark/12-h light cycle at a temperature of 23 ± 2 °C and humidity of 55 ± 5% and given free access to a laboratory diet (CE-2; CLEA Japan Inc., Tokyo, Japan) and filtered water. All procedures for animal experimentation were reviewed and approved by the Animal Care and Use Committee of the National Institutes of Natural Sciences (Nos. 15A102 and P09-070-A).
Pronuclear injection
Female rats at 4 weeks old were superovulated with 300 IU/kg equine chorionic gonadotropin (ASKA Pharmaceutical, Tokyo, Japan) and 300 IU/kg human chorionic gonadotropin (hCG; ASKA Pharmaceutical) at an interval of 46–50 h. Pronuclear stage zygotes were harvested from female rats that had been mated with a fertile male at 28–30 h after hCG administration. One of the following mixtures was injected into a male pronucleus: [1] Cas9 protein (100 ng/µl) with crRNA (25 ng/µl):tracrRNA (25 ng/µl), [2] Cas9 protein (60 ng/µl) with crRNA (15 ng/µl):tracrRNA (15 ng/µl), [3] Cas9 protein (30 ng/µl) with crRNA (12.5 ng/µl):tracrRNA (12.5 ng/µl), [4] plasmid DNA pX330 (5 ng/µl), [5] Cas9 mRNA (100 ng/µl) with Foxn1 sgRNA (50 ng/µl), or [6] Cas9 mRNA (100 ng/µl) with crRNA (25 ng/µl):tracrRNA (25 ng/µl). The injected zygotes were cultured for 16–19 h in modified Krebs–Ringer bicarbonate (mKRB) solution at 37 °C under 5% CO2 in air. The surviving zygotes were transferred into the oviductal ampullae of pseudopregnant Crlj:WI recipient rats (≥ 10 weeks old) anesthetized with isoflurane 2–2.5% in oxygen at 0.5 days post-coitum (dpc) (< 80 embryos per recipient). The recipient rats were sedated by carbon dioxide inhalation and sacrificed by cervical dislocation at 4.5 dpc. Blastocysts were harvested by oviduct-uterine flushing with the mKRB solution. In an additional experiment to determine the timing of genome editing, 1-cell stage embryos were collected at 3 or 6 h postinjection (hpi), and 2-cell stage embryos were collected at 19 hpi.
Genotyping
Genomic DNA was isolated from embryos at the 1-cell (3–6 hpi), 2-cell (19 hpi) or blastocyst stage (4.5 dpc) according to a previously described protocol [25] with a few modifications. Briefly, 1 µl of PBS containing a single embryo was placed on the wall near the bottom of a 200-µl PCR tube using a glass micropipette under a stereomicroscope. The samples were stored at − 80 °C until genotyping.
Whole-genome amplification of the DNA from a single blastocyst was performed using REPLI-g Single Cell Kit (QIAGEN, Hilden, Germany) according to the manufacturer’s manual. Briefly, 4 µl of crude DNA solution was mixed with 3 µl of Buffer D and incubated at 65 °C for 10 min. The reaction was interrupted by adding 3 µl of stop solution. Finally, 40 µl of REPLI-g solution was added to the mixture and further incubated at 30 °C for 16 h and at 60 °C for 3 min. For PCR using amplified genomic DNA, 2 µl of the genomic DNA solution was mixed with 0.4 µl each of 10 µM primers specific for the Foxn1 gene (Fwd: 5′-CAG GAC TGG GTG ATG GTG TC-3′, Rev: 5′-ACG GGG TTC CAT ATC TTG CC-3′), 10 µl of 2× AmpliTaq Gold 360® master mix (Life Technologies Japan, Tokyo, Japan) and 7.2 µl of ultra-pure water. The mixture was subjected to PCR under the following conditions: 5 min at 95 °C; 35 cycles of 30 s at 95 °C, 30 s at 60 °C, and 40 s at 68 °C; and 2 min at 68 °C.
To genotype the 1-cell and 2-cell stage embryos, nested PCR was performed using Tks Gflex™ DNA Polymerase Low DNA (Takara Bio Inc., Shiga, Japan). The sample in 1 µl PBS was mixed with 2 µl of distilled water for Low DNA, spun-down and then boiled at 95 °C for 20 min. The resulting crude genomic DNA solution (3 µl) was mixed with 0.2 µl each of 10 µM primers specific for the Foxn1 gene (Fwd: 5′-GTT GGC CAC ACC TAG ACG TT-3′, Rev: 5′-ACG GGG TTC CAT ATC TTG CC-3′), 5 µl of 2 × Tks Gflex™ DNA Polymerase Low DNA and 1.6 µl of ultra-pure water. The mixture was subjected to PCR under the following conditions: 1 min at 95 °C; 40 cycles of 10 s at 98 °C, 15 s at 60 °C, and 1 min at 68 °C; and 5 min at 68 °C. The second round of PCR was conducted as described above, except for the use of a different primer set (Fwd: 5′-CAG GAC TGG GTG ATG GTG TC-3′, Rev: 5′-ACG GGG TTC CAT ATC TTG CC-3′) and 2 µl of 1/20-diluted first-round PCR solution as the DNA template.
The PCR products were purified by ethanol precipitation and sequenced to screen for mutations using the primer (5′-ACG GGG TTC CAT ATC TTG CC-3′) by the Value Read sequence service (Eurofins Genomics, Tokyo, Japan). When this first screening was positive, the PCR products were cloned into the pCR2.1 vector using the TA Cloning® Kit (Invitrogen, CA, USA) and transformed into DH5α competent Escherichia coli (TOYOBO, Osaka, Japan). Ten plasmid clones from each PCR product were sequenced using the M13 primer. An embryo in which all 10 genotyped clones had the same mutations was defined as a homozygous mutant.
Pattern analysis of homozygous mutants
After induction of DSBs at the target Foxn1 site by the CRISPR/Cas9 system, a 1–3 bp deletion or 1 bp insertion is induced via the NHEJ repair pathway, or deletions using microhomologies of ≥ 2 bases are frequently induced via the MMEJ repair pathway [26]. Of 3/6 hpi 1-cell and 19 hpi 2-cell stage embryos and 4.5 dpc blastocysts, the homozygous induced mutations, in which all 10 of the clones have the same mutation as mentioned above, were first classified as deletion, insertion, or deletion plus insertion. Then, deletion mutations were classified as either NHEJ or MMEJ. In addition, the preference of the deletion direction from the Cas9 break site occurring between the 3rd and 4th bases upstream of the NGG PAM sequence was investigated in in-del mutants. In this context, the microhomology arms were equally divided in both the 5′ and 3′ directions. The maximum detectable size of the total, 5′, and 3′ deletions were 306, 61, and 245 bp, respectively.
Statistics
Fisher’s exact test followed by Benjamini and Hochberg false discovery rate correction was used to determine the significance of differences in the number of survived embryos, blastocysts, total mutant blastocysts, and homozygous blastocysts among groups [1]–[3] and [2], [4]–[6] in Tables 1 and 2, respectively. The same statistical test was used to determine the significance of differences in the number of total mutants and homozygous mutants in 3/6 hpi 1-cell and 19 hpi 2-cell stage embryos among groups [2], [4]–[6] in Table 3 (R version 4.1.3, https://www.r-project.org). Differences were considered to be significant when the P value was less than 0.05.
Results
Homozygous mutations of the Foxn1 locus in rat blastocysts
We firstly investigated the efficiency of producing rat blastocysts with homozygous mutations in the Foxn1 locus by pronuclear injection of Cas9 protein (group [1]–[3]: 30, 60, or 100 ng/µl) with crRNA:tracrRNA (Table 1). In this context, the form of guide RNA was fixed to crRNA:tracrRNA, because it is well-known crRNA:tracrRNA is more efficient in the genome-editing than sgRNA when Cas9 protein is used for pronuclear injection [14, 27]. Postinjection zygote survival (81.8–86.8%) did not differ among 30–100 ng/µl protein concentrations, while blastocyst harvest in the intermediate (60 ng/µl) and low (30 ng/µl) Cas9 protein concentration groups (16.7–17.2%) was higher than that in the high (100 ng/µl) concentration group (3.3%). Of the PCR-positive blastocysts, 69.2–100.0% and 30.8–63.6% were mutants and homozygous mutants, respectively. Overall, pronuclear injection of RNP solution at an intermediate concentration (60 ng/µl Cas9 protein + 15 ng/µl crRNA + 15 ng/µl tracrRNA) was likely optimal for producing rat blastocysts with homozygous Foxn1 mutations at high efficiency.
Next, we compared the efficiency of producing rat blastocysts with homozygous mutations in the Foxn1 locus by pronuclear injection of Cas9 protein (group [2]) with crRNA:tracrRNA, Cas9 mRNA with sgRNA or crRNA:tracrRNA (group [4] or [5]), or plasmid DNA pX330 (group [6]) (Table 2). Postinjection zygote survival in the protein and mRNA groups (86.8–95.1%) was higher than that in the plasmid DNA group (62.5%), while blastocyst harvest in the mRNA groups (29.3–46.6%) was higher than those in the protein and plasmid DNA groups (5.0–14.5%). Genotype analysis showed that 100.0, 75.0, 13.0, and 100.0% of the PCR-positive blastocysts were mutants in the protein, mRNA with sgRNA or crRNA:tracrRNA, and plasmid DNA groups, respectively. Of the PCR-positive blastocysts, 63.6, 20.0, 8.7, and 25.0% were homozygous mutants in the protein, mRNA with sgRNA or crRNA:tracrRNA, and plasmid DNA groups, respectively. The efficiency of producing rat blastocysts with homozygous mutations in the Foxn1 locus by pronuclear injection of Cas9 protein with crRNA:tracrRNA (blastocyst harvest rate: 14.5% × homozygous mutant blastocyst rate: 63.6% = 9.2%) and Cas9 mRNA with sgRNA (46.6% × 20.0% = 9.3%) was higher than Cas9 mRNA with crRNA:tracrRNA (29.3% × 8.7% = 2.5%) and plasmid DNA pX330 (5.0% × 25.0% = 1.3%). Considering the time and cost of genotyping analysis, pronuclear injection of RNP solution at an intermediate Cas9 protein concentration (group [2]) was likely optimal for producing homozygous Foxn1 mutant blastocysts at high efficiency, followed by mRNA with sgRNA injection (group [4]).
Initiation of genome editing in 1-cell/2-cell stage embryos
Genome editing of early stage embryos (1-cell; 3 or 6 hpi, 2-cell; 19 hpi) via plasmid DNA, mRNA (crRNA:tracrRNA and sgRNA), and protein injection was investigated (Table 3). The proportions of homozygous mutations in 2-cell stage embryos seemed to be correlated with those in blastocysts, as shown in Table 2. Some 1-cell stage zygotes (2.6–0.3%) were found to be homozygous mutants as early as 3 or 6 h after protein + crRNA:tracrRNA injection, while no homozygous mutations were observed in 1-cell stage zygotes after plasmid DNA or mRNA + crRNA:tracrRNA/sgRNA injection.
Pattern analysis of homozygous mutants in 1-cell/2-cell stage embryos and blastocysts
The homozygous induced mutations in 3/6 hpi 1-cell and 19 hpi 2-cell stage embryos (1 + 3 + 18 = 22 in group [2] and 0 + 0 + 7 = 7 in group [4], shown in Table 3) and 4.5 dpc blastocysts (7 in group [2] and 8 in group [4], shown in Table 2) were first classified as deletion, insertion, or deletion plus insertion (Fig. 1A). Regardless of the Cas9 delivery format (mRNA + sgRNA or protein + crRNA:tracrRNA), homozygous mutations were triggered by a deletion in most cases (Fig. 1B). The repair of DSBs at the target Foxn1 locus was induced dominantly via the NHEJ pathway in homozygous mutants in the mRNA + sgRNA group and to a lesser extent in those in the protein + crRNA:tracrRNA group (Fig. 1B). Based on the deletion length downstream versus upstream of the Cas9 break site, bias toward deletion in the 3' direction was detected in the deletion mutants, especially those with > 20 bp deletions (Fig. 1C). In the mRNA + sgRNA group and protein + crRNA:tracrRNA group, the deleted sequences in the 3' region occupied 73.0% and 85.1% of the total deletions (12.5/17.1 bp and 22.3/26.3 bp) on average, respectively.

Pattern analysis of homozygous mutations in rat 1-/2-cell and blastocyst stage embryos after pronuclear injection of Cas9 mRNA + sgRNA or Cas9 protein (60 ng/µl) + crRNA:tracrRNA. A Sequence analysis of homozygous mutations in 1-/2-cell and blastocyst stage embryos in Cas9 mRNA + sgRNA and Cas9 protein (60 ng/µl) + crRNA:tracrRNA groups. The 30 bases in a box indicate the target 20 bases (underlined) and 5 bases each before and after the target. Columns from left to right: sequence information, number of editing bases, deletion (Del or D) and/or insertion (Ins or I), and microhomology-mediated end joining (MMEJ) or nonhomologous end joining (NHEJ) for deletion. Red and blue letters indicate microhomology and inserted bases, respectively. *Microhomology of this mutant is the upstream CAGGCTTC. B Upper panel; Ins and/or Del frequency in homozygous mutants. Lower panel; NHEJ and MMEJ ratio in homozygous mutants. C 5′ and 3′ deletion lengths of deletion mutants. Closed symbols; Cas9 mRNA + sgRNA, open symbols; Cas9 protein (60 ng/ul) + crRNA:tracrRNA. Plots under the Y = X dot line represent the preferred deletion from the Cas9 break site in the 3′ direction rather than the 5′ direction. The maximum detectable size of the total, 5′, and 3′ deletions are 306, 61, and 245 bp, respectively. (Colour figure online)
Discussion
Our study demonstrates that homozygous Foxn1 mutation is effectively induced by delivering Cas9 endonuclease as a protein along with gRNA (assembled RNP complex) into pronuclear stage rat zygotes, and that RNP injection mainly induces homozygous in-del mutations via MMEJ before or during the first cleavage of injected zygotes. Some 1-cell zygotes are genome-edited shortly after pronuclear injection of the RNP, unlike the case for plasmid and mRNA injection (Table 3), suggesting the prompt availability of enzymatic activity of Cas9 protein when the transcription and translation processes can be bypassed. In previous studies, Cas9 protein evoked DNA cleavage in cultured human cells at 1 h posttransfection [28], and Cas9 protein coinjected with gRNA and single-stranded oligodeoxynucleotides (ssODN) was superior to plasmid plus ssODN in generating knock-in mice [16], both of which agree with our findings. Interestingly, homozygous mutations were observed in approximately half of both the Foxn1-edited blastocysts (56.5%, 13/23; Table 1) and 2-cell embryos (50.0%, 18/36; Table 3) that received Cas9 protein. On the other hand, the delivery of Cas9 mRNA resulted in low editing efficiencies in embryos at the 2-cell stage (13.3%, 10/75; Table 2) and a low proportion of homozygous mutant blastocysts among the total mutants (30.3%, 10/33; Table 1). This may suggest that delayed activity of the introduced CRISPR/Cas9 system leads to a higher frequency of monoallelic mutation or mosaicism. Moreover, it was reported that Cas9 protein/sgRNA introduction into human, mouse, and zebrafish cells reduced off-target effects but not on-target digestion [13, 28].
Optimization of the RNP injection is likely to be required for different animal species and strains, as well as individual target genes. Dose-dependent cytotoxicity of the injected RNP was observed in the blastocyst harvest of our study (Table 1). Ménoret et al. [15] reported that Cas9 protein (3 µM)/sgRNA (3 µM) injection into rat zygotes induced indel mutations at the Rosa26 locus more efficiently than Cas9 mRNA (50 ng/µl)/sgRNA (10 ng/µl) injection, but a higher concentration of Cas9 protein (6 µM)/sgRNA (6 µM) resulted in impaired fetal development after transfer. In the same study, Cas9 protein (1.5 µM)/sgRNA (3 µM) injection and Cas9 mRNA (20 ng/µl)/sgRNA (10 ng/µl) injection into mouse zygotes induced Rosa26 indel mutations at a similar efficiency, and genome-editing efficiency at the rat Foxp3 or Anks3 locus was comparable between the Cas9 protein and mRNA delivery groups [15]. Ma et al. [29] reported that the rat Dbndd1 locus was edited equally by pronuclear injection of Cas9 protein (30 ng/µl)/sgRNA (10 ng/µl) versus Cas9 mRNA (25 ng/µl)/sgRNA (10 ng/µl).
The type of gRNA (crRNA:tracrRNA or sgRNA) delivered via pronuclear injection could impact the efficiency of genome editing. The coinjection of crRNA:tracrRNA with Cas9 mRNA had a negative impact on the efficiency of genome editing in both 2-cell stage embryos and blastocysts (Tables 2, 3). Genome-editing efficiency in mouse offspring was influenced by the format of gRNA introduction in coinjection with Cas9 protein, as crRNA:tracrRNA resulted in higher genome-editing efficiency than sgRNA [14, 27], which is contradictory to our results when using Cas9 mRNA. No direct comparison of the gRNA format for rat genome editing was conducted by coinjection with Cas9 protein in our study. Zygotic genome activation in rodents occurs around the time of the first embryonic cleavage, and the associated dramatic change in gene expression may affect the translation of Cas9 mRNA and RNP formation.
Mutation pattern analysis in our study revealed that pronuclear RNP injection preferentially induced a homozygous deletion via the MMEJ pathway when compared with mRNA/sgRNA injection (Fig. 1). The pronuclear stage of 1-cell zygotes is morphologically classified as “PN3” [27, 30]; that is, the MMEJ pathway is predominant in these cells [31]. MMEJ has been considered more appropriate for precise genome editing to generate KO mutations than homology-directed repair or NHEJ [32, 33]. MMEJ has been used to generate high-throughput knock-in mice through precise integration into the target chromosome system, which is enhanced by a combination with exonuclease 1 [34]. The bias toward deletion in the 3′ direction from the Cas9 break site has been reported in the case of Cas9 mRNA and sgRNA injection into rat zygotes [35]. The current study adds to our knowledge, confirming the 3′ deletion preference for Cas9 delivered as both mRNA and protein.
In conclusion, homozygous Foxn1-mutated rat blastocysts can be produced efficiently by microinjection of CRISPR/Cas9 materials as RNPs (recombinant Cas9 protein + crRNA:tracrRNA) into pronuclear stage zygotes. Since the Cas9 protein becomes enzymatically active earlier than Cas9 mRNA or plasmid by bypassing the transcription and translation processes, DSB induction and editing at the Foxn1 locus before the first cleavage of injected zygotes may be facilitated. The optimal concentrations of the Cas9 protein and gRNA should be further investigated for different rodent species and target DNA loci.
References
Tong C, Huang G, Ashton C, Li P, Ying QL (2011) Generating gene knockout rats by homologous recombination in embryonic stem cells. Nat Protoc 6:827–844. https://doi.org/10.1038/nprot.2011.338
Behringer R, Gertsenstein M, Nagy KV, Nagy A (2014) Manipulating the mouse embryo: a laboratory manual, 4th edn. Cold Spring Harbor Laboratory Press, New York
Geurts AM, Cost GJ, Freyvert Y et al (2009) Knockout rats via embryo microinjection of zinc-finger nucleases. Science 325:433. https://doi.org/10.1126/science.1172447
Tesson L, Usal C, Ménoret S et al (2011) Knockout rats generated by embryo microinjection of TALENs. Nat Biotechnol 29:695–696. https://doi.org/10.1038/nbt.1940
Mashimo T, Kaneko T, Sakuma T et al (2013) Efficient gene targeting by TAL effector nucleases coinjected with exonucleases in zygotes. Sci Rep 3:1253. https://doi.org/10.1038/srep01253
Sung YH, Baek IJ, Kim DH et al (2013) Knockout mice created by TALEN-mediated gene targeting. Nat Biotechnol 31:23–24. https://doi.org/10.1038/nbt.2477
Li W, Teng F, Li T, Zhou Q (2013) Simultaneous generation and germline transmission of multiple gene mutations in rat using CRISPR-Cas systems. Nat Biotechnol 31:684–686. https://doi.org/10.1038/nbt.2652
Shen B, Zhang J, Wu H et al (2013) Generation of gene-modified mice via Cas9/RNA-mediated gene targeting. Cell Res 23:720–723. https://doi.org/10.1038/cr.2013.46
Wang H, Yang H, Shivalila CS et al (2013) One-step generation of mice carrying mutations in multiple genes by CRISPR/Cas-mediated genome engineering. Cell 153:910–918. https://doi.org/10.1016/j.cell.2013.04.025
Yeh CD, Richardson CD, Corn JE (2019) Advances in genome editing through control of DNA repair pathways. Nat Cell Biol 21:1468–1478. https://doi.org/10.1038/s41556-019-0425-z
Gasiunas G, Barrangou R, Horvath P, Siksnys V (2012) Cas9-crRNA ribonucleoprotein complex mediates specific DNA cleavage for adaptive immunity in bacteria. Proc Natl Acad Sci USA 109:E2579–E2586. https://doi.org/10.1073/pnas.1208507109
Jinek M, Chylinski K, Fonfara I, Hauer M, Doudna JA, Charpentier E (2012) A programmable dual-RNA-guided DNA endonuclease in adaptive bacterial immunity. Science 337:816–821. https://doi.org/10.1126/science.1225829
Sung YH, Kim JM, Kim HT et al (2014) Highly efficient gene knockout in mice and zebrafish with RNA-guided endonucleases. Genome Res 24:125–131. https://doi.org/10.1101/gr.163394.113
Aida T, Chiyo K, Usami T et al (2015) Cloning-free CRISPR/Cas system facilitates functional cassette knock-in in mice. Genome Biol 16:87. https://doi.org/10.1186/s13059-015-0653-x
Ménoret S, De Cian A, Tesson L et al (2015) Homology-directed repair in rodent zygotes using Cas9 and TALEN engineered proteins. Sci Rep 5:14410. https://doi.org/10.1038/srep14410
Nakagawa Y, Sakuma T, Nishimichi N et al (2016) Ultra-superovulation for the CRISPR-Cas9-mediated production of gene-knockout, single-amino-acid-substituted, and floxed mice. Biol Open 5:1142–1148. https://doi.org/10.1242/bio.019349
Yoshimi K, Kaneko T, Voigt B, Mashimo T (2014) Allele-specific genome editing and correction of disease-associated phenotypes in rats using the CRISPR-Cas platform. Nat Commun 5:4240. https://doi.org/10.1038/ncomms5240
Li D, Qiu Z, Shao Y et al (2013) Heritable gene targeting in the mouse and rat using a CRISPR-Cas system. Nat Biotechnol 31:681–683. https://doi.org/10.1038/nbt.2661
Mashiko D, Fujihara Y, Satouh Y, Miyata H, Isotani A, Ikawa M (2013) Generation of mutant mice by pronuclear injection of circular plasmid expressing Cas9 and single guided RNA. Sci Rep 3:3355. https://doi.org/10.1038/srep03355
Goto T, Hara H, Sanbo M et al (2019) Generation of pluripotent stem cell-derived mouse kidneys in Sall1-targeted anephric rats. Nat Commun 10:451. https://doi.org/10.1038/s41467-019-08394-9
Yen ST, Zhang M, Deng JM et al (2014) Somatic mosaicism and allele complexity induced by CRISPR/Cas9 RNA injections in mouse zygotes. Dev Biol 393:3–9. https://doi.org/10.1016/j.ydbio.2014.06.017
Oliver D, Yuan S, McSwiggin H, Yan W (2015) Pervasive genotypic mosaicism in founder mice derived from genome editing through pronuclear injection. PLoS ONE 10:e0129457. https://doi.org/10.1371/journal.pone.0129457
Nowell CS, Bredenkamp N, Tetelin S et al (2011) Foxn1 regulates lineage progression in cortical and medullary thymic epithelial cells but is dispensable for medullary sublineage divergence. PloS Genet 7:e1002348. https://doi.org/10.1371/journal.pgen.1002348
Goto T, Hara H, Nakauchi H, Hochi S, Hirabayashi M (2016) Hypomorphic phenotype of Foxn1 gene-modified rats by CRISPR/Cas9 system. Transgenic Res 25:533–544. https://doi.org/10.1007/s11248-016-9941-9
Sakurai T, Watanabe S, Kamiyoshi A, Sato M, Shindo T (2014) A single blastocyst assay optimized for detecting CRISPR/Cas9 system-induced indel mutations in mice. BMC Biotechnol 14:69. https://doi.org/10.1186/1472-6750-14-69
Bae S, Kweon J, Kim HS, Kim JS (2014) Microhomology-based choice of Cas9 nuclease target sites. Nat Methods 11:705–706. https://doi.org/10.1038/nmeth.3015
Abe T, Inoue KI, Furuta Y, Kiyonari H (2020) Pronuclear microinjection during S-phase increases the efficiency of CRISPR-Cas9-assisted knockin of large DNA donors in mouse zygotes. Cell Rep 31:107653. https://doi.org/10.1016/j.celrep.2020.107653
Kim S, Kim D, Cho SW, Kim J, Kim JS (2014) Highly efficient RNA-guided genome editing in human cells via delivery of purified Cas9 ribonucleoproteins. Genome Res 24:1012–1019. https://doi.org/10.1101/gr.171322.113
Ma Y, Chen W, Zhang X et al (2016) Increasing the efficiency of CRISPR/Cas9-mediated precise genome editing in rats by inhibiting NHEJ and using Cas9 protein. RNA Biol 13:605–612. https://doi.org/10.1080/15476286.2016.1185591
Santos F, Hendrich B, Reik W, Dean W (2002) Dynamic reprogramming of DNA methylation in the early mouse embryo. Dev Biol 241:172–182. https://doi.org/10.1006/dbio.2001.0501
Taleei R, Nikjoo H (2013) Biochemical DSB-repair model for mammalian cells in G1 and early S phases of the cell cycle. Mutat Res 756:206–212. https://doi.org/10.1016/j.mrgentox.2013.06.004
Grajcarek J, Monlong J, Nishinaka-Arai Y et al (2019) Genome-wide microhomologies enable precise template-free editing of biologically relevant deletion mutations. Nat Commun 10:4856. https://doi.org/10.1038/s41467-019-12829-8
Vu TV, Doan DTH, Kim J et al (2021) CRISPR/Cas-based precision genome editing via microhomology-mediated end joining. Plant Biotechnol J 19:230–239. https://doi.org/10.1111/pbi.13490
Aida T, Nakade S, Sakuma T et al (2016) Gene cassette knock-in in mammalian cells and zygotes by enhanced MMEJ. BMC Genomics 17:979. https://doi.org/10.1186/s12864-016-3331-9
Yoshimi K, Kunihiro Y, Kaneko T, Nagahora H, Voigt B, Mashimo T (2016) ssODN-mediated knock-in with CRISPR-Cas for large genomic regions in zygotes. Nat Commun 7:10431. https://doi.org/10.1038/ncomms10431
Acknowledgements
The authors thank Reiko Terada, Fumika Yoshida, Mika Douki and Keiko Yamauchi (National Institute for Physiological Sciences) for their assistance with animal care and PCR sample preparations.
Funding
This work was supported by a grant from LEAP-AMED (JP18gm0010002) to M.H. and by the NINS program for Cross-Disciplinary Science study to T.G.
Author information
Authors and Affiliations
Contributions
TG: Conceptualization, Investigation, Data Curation, Writing—Original Draft, Funding Acquisition. KY: Investigation. SH: Data Curation, Validation, Visualization, Writing—Review & Editing. MH: Investigation, Data Curation, Writing—Review & Editing, Funding Acquisition, Supervision, Project Administration.
Corresponding author
Ethics declarations
Competing interests
The authors have no relevant financial or non-financial interests to disclose.
Ethical approval
All procedures for animal experimentation were reviewed and approved by the Animal Care and Use Committee of the National Institutes of Natural Sciences (Date: 2015.3.27/No. 15A102, Date: 2015.1.23/No. P09-070-A).
Consent to publish
All the authors have approved the manuscript being submitted for publication in the journal Molecular Biology Reports.
Consent to participate
Not applicable.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Springer Nature or its licensor (e.g. a society or other partner) holds exclusive rights to this article under a publishing agreement with the author(s) or other rightsholder(s); author self-archiving of the accepted manuscript version of this article is solely governed by the terms of such publishing agreement and applicable law.
About this article

Cite this article
Goto, T., Yogo, K., Hochi, S. et al. Characterization of homozygous Foxn1 mutations induced in rat embryos by different delivery forms of Cas9 nuclease. Mol Biol Rep 50, 1231–1239 (2023). https://doi.org/10.1007/s11033-022-08054-0
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s11033-022-08054-0