
Abstract
Objective
To investigate the effect and mechanisms of Andira anthelmia lectin in rat models of acute inflammation.
Material
AAL anti-inflammatory activity was evaluated in Wistar rat models of paw edema and peritonitis.
Methods
AAL (0.01–1 mg/kg i.v.) was injected 30 min before stimulation with carrageenan and with initial and late phase inflammatory mediators into the animals paw or peritoneum for evaluation of cell migration (optical and intravital microscopy), paw edema (plethysmometry and histopathology); hyperalgesia (analgesimetry).
Results
AAL inhibited leukocyte migration induced by carrageenan, mainly neutrophils to the peritoneal fluid, decreasing leukocyte adhesion. In the peritoneal fluid, AAL reduced the gene expression of TNF-α and cyclooxygenase, as well the levels of PGE2. AAL inhibited the paw edema induced by carrageenan, serotonin, histamine, TNF-α, PLA2 and PGE2, but not by L-arginine. In this model, AAL also inhibited mechanical hypernociception induced by TNF-α, PGE2, db-cAMP and capsaicin, and the activity of myeloperoxidase in the paw tissues.
Conclusion
AAL presents anti-inflammatory effect in acute models of rat inflammation involving the participation of prostaglandins, TNF-α and lectin domain.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
The leukocyte adhesion to activated endothelial cells is a sequential, multistep process consisting of tethering, rolling, firm adhesion and transmigration [1] that involves a series of activation mediated by inflammatory agents, such as histamine, cytokines, chemokines and prostaglandins [2, 3].
Recognition between proteins and carbohydrates is critical in many biological processes, such as viral, bacterial and parasitic infections, cell differentiation, cancer and leukocyte adhesion to the vascular endothelium [4]. In this context, there is a growing number of papers using lectins as tools for studying biological processes since lectins are proteins or glycoproteins capable of recognition and binding to carbohydrates in a reversible and highly specific way [5].
Plant lectins are considered a heterogeneous group of proteins regarding its biochemical/physicochemical properties, molecular structure, binding specificity for carbohydrates and biological activities. Studies involving lectins belonging to the plants of Dalbergieae tribe are scarce, being currently close to 30 purified lectins [6].
The interest in studying the effects of plant lectins has increased because glycoconjugate interactions are implied in inflammation pathways. Therefore, these proteins have similar structures to each other, but possess different carbohydrate binding sites, making them potential tools for studies in biotechnology or immunotherapies [7].
Lectins isolated from seeds of leguminous plants belonging to Dalbergieae tribe, genus Lonchocarpus (L. sericeus, L. araripensis, L. campestris) have been described for its anti-inflammatory properties in animal models via interaction with carbohydrates [6].
The lectin of Andira anthelmia (AAL), a native plant of the Brazilian Amazon (tribe Dalbergieae), was isolated by affinity chromatography (sepharose-mannose) and partially characterized. AAL presents glycosylation and double band of molecular weight about 20 kDa and other minor bands of 17, 15, and 13 kDa. The AAL hemagglutinating activity was inhibited by D-mannose and D-glucose and was shown to be dependent on divalent cations. AAL presents antinociceptive effect in behavioral tests (writhing and formalin), involving the lectin domain and peripheral mechanisms of inflammatory nociception [8]. However, up to date, there is no report in the scientific literature showing the anti-inflammatory effect of a lectin mannose/glucose-binding belonging to plants of Dalbergieae tribe.
This study aimed to investigate the underlying mechanism of the anti-inflammatory effect of Andira anthelmia lectin in rat models of acute inflammation.
Materials and methods
Drugs and reagents
Ketamine and xylazine were supplied by König S/A (Buenos Aires, province of Buenos Aires, Argentina). α-D-mannose, Carrageenan λ, histamine, serotonin (5-hydroxytryptamine), L-arginine, 1H-[1,2,4]oxadiazolo[4,3-a]-quinoxalin-1-one (ODQ), N-nitro-L-arginine methyl ester (L-NAME), hexadecyltrimethylammonium bromide, tetramethylbenzidine (TMB), hydrogen peroxide, phospholipase A2 (PLA2), prostaglandin E2 (PGE2), tumor necrosis factor alpha (TNF-α), N6,2′-O-Dibutyryladenosine 3′,5′-cyclic monophosphate sodium salt (db-cAMP) and dexamethasone were obtained from Sigma Chemical Company (St. Louis, Missouri, USA). Brazol kit from LGC Biotecnologia (Cotia—SP, Brazil); DNAse and reverse transcriptase SuperScriptTM IV First-Strand Synthesis System from Invitrogen® (Carlsbad—California, USA); GelRed from Biotium (Fremont—California, USA); primers Oligo (dT) and dNTP from Thermo Fisher (Waltham—Massachusetts, USA); GoTaq® qPCR Master Mix kit from Promega (Madsison—Wisconsin, USA) and primers for β-actin, COX-1, COX-2 and TNF-α from Integrated DNA Technology-IDT (Coralville—Iowa, USA). Kit ELISA PGE2 was purchased from Abcam® (Cambridge, MA-EUA). All other chemicals were of analytical grade.
Lectin isolation and treatment
Andira anthelmia lectin (AAL) was isolated by sepharose–mannose affinity chromatography, presenting binding affinity for mannose and glucose [8]. Animals received AAL (0.01; 0.1; 1 mg/kg) or 0.9% NaCl (sterile saline) by intravenous (i.v.) route 30 min before injection of the inflammatory stimuli, and the anti-inflammatory effect of AAL evaluated in models of acute inflammation (peritonitis, paw edema).
Animals
Wistar rats (200–300 g) were maintained in cages (six in each) in a controlled environment (circadian cycle, 25 °C, food and water ad libitum). Experimental protocols were previously approved by the Institutional Animal Care and Use Committee of the State University of Ceará (No. 2126961/2015) following the recommendations of the Brazilian College of Animal Experimentation (COBEA), in accordance to NIH guidelines (publication nº 85-23, revised 2011).
Peritonitis model
Peritonitis was induced by intraperitoneal (i.p.) injection of carrageenan (500 µg/cavity) 30 min after the treatment with AAL (0.01–1 mg/kg; i.v.). In order to evaluate the involvement of the lectin domain, animals were also treated with AAL associated to its binding sugar mannose (0.1 M) before carrageenan. After 4 h, peritoneal fluid was collected in 5 ml of saline (5 IU heparin) for total and differential leukocyte counts [9], dosage of PGE2 levels and analysis of the relative gene expression of cyclooxygenase enzymes and TNF-α. Leukocyte adhesion and rolling were also evaluated by intravital microscopy 4 h after peritonitis induction. For this, animals were anesthetized (xylazine 5 mg/kg + ketamine 50 mg/kg, i.m.) before exposition of the mesenteric bed for in situ microscopic examination. Leukocyte rolling was defined as white blood cells that move at a velocity significantly slower than erythrocytes in a given microvessel. Leukocytes were considered to be adherent to the venular endothelium if they remained stationary for more than 30 s. Rolling and adherent cells were counted for 10 min and expressed as the number of rolled leukocytes/10 min and adherent cells/10 min [10].
Paw edema model
Animals received AAL (1 mg/kg; i.v) 30 min before intraplantar (s.c.) administration of carrageenan (300 µg), serotonin (100 µg/paw), histamine (1 µg/paw), L-arginine (15 nmol/paw), TNF-α (5 ng/paw), PLA2 (30 µg/paw) or PGE2 (30 µg/paw). Control animals received saline (s.c.) in the contralateral paw. The edema was measured by hydroplethysmometry (Panlab, LE 7500, Barcelona, Spain) before (zero time) and after (30 min–4 h) inflammatory stimuli, being expressed as the difference of paw volume displacement (ml) or area under curve-AUC (arbitrary units) [11]. L-NAME (30 mg/kg; s.c.), ODQ (8 µg/kg; s.c.), thalidomide (45 mg/kg; s.c.), dexamethasone (0.5 mg/kg; s.c.) or indomethacin (5 mg/kg; s.c.) were used as reference inhibitory drugs of nitric oxide synthesis, guanylate cyclase, cytokines, NF-kB or cyclooxygenases. Paw tissues were collected at the fourth hour after carrageenan administration for evaluation of myeloperoxidase (MPO) activity, total protein dosage and histopathology. The MPO activity was determined by spectrometry (A450 nm) and expressed as MPO U/mg tissue [12]. Total protein dosage was performed using the Bradford method [13]. For histopathology, longitudinal paw segments were fixed overnight in 10% buffered formalin, embedded in paraffin, sliced (5 µm) and stained by HE. Slices were observed by light microscopy coupled to image acquisition systems. The intensity of tissue inflammatory alterations was evaluated counted in total polymorphonuclears and according to the following scores: (0) normal tissue—absence of edema and inflammatory cell infiltrate; (1) discrete tissue changes—mild edema and cellular infiltration; (2) moderate tissue alteration—significant edema and cellular infiltration; (3) intense tissue alteration—wide area of edema and cellular infiltration [14].
Hypernociception evaluation
Animals were individually placed into plexiglas boxes with malleable mesh net floor. A large probe coupled to electronic algesimeter was used to apply a force on the footpad center in order to cause paw withdrawal reflex from mechanical stimulation. The reduced intensity force required to evoke paw withdrawal is indicative of hypernociception [15]. The paw withdrawal was quantified (g) before (basal value) and up to 4 h thereafter stimuli. The antinociceptive effect of AAL (1 mg/kg i.v.) was evaluated 30 min before s.c. administration of TNF-α (5 ng/paw), PGE2 (30 µg/paw), db-cAMP (100 µg/paw) or capsaicin (1 µg/paw). Indomethacin (5 mg/kg, s.c., 30 min prior PGE2) or saline 0.9% were used as controls.
Measurement of inflammatory mediators: Gene Expression and ELISA
Gene sequences from Rattus norvegicus were obtained from GeneBank database. Genes specific primer for COX-1 (1F 5′ TGA GCT ACT ATA CTC GCA TTC TG 3′; 2R 5′ TGG TAA CTG TTT CTT CCC TTT GG 3′), COX-2 (1F 5′ TTT CTC CAA CCT CTC CTA CTA CAC 3′; 2R 5′ TCC TTA TTT CCT TTC ACA CCC A 3′) and TNF-α (1F 5′ TTCTCATTCCTGCTTGTGGC 3′; 2R 5′CCATTTGGGAACTTCTCATCC 3′) reference gene primer for β-actin gene (1F 5′ GCA CCA CAC CTT CTA CAA TGA G 3′; 2R 5′ GGT CTC AAA CAT GAT CTG GGT C 3′) were obtained using Primer3 (http://primer3.ut.ee). All primers were synthesized by IDT with 25 nt lenght. The negative control of PCR reactions consisted of 0.3 μM of each primer pair, 10 μL of GoTaq® qPCR Master Mix kit (Promega) and 2 μL of RNase-free water, in a final volume of 20 μL. From resident cells of peritoneal fluid total RNA was extracted using Brazol kit and treated with DNase (Invitrogen). Total RNA was resuspended in RNase-free water and its concentration determined by spectrophotometry (NanoVue) at 260 nm. RNA purity was checked at optical density ratio (OD260nm/OD280nm) between 1.8 and 2.0. RNA quality and integrity were analyzed in 1.2% agarose gel staining with GelRed. The cDNA was synthesized using reverse transcriptase SuperScript™ IV First-Strand Synthesis System (Invitrogen), 1000 ng of total RNA was used and the reaction was performed according to manufacturer’s recommendations. At the final, the cDNA concentration was determined by spectrophotometry at 260 nm. qRT-PCr was performed using termocycler Bioer LineGene 9660. Genes amplification were performed in a reaction including 0.3 µM of each primer (forward and reverse), 2 µL cDNA and 10 µL of GoTaq® qPCR Master Mix kit (Promega) in a total volume reaction of 20 µL. A initial cycle was performed at 95 °C for 10 min, followed by 40 cycles at 95 °C for 15 s, and 60 °C for 15 s and 60 °C for 30 s. Melting curve analysis was performed to evaluated primer dimers and other artifacts. The gene expression was determined using the 2−∆∆CT method [16]. Additionally, the peritoneal fluid was centrifuged (3000g × 15 min), and the obtained supernatant was assessed for the content of PGE2 (pg/ml) by enzyme-linked immunosorbent assay (A405nm).
Statistical analysis
Statistical differences were determined by analysis of variance (one-way or two-way ANOVA), followed by Bonferroni’s test. The results were expressed as mean ± SEM (n = 8 or n = 3/group for gene expression). Histopathological data was expressed as Median (maximum and minimum) and analyzed by the Mann–Whitney test. p < 0.05 was considered significant.
Results
AAL inhibits leukocyte migration induced by carrageenan
The inflammatory parameter of leukocyte migration was investigated in peritonitis models induced by carrageenan. AAL inhibited the number of leukocyte migration induced by carrageenan (5908 ± 490 cells/ml) by 32% (0.01: 3960 ± 353 cells/ml), 48% (0.1: 3460 ± 309 cells/ml) and 78% (1 mg/kg: 2291 ± 241 cells/ml). This inhibition was mainly due to the decrease in neutrophils by 31% (0.01: 2563 ± 240 cells/ml), 47% (0.1: 1967 ± 172 cells/ml) and 78% (1 mg/kg: 803 ± 147 cells/ml) (carrageenan: 3749 ± 374 cells/ml) (Fig. 1a). The inhibitory effect of AAL (1 mg/kg) on leukocyte migration (2825 ± 229 cells/ml) was completely blocked by its association with mannose (5643 ± 742 cells/ml). Mannose per se did not induce any effect (6513 ± 243 cells/ml) (Fig. 1b). The intravital microscopy revealed that carrageenan increased the endothelial leukocyte rolling (403.0 ± 13.3 cells/10 min) and adhesion (15.1 ± 1.8 cells/10 min) compared to saline (107.8 ± 12.42 rolling cells/10 min; 6.8 ± 0.8 adhesion cells/10 min). AAL significantly decreased leukocyte rolling by 37% (259.8 ± 22.7 cells/10 min) and adhesion by 50% (7.5 ± 0.6 cells/10 min) (Fig. 1 c, d).

AAL reduces leukocyte-migration induced by carrageenan. A Animals received AAL (0.01–1 mg/kg; i.v.), B mannose (0.1 M; i.v.) or AAL (1 mg/kg i.v.) associated to mannose 30 min before peritonitis induction by carrageenan (500 µg/cavity). Control animals received 0.9% saline (0.1 ml/100 g; s.c.). Total and differential cell counts of peritoneal fluid was performed 4 h after peritonitis induction. C Rolled and D adhered leukocytes were evaluated by intravital microscopy for 10 min. Mean ± S.E.M. (n = 6). ANOVA and Bonferroni tests. #p < 0.05 vs. saline, *p < 0.05 vs. carrageenan, @p. < 0.05 vs. AAL 0.01
AAL inhibits inflammatory parameters induced by carrageenan in the paw edema model
In order to investigate AAL effect on swelling cardinal sign of inflammation, AAL was evaluated in paw edema model. AAL at 1 mg/Kg inhibited the paw edema time-course induced by carrageenan from 0 to 4 h (Fig. 2a). The inhibitory effect was observed in the edema initial phase (0–2 h) by 26% (47 ± 3.7 AUC) compared to carrageenan (67.3 ± 1.8 vs. saline: 18 ± 1.7 AUC) (Fig. 2b), and in the late phase (2–4 h) by 35% (62.6 ± 8.5 AUC) compared to carrageenan (96.2 ± 8.5 vs. saline: 6.3 ± 2 AUC) (Fig. 2c). AAL also reduced the total protein concentration by 37% (0.72 ± 0.14 mg/ml vs. carrageenan: 1.15 ± 0.13 vs. saline: 0.59 ± 0.08 mg/ml) (data not shown) and the myeloperoxidase activity by 62% (0.11 ± 0.01 U/mg vs. carrageenan: 0.29 ± 0.02 vs. saline: 0.13 ± 0.01 U/mg) (Fig. 2d) in the paw tissues.

AAL inhibits paw edema and the increased myeloperoxidase activity induced by carrageenan. Animals received AAL (1 mg/kg; i.v.) 30 min before injection of carrageenan (300 µg/paw) or 0.9% saline (100 µl/paw) (A) Paw edema was measured by hydroplethysmometry at 0–240 min after carrageenan and expressed as the variation in paw volume displacement (ml) or area under curve (B, C). After 4 h, samples of subcutaneous plantar tissue were collected for evaluation of (D) myeloperoxidase activity (A405nm). ANOVA and Bonferroni tests. #p < 0.05 vs. saline, *p < 0.05 vs. carrageenan
The histopathological evaluation showed that carrageenan induced severe alterations (leucocyte infiltration, edema) in the animal paw-tissues [median score: 3 (+ 3; + 2)] (Fig. 3b,e,g) compared to saline [score: 0 (+ 1; 0)] (Fig. 3a,d,g). AAL treatment reduced these alterations [score: 0 (+ 1; 0)] (Fig. 3c,f,g). The macroscopic alteration is seen in Fig. 3h.

AAL attenuates the histopathological profile of carrageenan-induced paw edema. Cross section; HE (100X a, b, c; 400X d, e, f). A/D Saline: absence of inflammation; B/E Carrageenan: severe cellular infiltrate; C/F AAL: reduction of inflammatory parameters; G Image quantification of recruited polimorfonuclear (PMN). H) Whole paw images. Arrows: cellular infiltrate; Arrowheads: edema formation. *p < 0.05 vs. stimuli
AAL inhibits paw edema induced by initial and late-phase inflammatory mediators
In the paw edema model, it was also investigated the participation of inflammatory mediators. AAL (1 mg/kg) inhibited by 40% (105.5 ± 25.6 AUC) the paw edema induced by serotonin (177.1 ± 18.3 vs. saline: 45.4 ± 9.1 AUC) and by 32% (93.2 ± 7) that of histamine (137 ± 8.4 vs. saline: 29 ± 1.7 AUC) (Fig. 4a). However, the edema elicited by L-arginine (71 ± 7.8 AUC) was unaltered by AAL (66 ± 8.6 vs. L-NAME: 43.5 ± 4.3 AUC) (Fig. 4b). Animals that received ODQ associated with AAL (98.2 ± 13.3) did not show different responses from those that received both isolated drugs (ODQ: 103.5 ± 10.7 vs. AAL: 93.1 ± 16 AUC) compared to carrageenan (150.2 ± 6.7 AUC) (Fig. 4b). Moreover, the edema induced by TNF-α (90.5 ± 10.6 AUC), PLA2 (117.5 ± 4.5 AUC) and PGE2 (102.9 ± 14.9 AUC) were notably reduced by AAL by 73% (24 ± 4 AUC), 77% (26 ± 5 AUC) and 38% (64.5 ± 5.1 AUC), respectively (Fig. 4c, d). Similar effects were observed the classical inhibitors of TNF-α (thalidomide: 41 ± 1.5), PLA2 (dexamethasone: 51 ± 4.8 AUC) and PGE2 (indomethacin: 54.5 ± 3.5 AUC).

AAL inhibits paw edema induced by serotonin, histamine, TNF-α, phospholipase A2 (PLA2) and PGE2. Animals received AAL (1 mg/kg i.v.) 30 min before stimuli injection, and paw edema measured by hydroplethysmometry and expressed as area under curve. A Serotonin (5-HT: 100 µg/paw), histamine (1 µg/paw), B L-arginine (15 nmol/paw), carrageenan (300 µg/paw), C TNF-α (5 ng/paw), D PLA2 (30 µg/paw) and PGE2 (30 µg/paw). L-NAME (30 mg/kg; s.c.), ODQ (8 µg/kg; s.c.), thalidomide (45 mg/kg; s.c.), dexamethasone (0.5 mg/kg; s.c.) and indomethacin (5 mg/kg; s.c.) were used as anti-inflammatory control. ANOVA and Bonferroni tests. #p < 0.05 vs. saline, *p < 0.05 vs. stimuli
AAL inhibits hypernociception elicited by mediators involved in the TNF-α and prostaglandins pathway
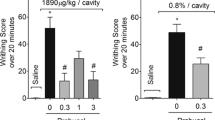
The effect of AAL on nociceptive mediators was investigated by the protocol of mechanical hypernociception. TNF-α, PGE2, db-cAMP and capsaicin increased animals paw withdrawal in response to mechanical stimulation with electronic analgesimeter. At the first hour, AAL (1 mg/kg) recovered (32.9 ± 3.1 vs. saline: 35.1 ± 2 g) the response elicited by TNF-α (22.2 ± 0.7 g). At the third hour, AAL abolished (31.9 ± 0.9 vs. saline: 37.6 ± 1.8 g) the hypernociceptive response induced by TNF-α (23.1 ± 3.1 g) (Fig. 5a). The hypernociceptive effect of PGE2 (41.3 ± 1.7 vs. AAL: 54 ± 1.3 vs. indomethacin: 54 ± 3.9 g) and db-cAMP (17.8 ± 1.7 vs. AAL: 30.2 ± 1.4 vs. saline: 31.2 ± 0.8 g) was also fully recovered by AAL at the first and third hour, respectively (Fig. 5b, c). AAL also inhibited the hypernociceptive response (50.5 ± 1.9 g) induced by capsaicin (38.1 ± 3 vs. saline: 57.6 ± 4.6 g) at the second hour (Fig. 5d).

AAL inhibits mechanical hypernociception induced by TNF-α, PGE2, db-cAMP and capsaicin. Animals received AAL (1 mg/kg; i.v.) 30 min before injection of the stimuli and evaluated for paw withdrawal response (g) analgesimetry. A TNF-α (5 ng/paw), B PGE2 (30 µg/paw), C db-cAMP (100 µg/paw) and D Capsaicin (1 µg/paw). Indomethacin (5 mg/kg; s.c.) was used as antihypernociceptive control. ANOVA and Bonferroni tests. #p < 0.05 vs. saline, *p < 0.05 vs. stimuli
AAL alters the gene expression of cyclooxygenases and TNF-α; and the levels of PGE2
The qRT-PCR was performed to investigate the effect of AAL on inflammatory gene expression. The carrageenan group significant increase the gene relative expression for COX-1 (3.1 ± 0.01 vs. saline: 1), COX-2 (1 vs. saline 0) and TNF-α (1 vs. saline: 0). AAL reduced the relative expression in 16.3 × of COX-1 (0.19 ± 0.01) (Fig. 6a), 13.8 × of COX-2 (0.07 ± 0.01) (Fig. 6b) and 3.2 × in TNF-α (0.31 ± 0.03) (Fig. 6c). Carrageenan increased the PGE2 levels in peritoneal fluid (0.23 ± 0.04) and AAL (0.06 ± 0.007 pg/ml) reduced by 73% (Fig. 6d). This effect was completely blocked by the association of lectin with mannose (0.18 ± 0.02 pg/ml).

AAL modulates gene expression of cyclooxygenases, TNF-α and prostaglandin E2. Animals received AAL (1 mg/kg i.v.) isolated or associated to mannose (0.1 M) 30 min before injection of carrageenan (500 µg/cavity). Peritoneal fluid was collected after 4 h for qRT-PCR A) COX-1, B COX-2 and C TNF-α. D PGE2 levels were determined by enzyme-linked immunosorbent assay. ANOVA and Bonferroni tests. #p < 0.05 vs. saline, *p < 0.05 vs. stimuli
Discussion
This study demonstrated the modulator effect of the purified Andira anthelmia lectin (AAL) on vascular and cellular events of acute inflammation in rats, that was shown to be dependent on the lectin domain, prostaglandins and TNF-α.
Pre-treatment with AAL showed high efficacy to reduce leukocyte migration induced by carrageenan, a flogistic agent that stimulates resident macrophages to release primary cytokines, such as TNF-α, a powerful chemotactic agent to neutrophils [17]. The differential leukocyte count showed that neutrophils are the principal target cells involved in the AAL anti-inflammatory effect. Similar results have been observed in rats for the lectins isolated from Lonchocarpus sericeus [18, 19] and L. araripensis [20], presenting binding affinity for N-acetylglucosamine. In addition, the interference of AAL in neutrophil migration was also confirmed by the reduced MPO activity measured directly in the paw, a heme-containing enzyme that is highly expressed in neutrophils. Excessive generation of MPO-derived oxidants has been associated to tissue damage in acute and chronic inflammation [21].
AAL is a pioneer lectin, isolated from plants of the Dalbergieae tribe presenting affinity for mannose/glucose, to inhibit leukocyte migration. Intravital microscopy showed the reduction in of leukocyte rolling and adhesion to the endothelium of mesenteric vessels after AAL treatment. The association of mannose with AAL reversed the lectin inhibitory effect on leukocyte migration, probably by competitive interactions on selectins–sugar binding sites of endothelial and leukocyte cell membranes, as suggested before [22, 23]. In general, lectins that interact to mannose and glucose residues present anti-inflammatory effect by systemic administration [22, 23] and inflammatory effect by local administration [24, 25]. Despite the reduction in neutrophil migration, it is not possible to rule out the participation of AAL on macrophages, since they are important cells of acute inflammation for the release of pro-inflammatory mediators such as TNF-α, IL-1 and prostaglandins [17].
The edema induced by carrageenan in rodents is biphasic characterized by intense leukocyte influx involving a complex network of inflammatory mediators [26]. The initial phase (0–2 h) is predominantly vascular, triggered by the release of biogenic amines (histamine, bradykinin and serotonin) [27], whereas the second phase (2–4 h) is maintained by neutrophil infiltration and production of prostaglandins, nitric oxide and cytokines [26, 28, 29]. AAL inhibited the entire time-course of the paw edema induced by carrageenan, including the cell infiltrate phase, being in accordance to the anti-inflammatory effect of other leguminous lectins [22, 30, 31]. Lectins isolated from Dalbergieae plants also showed inhibitory effect on carrageenan-induced paw edema such as that of L. sericeus [18, 19], L. araripensis [31] and L. campestris [20].
The cellular nature of AAL anti-inflammatory effect was supported either by the paw histopathological data, inhibition on MPO activity and evaluation of the edema induced by late phase mediators of carrageenan edema (phospholipase A2, prostaglandin E2, TNF-α). However, it cannot be excluded the vascular effect of AAL, since it inhibited the increased vascular permeability elicited by carrageenan and the paw edema elicited by histamine and serotonin. Histamine and serotonin (5-HT) are mediators released by inflammatory cells that mediate the vascular events of acute inflammation induced by carrageenan in rats [32]. Thus, our data suggest that the anti-edematogenic effect of AAL has the participation of biogenic amines, cytokines (TNF-α) and prostaglandins.
Moreover, AAL inhibited the inflammatory process and hypernociception induced by TNF-α and PGE2. Accordingly, the antinociceptive effect of AAL had been demonstrated in two inflammatory nociception model (acetic acid induced contortions and formalin test), an effect that was reversed by mannose [8]. The enhanced sensitivity by nociceptive stimuli is one of the characteristics of the inflammatory response and results from an increased excitability of primary nociceptive neurons [33]. There is experimental evidence demonstrating that prostaglandins, among other mediators (sympathetic amines, endothelins, cytokines) may sensitize the primary nociceptive neurons to innocuous mechanical stimuli [34, 35]. It has been demonstrated that the intraplantar administration of carrageenan in rats induce hypernociception mediated initially by TNF-α [36] and in our study, the hypernociception induced by TNF-α was inhibited by AAL, suggesting a direct involvement of this pathway in the sensitization of nociceptors. It is well established that inflammatory hypernociception, such as that induced by PGE2 in EP receptors, depends on signalling pathways, requiring activation of PKA and PKC in neurons, in presence of cAMP [33, 37,38,39]. In the present study, AAL prevented hypernociception induced by the cAMP analog db-cAMP that elicits hypernociception via PGE2 pathway [33] and also directly by PGE2, suggesting that the lectin presents antinociceptive property via direct action on PGE2 receptor or by intracellular mechanisms involving the pathway PGE2/PKA/PKC.
PKA and PKC mediate hypernociception following activation of EP receptors (4 and 1) in peripheral nociceptors [33, 35]. Studies suggest that PGE2 involves sensitization of the potencial transient receptor vanilloid-1 (TRPV1) channels by PKA and PKC activation [35]. The sensitization of TRPV1 via PGE2/EP1/PKC and PGE2/EP4/PKA cascades is considered to be involved in inflammatory processes [40]. In rats, the local injection of capsaicin into the plantar hind paw produces thermal and mechanical hypernociception at the site of injection [41]. The capsaicin model is well understood in the literature due to the known molecular events (activation of TRPV1 receptors, cation entry, C-fibers depolarization) [42]. Thus, it is possible to be speculated that the TRPV1 channel is the target of the lectin antinociceptive effect since it inhibited nociception induced by PGE2 and capsaicin.
Our data support the idea that AAL may inhibit several stages of the prostanoids formation, either directly on the enzyme cyclooxygenase or direct on PGE2 receptors. This result allows us to speculate that AAL may be inhibiting other prostanoids that participate in acute inflammation such as PGD2 and PGI2. It is also possible that the lectin is modulating the expression of inflammatory pathways as COX and TNF-α via the nuclear factor kappa B (NF-kB). Other Dalbergieae lectin also show modulator effect on the gene expression of inflammatory pathways such as Lonchocarpus araripensis [43].
Plant lectins are also capable to interact with Toll-like receptors, responsible for activating the NF-kB pathway [44], which regulates the expression of several cytokines, such as TNF-α, IL-1, IL-6, IL-8, IL-12 and chemokines. These data allow us to open perspectives for even more to be discovered about the interactions of lectins with cellular structures involved in inflammation. It has been observed by molecular docking the ability of lectins isolated from the Dalbergieae tribe (possessing mannoside affinity) to bind to glycan structures involved in cell–cell recognition process [45,46,47]. Studies focusing a lectin-like domain of TNF-α have shown to improve lung function in transplanted rats, including the reduction in reactive oxygen species [48]. Therefore, the presence of this domain in TNF-α opens the possibility that AAL can occupy the carbohydrate site preventing the binding of TNF-α by competitive mechanisms. Another potential target that could be explored in the future is the inhibitory effect of AAL on the expression of E and P selectins and integrin B2, since they are glycoproteins involved in the leukocytes rolling and adhesion [49, 50].
In conclusion, Andira anthelmia lectin demonstrates anti-inflammatory effect, via lectin domain, with the participation of TNF-α and PGE2/cAMP/TRVP1 pathway.
References
Kameritsch P, Renkawitz J (2020) Principles of leukocyte migration strategies. Trends Cell Biol 10:818–832. https://doi.org/10.1016/j.tcb.2020.06.007
Nourshargh S, Alon R (2014) Leukocyte migration into inflamed tissues. Immunity 41(5):694–707. https://doi.org/10.1016/j.immuni.2014.10.008
Kalinski P (2012) Regulation of immune responses by prostaglandin E2. J Immunol 188(1):21–28. https://doi.org/10.4049/jimmunol.1101029
Sharon N, Lis H (2004) History of lectins: from hemagglutinins to biological recognition molecules. Glycobiology 14(11):53–62. https://doi.org/10.1093/glycob/cwh122
Wiederschain GT (2013) Glycobiology: progress, problems, and perspectives. Biochemistry (Mosc) 78(7):679–696. https://doi.org/10.1134/S0006297913070018
Nascimento KS, Silva MTL, Oliveira MV, Lossio CF, Pinto-Junior VR, Osterne VJS, Cavada BS (2020) Dalbergieae lectins: a review of lectins from species of a primitive Papilionoideae (leguminous) tribe. Int J Biol Macromol 144:509–526. https://doi.org/10.1016/j.ijbiomac.2019.12.117
Rüdiger H, Gabius HJ (2001) Plant lectins: occurrence, biochemistry, functions and applications. Glycoconj J 18(8):589–613. https://doi.org/10.1023/a:1020687518999
Nascimento KS, Nascimento FLF, Silva MTL, Nobre CB, Moreira CG, Brizeno LAC, da Ponte EL, Assreuy AMS, Cavada BS (2015) Purification of a thermostable antinociceptive lectin isolated from Andira anthelmia. J Mol Recognit 29(6):248–252. https://doi.org/10.1002/jmr.2523
Souza GE, Ferreira SH (1985) Blockade by antimacrophage serum of the migration of PMN neutrophils into the inflamed peritoneal cavity. Agents Actions 17(1):97–103. https://doi.org/10.1007/BF01966691
Fortes ZB, Farsky SP, Oliveira MA, Garcia-Leme J (1991) Direct vital microscopic study of defective leukocyte–endothelial interaction in diabetes mellitus. Diabetes 40(10):1267–1273. https://doi.org/10.2337/diab.40.10.1267
Landucci EC, Antunes E, Donato JL, Faro R, Hyslop S, Marangoni S, Oliveira B, Cirino G, de Nucci G (1995) Inhibition of carrageenin-induced rat paw oedema by crotapotin, a polypeptide complexed with phospholipase A2. Br J Pharmacol 114(3):578–583. https://doi.org/10.1111/j.1476-5381.1995.tb17178.x
Bradley PP, Christensen RD, Rothstein G (1982) Cellular and extracellular myeloperoxidase in pyogenic inflammation. Blood 60(3):618–622
Bradford MM (1976) A rapid and sensitive method for the quantitation of microgram quantities of protein utilizing the principle of protein-dye binding. Anal Biochem 7(72):248–254. https://doi.org/10.1006/abio.1976.9999
Teixeira CS, Assreuy AMS, Osterne VJS, Amorim RMF, Brizeno LAC, Debray H, Nagano CS, Delatorre P, Sampaio AH, Rocha BAM, Cavada BS (2014) Mannose-specific legume lectin from the seeds of Dolichos lablab (FRIL) stimulates inflammatory and hypernociceptive processes in mice. Process Biochem 49(3):529–534. https://doi.org/10.1016/j.procbio.2013.12.020
Vivancos GG, Verri WA Jr, Cunha TM, Schivo IRS, Parada CA, Cunha FQ, Ferreira SH (2004) An electronic pressure-meter nociception paw test for rats. Braz J Med Biol Res 37(3):391–399. https://doi.org/10.1590/s0100-879x2004000300017
Livak KJ, Schmittgen TD (2001) Analysis of relative gene expression data using real-time quantitative PCR and the 2(-Delta Delta C(T)) Method. Methods 25(4):402–408. https://doi.org/10.1006/meth.2001.1262
Souza GE, Cunha FQ, Mello R, Ferreira SH (1988) Neutrophil migration induced by inflammatory stimuli is reduced by macrophage depletion. Agents Actions 24(3–4):377–380. https://doi.org/10.1007/BF02028296
Alencar NMN, Cavalcante CF, Vasconcelos MP, Leite KB, Aragão KS, Assreuy AMS, Nogueira NAP, Cavada BS, Vale MR (2005) Anti-inflammatory and antimicrobial effect of lectin from Lonchocarpus sericeus seeds in an experimental rat model of infectious peritonitis. J Pharm Pharmacol 57(7):919–922. https://doi.org/10.1211/0022357056352
Napimoga MH, Cavada BS, Alencar NMN, Mota ML, Bittencourt FS, Alves-Filho JC, Grespan R, Gonçalves RB, Clemente-Napimoga JT, de Freitas A, Parada CA, Ferreira SH, Cunha FQ (2007) Lonchocarpus sericeus lectin decreases leukocyte migration and mechanical hypernociception by inhibiting cytokine and chemokines production. Int Immunopharmacol 7(6):824–835. https://doi.org/10.1016/j.intimp.2007.02.001
Pires AF, Marques GF, Alencar N, Martins MG, Silva MTD, Nascimento KS, Cavada BS, Assreuy A, Maria S (2019) Inhibitory effect of Lonchocarpus araripensis lectin in rat acute models of inflammation. An Acad Bras Cienc. https://doi.org/10.1590/0001-3765201920180991
van der Veen BS, de Winther MP, Heeringa P (2009) Myeloperoxidase: molecular mechanisms of action and their relevance to human health and disease. Antioxid Redox Signal 11(11):2899–2937. https://doi.org/10.1089/ars.2009.2538
Assreuy AM, Shibuya MD, Martins GJ, De Souza ML, Cavada BS, Moreira RA, Oliveira JT, Ribeiro RA, Flores CA (1997) Anti-inflammatory effect of glucose-mannose binding lectins isolated from Brazilian beans. Mediators Inflamm 6(3):201–210. https://doi.org/10.1080/09629359791695
Assreuy AM, Martins GJ, Moreira ME, Brito GA, Cavada BS, Ribeiro RA, Flores CA (1999) Prevention of cyclophosphamide-induced hemorrhagic cystitis by glucose-mannose binding plant lectins. J Urol 161(6):1988–1993
Assreuy AM, Fontenele SR, Pires AF, Fernandes DC, Rodrigues NV, Bezerra EH, Moura TR, do Nascimento KS, Cavada BS (2009) Vasodilator effects of Diocleinae lectins from the Canavalia genus. Naunyn-Schmiedeberg’s Arch Pharmacol 380(6):509–521. https://doi.org/10.1007/s00210-009-0465-1
Rocha BA, Delatorre P, Oliveira TM, Benevides RG, Pires AF, Sousa AA, Assreuy AM, Debray H, Azevedo WFJR, Sampaio AH, Cavada BS (2011) Structural basis for both pro- and anti-inflammatory response induced by mannose-specific legume lectin from Cymbosema roseum. Biochimie 93(5):806–816. https://doi.org/10.1016/j.biochi.2011.01.006
Di Rosa M, Giroud JP, Willoughby DA (1971) Studies on the mediators of the acute inflammatory response induced in rats in different sites by carrageenan and turpentine. J Pathol 104(1):15–29. https://doi.org/10.1002/path.1711040103
Ferreira SH, Moncada S, Parsons M, Vane JR (1974) Proceedings: the concomitant release of bradykinin and prostaglandin in the inflammatory response to carrageenin. Br J Pharmacol 52(1):108–109
Boughton-Smith NK, Deakin AM, Follenfant RL, Whittle BJ, Garland LG (1993) Role of oxygen radicals and arachidonic acid metabolites in the reverse passive Arthus reaction and carrageenin paw oedema in the rat. Br J Pharmacol 110(2):896–902. https://doi.org/10.1111/j.1476-5381.1993.tb13897.x
Di Rosa M, Sorrentino L (1968) The mechanism of the inflammatory effect of carrageenin. Eur J Pharmacol 4(3):340–342. https://doi.org/10.1016/0014-2999(68)90103-9
Pinto NV, Cavada BS, Brito LF, Pereira RI, da Silva MT, Castro RR, de Freitas PA, Assreuy AM (2013) Effects of Canavalia lectins on acute inflammation in sensitized and non-sensitized rats. Inflammation 36(3):713–722. https://doi.org/10.1007/s10753-013-9596-0
Pires AF, Rodrigues NV, Soares PMG, Ribeiro RDA, Aragao KS, Marinho MM, da Silva MT, Cavada BS, Assreuy AMS (2016) A novel N-acetyl-glucosamine lectin of Lonchocarpus araripensis attenuates acute cellular inflammation in mice. Inflamm Res 65(1):43–52. https://doi.org/10.1007/s00011-015-0889-7
Vinegar R, Schreiber W, Hugo R (1969) Biphasic development of carrageenin edema in rats. J Pharmacol Exp Ther 166(1):96–103
Sachs D, Villarreal CF, Cunha FQ, Parada CA, Ferreira SH (2009) The role of PKA and PKCepsilon pathways in prostaglandin E2-mediated hypernociception. Br J Pharmacol 156(5):826–834. https://doi.org/10.1111/j.1476-5381.2008.00093.x
Mizumura K, Minagawa M, Tsujii Y, Kumazawa T (1993) Prostaglandin E2-induced sensitization of the heat response of canine visceral polymodal receptors in vitro. Neurosci Lett 161(1):117–119. https://doi.org/10.1016/0304-3940(93)90154-d
Moriyama T, Higashi T, Togashi K, Iida T, Segi E, Sugimoto Y, Tominaga T, Narumiya S, Tominaga S (2005) Sensitization of TRPV1 by EP1 and IP reveals peripheral nociceptive mechanism of prostaglandins. Mol Pain 1:13. https://doi.org/10.1186/1744-8069-1-3
Cunha FQ, Poole S, Lorenzett BB, Ferreira SH (1992) The pivotal role of tumour necrosis factor alpha in the development of inflammatory hyperalgesia. Br J Pharmacol 107(3):660–664. https://doi.org/10.1111/j.1476-5381.1992.tb14503.x
Ferreira SH, Nakamura M (1979) I—Prostaglandin hyperalgesia, a cAMP/Ca2+ dependent process. Prostaglandins 18(2):179–190. https://doi.org/10.1016/0090-6980(79)90103-5
Cunha FQ, Teixeira MM, Ferreira SH (1999) Pharmacological modulation of secondary mediator systems–cyclic AMP and cyclic GMP–on inflammatory hyperalgesia. Br J Pharmacol 127(3):671–678. https://doi.org/10.1038/sj.bjp.0702601
Kassuya CAL, Ferreira J, Claudino RF, Calixto JB (2007) Intraplantar PGE2 causes nociceptive behaviour and mechanical allodynia: the role of prostanoid E receptors and protein kinases. Br J Pharmacol 150(6):727–737. https://doi.org/10.1038/sj.bjp.0707149
Kawabata A (2011) Prostaglandin E2 and pain–an update. Biol Pharm Bull 34(8):1170–1173. https://doi.org/10.1248/bpb.34.1170
Zhang X, Wu J, Fang L, Willis WD (2003) The effects of protein phosphatase inhibitors on nociceptive behavioral responses of rats following intradermal injection of capsaicin. Pain 106(3):443–451. https://doi.org/10.1016/j.pain.2003.09.002
Sawynok J, Reid A, Meisner J (2006) Pain behaviors produced by capsaicin: influence of inflammatory mediators and nerve injury. J Pain 7(2):134–141. https://doi.org/10.1016/j.jpain.2005.09.013
Assreuy AMS, Amorim RMF, Martins SL, Martins MGQ, Cajazeiras JB, da Silva MTL, Pires AF, Nascimento KS, Cavada BS, Mota MRL (2020) Antinociceptive effect of Lonchocarpus araripensis lectin: activation of L-arginine/NO/cGMP/K+ ATP signaling pathway. Inflammopharmacology 28(6):1623–1631. https://doi.org/10.1007/s10787-020-00729-z
Unitt J, Hornigold D (2011) Plant lectins are novel Toll-like receptor agonists. Biochem Pharmacol 81(11):1324–1328. https://doi.org/10.1016/j.bcp.2011.03.010
Almeida AC, Osterne VJS, Santiago MQ, Pinto-Junior VR, Silva-Filho JC, Lossio CF, Nascimento FLF, Almeida RPH, Teixeira CS, Leal RB, Delatorre P, Rocha BAM, Assreuy AMS, Nascimento KS, Sousa Cavada BS (2016) Structural analysis of Centrolobium tomentosum seed lectin with inflammatory activity. Arch Biochem Biophys 596:73–83. https://doi.org/10.1016/j.abb.2016.03.001
Cavada BS, Araripe DA, Silva IB, Pinto-Junior VR, Osterne VJS, Neco AHB, Laranjeira EPP, Lossio CF, Correia JLA, Pires AF, Assreuy AMS, Nascimento KS (2018) Structural studies and nociceptive activity of a native lectin from Platypodium elegans seeds (nPELa). Int J Biol Macromol. https://doi.org/10.1016/j.ijbiomac.2017.08.174
Neco AHB, Pinto-Junior VR, Araripe DA, Santiago MQ, Osterne VJS, Lossio CF, Nobre CAS, Oliveira MV, Silva MTL, Martins MGQ, Cajazeiras JB, Marques GFO, Costa DR, Nascimento KS, Assreuy AMS, Cavada BS (2018) Structural analysis, molecular docking and molecular dynamics of an edematogenic lectin from Centrolobium microchaete seeds. Int J Biol Macromol 117:124–133. https://doi.org/10.1016/j.ijbiomac.2018.05.166
Hamacher J, Stammberger U, Roux J, Kumar S, Yang G, Xiong C, Schmid RA, Fakin RM, Chakraborty T, Hossain HMD, Pittet JF, Wendel A, Black SM, Lucas R (2010) The lectin-like domain of tumor necrosis factor improves lung function after rat lung transplantation–potential role for a reduction in reactive oxygen species generation. Crit Care Med 38(3):871–878. https://doi.org/10.1097/CCM.0b013e3181cdf725
Yixin G, Yan Z, Shiliang F, Xiaofeng L, Shouqin L, Mian L (2017) Dynamic contributions of P- and E-selectins to β2-integrin induced neutrophil transmigration. FASEB J 31(1):212–223. https://doi.org/10.1096/fj.201600398RRR
Matthias K, Sylvain L, Veronica A, Richard DC, Asma N, Charles AP, Jennifer CB (2020) Regulation of neutrophil function by selective targeting of glycan epitopes expressed on the integrin CD11b/CD18. FASEB J 34(2):2326–2343. https://doi.org/10.1096/fj.201902542R
Acknowledgements
The authors thank Conselho Nacional de Desenvolvimento Científico e Tecnológico-CNPq. Cavada BS, Nascimento KS and Assreuy AM are senior investigators of CNPq.
Funding
Conselho Nacional de Desenvolvimento Científico e Tecnológico,308433/2017-3,Ana Maria Sampaio Assreuy
Author information
Authors and Affiliations
Corresponding authors
Ethics declarations
Conflict of interest
The authors declare no conflict of interest.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
About this article

Cite this article
do Nascimento, F.L.F., de Freitas Pires, A., Mota, M.R.L. et al. The anti-inflamatory effect of Andira anthelmia lectin in rats involves inhibition of the prostanoid pathway, TNF-α and lectin domain. Mol Biol Rep 49, 8847–8857 (2022). https://doi.org/10.1007/s11033-022-07735-0
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s11033-022-07735-0