
Abstract
Background
S100A12, also known as Calgranulin C, is a ligand for the receptor for advanced glycation end products (RAGE) and plays key roles in cardiovascular and other inflammatory diseases. Interactions between S100A12 and RAGE initiate downstream signaling activating extracellular signal-regulated kinases (ERK1/2), mitogen activated protein kinases (MAPK), and transcription factor NF-κB. This increases the expression of pro-inflammatory cytokines to induce the inflammatory response. S100A12, and RAGE play a critical role in the development and progression of atherosclerosis. There is a well-known relationship between the bacterial endotoxin lipopolysaccharide (LPS) and the lipid antigens oxidized low-density lipoprotein (oxLDL) in driving the immune response in atherosclerosis.
Methods and results
Our study aimed to compare the potential of LPS and oxLDL in regulating the expression of S100A12 and RAGE in atherosclerosis. The expression of these proteins was assessed in the harvested carotid arteries from LPS- and oxLDL-treated atherosclerotic Yucatan microswine. Tissues were collected from five different treatment groups: (i) angioplasty alone, (ii) LPS alone, (iii) oxLDL alone, (iv) angioplasty with LPS, and (v) angioplasty with oxLDL. Immunohistochemical findings revealed that angioplasty with LPS induced higher expression of S100A12 and RAGE compared to other treatment groups. The results were further corroborated by testing their gene expression through qPCR in cultured vascular smooth muscle cells (VSMCs) isolated from control carotid arteries and LPS- and oxLDL-treated arteries.
Conclusions
The results of this study suggest that LPS induces the expression of S100A12 and RAGE more than oxLDL in atherosclerotic artery and both S100A12 and RAGE could be therapeutic targets.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Cardiovascular disease (CVD) is the number one cause of death and today is a major clinical condition worldwide. It has been reported that 80% mortality in men and 75% in women have resulted from CVD [1, 2]. According to the World Health Organization, approximately 17.9 million people died from CVD in 2019, amongst which 85% were due to heart attack and stroke [3]. Most CVD-related deaths are the direct result of atherosclerosis, which is characterized by the narrowing of the arteries due to subendothelial accumulation of lipid-laden cells and the formation of atherosclerotic plaques. The rupture of these plaques induces the formation of a thrombus that results in a heart attack or stroke, ultimately leading to increased morbidity and mortality [4]. The current treatment regimen for CVD is by changing the lifestyle and using lipid-lowering drugs like statins. Even with the current treatment regimen, the number of CVD cases is expected to increase almost by 24 million in 2030 [5, 6]. This highlights the importance of understanding the underlying mechanism(s) of atherosclerosis to pave the way for novel therapeutic strategies.
In the early 20th century, it was observed that atherosclerotic plaques consist of calcified connective tissue and cholesterol. These findings led to the development of an animal model of atherosclerosis, specifically by feeding the rabbits a high cholesterol diet [7]. These studies not only provided the information that cholesterol (lipids) is an important risk factor for the development of atherosclerosis but also provided the idea that lipids are the key players in atherosclerotic lesion formation. However, the complete cellular mechanisms underlying atherosclerosis remained unclear until approximately four decades ago when it became clear that most of the cells present in the plaque were macrophages [8]. Further immunohistochemical (IHC) analyses of human atherosclerotic plaques from carotid arteries revealed the presence of many different immune cells in the plaque, implicating the role of immune response in atherosclerosis [9,10,11]. It has been previously reported that lipopolysaccharide (LPS) and oxidized low-density lipoprotein (oxLDL) are two lipid antigens in atherosclerosis that can drive the immune response in atherosclerosis. Additional studies showing that T-cell clones isolated from atherosclerotic plaques became activated by these two lipids support the notion that LPS and oxLDL are immunological antigens [12,13,14,15,16].
Besides these immune cells, the number of systemic markers of inflammation, such as C-reactive protein (CRP), interleukin (IL)-1β, IL-6, nuclear factor-kappa B (NF-κB), LDL, S100 proteins (S100A8, S100A9, and S100A12), have been associated with the prediction of the risk of cardiovascular disease [17, 18]. S100A12 (calgranulin C) is a member of the S100 family of calcium-binding proteins and is highly expressed endogenously in neutrophilic granulocytes and lesser in other cells [19]. Accordingly, elevated serum concentration of S100A12 is an emerging biomarker of atherosclerosis [20]. Due to its ability to activate RAGE (receptor for the advanced glycation end products), a multi-ligand cell surface receptor, there is growing evidence that S100A12 chronically modulates atherosclerotic inflammation [21,22,23,24]. S100A12 is endogenously expressed on myeloid cells and has pleiotropic effects, thereby playing a key role in host defense [24,25,26]. Once S100A12 activates RAGE, it initiates a downstream signaling pathway to induce the inflammatory process by activating extracellular signal-regulated kinases (ERK1/2), mitogen activated protein kinases (MAPK), and transcription factor NF-κB that further initiate the secretion of pro-inflammatory cytokines like interleukin (IL)-1β, IL-6, and tumor necrosis factor (TNF)-α [27]. LPS increases the expression of S100A12 in the cultured cells and oral tissue cultures [28].
Since inflammation plays a key role in chronic inflammatory atherosclerotic process, in this study, we evaluated the effect of LPS and oxLDL on the expression of S100A12 and RAGE in an atherosclerotic animal model of hyperlipidemic Yucatan microswine. To evaluate the expression of S100A12 and RAGE, we administered LPS and oxLDL in the carotid arteries with and without angioplasty. Angioplasty was performed to induce intimal injury and initiation of atherosclerotic lesions in denuded arteries. The carotid arteries were assessed for the expression of the proteins using immunohistochemistry. The results were further confirmed by the mRNA expression of S100A12 and RAGE in LPS and oxLDL-treated vascular smooth muscle cells (VSMCs) extracted from the control (contralateral side) carotid arteries of the microswine.
Materials and methods
Animal model
All microswine weighing 25–30 kg were kept at the animal facility of Western University of Health Sciences, Pomona, California, with a 12 h light and dark cycle at 72–74 °F and cared for per National Institute of Health standards. The research protocol (R19IACUC026) was approved by the Institutional Animal Care and Use Committee of Western University of Health Sciences. The swine were fed with an atherogenic diet purchased from Research Diets Inc (RDI) (NJ, USA) at Sinclair Bio-resources before transferring to our animal facility. All microswine were fed with RDI high-cholesterol diet (HC) D17012601 that contains 51% carbohydrates, 20% protein, 10% fat, and 4% cholesterol. For these experiments, the microswine were divided randomly into five different experimental groups; angioplasty alone, lipopolysaccharide (LPS; 100 µg/3 mL of PBS) alone, oxidized low-density lipoprotein (oxLDL; 600 µg/3 mL of PBS) alone, angioplasty + LPS (100 µg/3 mL of PBS), and angioplasty + oxLDL (600 µg/3 mL of PBS) consisting of 6–7 animals in each group with similar body weights and age. All the surgical procedures and tissue harvesting were performed as discussed previously by our group [29]. Specifically, balloon angioplasty for the carotid arteries was performed through the femoral artery of either side. The femoral artery was punctured with ultrasound guidance and a 4 F sheath was introduced via femoral artery, followed by a 6 F sheath to insert the guiding catheters typically used for human angiography. A guidewire was inserted using the guiding catheter to the carotid artery by injecting the iodine contrast media simultaneously to visualize the guidewire. A balloon angioplasty catheter (Abbott Vascular, USA) was inserted through the guidewire to reach the bifurcation of the selected common carotid artery (CCA) into ascending pharyngeal artery (APA) and inflated to at least 1.1 times the measured internal diameter of the artery (measured by optical coherence tomography; OCT). The inflated balloon was moved up and down to induce intimal injury and plaque formation in the carotid artery. In the angioplasty alone group, after intimal injury, the catheter was removed without any further intervention. In the experimental groups of angioplasty + LPS and angioplasty + oxLDL, the angioplasty balloon was deflated and retracted. LPS (100 µg/3 mL of PBS) or oxLDL (600 µg/3 mL of PBS) was administered intraluminally at the site of angioplasty through the OTW (over the wire) catheter after removing the guidewire while holding the blood flow by keeping the balloon inflated for 2 min to induce plaques inside the artery. Similarly, for the LPS only and oxLDL only groups, the treatment was given intraluminally through the OTW catheter without injuring the carotid artery with angioplasty [29].
Tissue harvesting and processing
After the surgical procedure, euthanasia was performed by i.v administration of a single dose of pentobarbital sodium (85 mg/kg) and phenytoin sodium (11 mg/kg). Tissues were harvested and kept in formalin solution for long storage. Collected carotid tissues fixed in formalin were dehydrated with a series of ethanol solutions of increasing concentration until water-free ethanol was reached. Typical dehydration was done with different concentrations of ethanol as, 70% ethanol for 15 min, 90% ethanol for 15 min, two times in 100% ethanol for 15 min, 100% ethanol for 30 min, and 100% ethanol for 45 min. After dehydration, clearing of the tissues was done by immersing the tissues in xylene followed by wax infiltration. All the processed tissues were then embedded in paraffin wax blocks and thin sections of 5 μm were cut using a microtome and sections were fixed on glass slides for staining.
Hematoxylin and eosin (H&E) staining
For H&E staining, the tissue sections were de-paraffinized, rehydrated in ethanol, rinsed in double-distilled water, and stained with hematoxylin and eosin for 45 and 30 s, respectively. The stained sections were mounted with a xylene-based mounting medium and a coverslip was placed over the tissue. The stained sections were examined under a light microscope (Leica DM6 at ×100 magnification). We scanned at least three adjacent sections from each tissue.
Immunohistochemistry (IHC)
IHC was performed using the peroxidase anti-peroxidase method using a secondary antibody conjugated to horseradish peroxidase (HRP). For immunohistochemistry, the slides were deparaffinized and rehydrated. The antigen retrieval was carried out by heating the slides with 1% citrate buffer (C9999; Sigma) in a steamer for 45 min. After 1 h cool-down period, the slides were washed with phosphate-buffered saline (PBS) for 5 min. The tissue sections were circled with a PAP pen and endogenous peroxidases were blocked with 3% hydrogen peroxide (H1009; Sigma) for 15 min and rinsed again in PBS for 5 min. Blocking was done by using the blocking solution (150 µL of stock in 10 mL of buffer) corresponding to the primary antibody from the Vectastain Elite ABC kit (Vector Labs PK-6101: rabbit; and PK-6105: goat) and the tissues were incubated for 1 h at room temperature. The blocking solution was tipped off and the tissue sections on the slides were incubated with the primary antibodies anti-S100A12 (MBS2026249) and anti-RAGE (sc8230; validated in swine) at an empirically optimized dilution and incubated at 4 °C overnight. The next day, the slides were washed 3 times for 5 min each with 1× PBS, and the slides were incubated with the secondary antibody (50 µL of biotinylated secondary antibody stock in 10 mL of blocking solution) from the corresponding Vectastain Elite ABC kit for 1 h at room temperature. The slides were rinsed 3 times with 1× PBS, followed by incubation of the tissue sections with the Vectastain ABC horseradish peroxidase (HRP) for 30 min at RT. The tissue sections on the slides were then rinsed with 1× PBS followed by incubation with 3,3′-diaminobenzidine (DAB) (34002; Thermo Scientific) for 2 to 5 min until the development of brown color. After the DAB had developed sufficiently, slides were washed with water, stained with hematoxylin for 20 s, rinsed in running tap water for 5 min, and mounted with a xylene-based mounting medium. The stained slides were imaged with a Leica DM6 microscope. The high magnification images from each tissue section were manually analyzed using Image J and the average intensity of the positively stained cells (% area stained) was analyzed from each group using three sections for each tissue for statistical analysis.
Oxidized low-density lipoprotein (ox-LDL) preparation
The ox-LDL used for this study was prepared by the controlled oxidation of LDL which was extracted from the plasma of atherosclerotic pigs using a single-step discontinuous gradient ultracentrifugation method [28]. The density gradient was created using potassium bromide (KBr) and the density was adjusted to 1.063 g/mL [using the formula plasma (mL) × 0.0834 = KBr (g)] followed by ultra-centrifugation in a Beckman L8 80 M Ultracentrifuge using an SW55 Ti swinging bucket rotor at 100,000 g for 24 h. The LDL-containing band was carefully removed, and the lipoprotein concentration was measured by Pierce™ BCA Protein Assay Kit. The dialysis-purified LDL was oxidized using 5 µM copper sulfate (CuSO4) for 2 h and was stopped with ethylene diamine tetra acetic acid (EDTA) and was further purified by dialysis to remove excess EDTA. The level of oxidation was quantified by TBARS assay using a commercially available kit (Cayman chemical, Cat#: 10009055) following the manufacturer’s protocol and was detected to be > 2 µM.
Cell culture
Primary vascular smooth muscle cells were harvested from the common carotid arteries collected from the contralateral side of the swine of angioplasty group euthanized after the completion of the experiments. Briefly, smooth muscle cells were grown after mincing the arteries and digesting with Collagenase I (Sigma: Cat#: C0130) in a smooth muscle cell-specific growth medium (SMCM 1101; ScienCell), and cells up to passage 4 were used for the experiment. The characterization of VSMCs was done by staining the cells for α-smooth muscle actin using immunofluorescence assay (data not shown). In six-well culture plates, cells were grown up to 80–90% confluency confirmed under a light microscope. The cells were incubated with two different concentrations of LPS (100 ng/mL and 500 ng/mL) and oxLDL (50 µg/mL and 100 µg/mL) for 24 h in a humidified incubator with 5% CO2 at 37 °C. After 24 h, cells were washed with PBS and total RNA was extracted.
RNA extraction and real-time PCR
Total RNA was extracted from treated and untreated cultured smooth muscle cells using Trizol reagent (Invitrogen, Carlsbad, CA), and 1.5 µg of total RNA was used for cDNA synthesis with ImProm-II Reverse Transcription System from Promega following the manufacturer’s instructions. Real-time PCR was carried out using a CFX96 Touch Real-Time PCR Detection System from BioRad. The PCR cycling conditions were 3 min at 95 °C for initial denaturation, 42 cycles of 15 s at 95 °C, 15 s at 55–60 °C (according to the primer annealing temperatures) followed by melting curve analysis. Reactions were run in triplicate for three independent experiments. The primers for S100A12 and RAGE genes in swine were obtained from Integrated DNA Technologies (Coralville, IA, USA) for which sequences were as follows: S100A12 (Fwd-5′-GTACTCAGTTCGGTTGGGGC-3′, Rev-5′-ATCAGCACATCAGTCACCAGG-3′), RAGE (Fwd-5′-ACCGAGTCCAAGTCTACCGT-3′, Rev-5′-GGTGGAAGGCTTTTCCTGA-3′). The expression data were normalized to the geometric mean of housekeeping gene GAPDH to control the variability in expression levels. The sequences of the forward and reverse primer of GAPDH were (Fwd-5′-CGGAGTGAACGGATTTGGCCG-3′, Rev-5′-GGAACTTGCCGTGGGTGGAA-3′). Fold change in expression of mRNA transcripts relative to controls was determined using the 2−ΔΔCT after normalizing the CT values with GAPDH [30].
Statistical analysis
Data are presented as the mean ± SD. Data were analyzed using GraphPad Prism 9. The comparison between groups for the expression of the protein of interest was performed using One-way ANOVA with Bonferroni’s posthoc test. The probability (p) value < 0.05 was accepted as statistically significant. *p < 0.05, **p < 0.01, ***p < 0.001 and ****p < 0.0001.
Results
Hematoxylin and eosin (H&E) staining showed significantly increased neointima formation in angioplasty with oxLDL carotid arteries
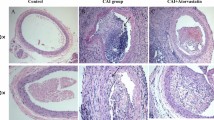
H&E-stained sections revealed obliterated carotid arteries lumen with an atherosclerotic plaque in the animal groups of angioplasty with LPS and angioplasty with oxLDL. (Fig. 1) Additionally, a minimal neointimal hyperplasia was observed in angioplasty group alone and inflammation was present in the neointima and the adjoining medial layer. (Fig. 1a) while in the other treatment groups such as LPS and oxLDL alone, we did not observe inflammation and neointima formation which justifies the potential of LPS and oxLDL with angioplasty to induce significant atherosclerotic plaques. In the angioplasty alone group, a thin-sized atherosclerotic plaque was observed (Fig. 1a). In the LPS- and oxLDL-treated swine without angioplasty, no plaque was observed. While in the experimental groups of angioplasty with LPS and angioplasty with oxLDL, a significant plaque size (approximately 30–50% of the luminal surface) was observed. More specifically, about 50% of the luminal surface was covered with plaque and the lumen was found blocked in angioplasty with oxLDL group with dense inflammation with atherosclerotic plaque (Fig. 1e, e′) and about 30% of the luminal surface was found with the plaque formation in angioplasty with LPS group (Fig. 1d, d′). But in the angioplasty with the oxLDL group, one section showed the presence of ruptured plaque while others suggest vulnerable plaque with characteristics such as macrophage infiltration, necrotic core, thin capped atheroma [31]. (Fig. 1e′) The light necrotic core was present in the angioplasty with LPS-treated carotid arteries (Fig. 1d, d′) but was found significantly lower than that of the oxLDL-treated group accompanied with the angioplasty. We speculated that the expression of S100A12 and RAGE proteins might be dependent on the size and severity of the lesion formed in the carotid arteries and thus proceeded to perform IHC with S100A12 and RAGE antibodies.

H&E staining of carotid artery on the intervention side in angioplasty alone (panel a), LPS alone (panel b), oxLDL alone (panel c) angioplasty with LPS (panel d), and angioplasty with oxLDL (panel e); Panels a′–e′ are the higher magnification images of the corresponding panels. H&E hematoxylin and eosin, LPS lipopolysaccharide, oxLDL oxidized low-density lipoprotein
Immunohistochemistry revealed significantly increased expression of S100A12 and RAGE expression in angioplasty with the LPS group
Immunostaining of the carotid arteries revealed that the expression of S100A12 and RAGE proteins completely depends on the treatment given to the carotid arteries. The angioplasty with LPS and angioplasty with oxLDL group produced significant lesion size. S100A12 and RAGE expression in the contralateral side carotid (control) artery of angioplasty alone group was lower than that of the intervention side carotid artery but the differential expression was statistically non-significant (Figs. 2 and 3). The expression on the intervention was significantly increased in the carotid arteries of angioplasty with LPS and angioplasty with oxLDL groups (Figs. 2 and 3). When we compared the data of angioplasty alone group with LPS alone and oxLDL alone treated groups, it was observed that the positive staining of S100A12 and RAGE was higher than that of LPS- and oxLDL-treated swine groups (Table 1). Particularly, S100A12 expression in the angioplasty group was 6.38% while in the case of LPS- and oxLDL-treated groups the expression was slightly lesser than the angioplasty group i.e., 3.91% and 5.40%, respectively (Table 1). The significant difference in the expression of S100A12 was observed in the angioplasty with LPS when compared to the angioplasty alone group which indicated that S100A12 expression increased by two-folds by angioplasty with LPS treatment i.e., 11.90% (Fig. 2e, h; Table 1). The angioplasty with oxLDL group did not show significant enhancement in the expression of S100A12 when compared with angioplasty alone (Fig. 2i). These results suggest that LPS potentially induces S100A12 expression in carotid arteries compared to oxLDL when accompanied with angioplasty.

IHC staining of S100A12 in the carotid artery on the contralateral side in angioplasty alone as control (panel a), intervention side in angioplasty alone (panel b), LPS alone (panel c), oxLDL alone (panel d) angioplasty with LPS (panel e), and angioplasty with oxLDL (panel f); Panels a′–f′ are the higher magnification images of the corresponding panels; Panels g–l show the percent expression of S100A12 and statistical difference between the groups. *p < 0.05, **p < 0.01, ***p < 0.001 and ****p < 0.0001. IHC immunohistochemistry, LPS lipopolysaccharide, oxLDL oxidized low-density lipoprotein

IHC staining of RAGE in the carotid artery on the contralateral side in angioplasty alone as control (panel a), intervention side in angioplasty alone (panel b), LPS alone (panel c), oxLDL alone (panel d) angioplasty with LPS (panel e), and angioplasty with oxLDL (panel f); Panels a′–f′ are the higher magnification images of the corresponding panels; Panels g–l show the percent expression of RAGE and statistical difference between the groups. *p < 0.05, **p < 0.01, ***p < 0.001 and ****p < 0.0001. IHC immunohistochemistry, RAGE receptor for advanced glycation end products, LPS lipopolysaccharide, oxLDL oxidized low-density lipoprotein
Similarly, immunohistochemistry revealed comparable RAGE expression and was almost 3-folds more in angioplasty with LPS (9.75%) than that of angioplasty alone (3.14%) on the side of intervention (Fig. 3b, e, h; Table 1). A two-fold increase in RAGE expression was observed in angioplasty with oxLDL (6.56%) as compared to angioplasty alone but was statistically significant (Fig. 3b, f, i; Table 1). The RAGE expression in angioplasty with LPS was significantly higher than in the angioplasty alone group. There was no statistically significant difference between the change in RAGE expression in angioplasty, LPS, and oxLDL alone treated groups.
Real-time PCR showed increased S100A12 and RAGE mRNA expression with LPS and oxLDL
Quantitative Real-time polymerase chain reaction (RT-qPCR) results of the VSMCs isolated from the contralateral carotid arteries of the angioplasty group Yucatan microswine treated with different concentrations of LPS (0, 100, and 500 ng/mL) and oxLDL (0, 50 and 100 µg/mL) for 24 h showed significant change in the mRNA expression of S100A12 and RAGE (Fig. 4a, b). In these cells, LPS significantly increased the mRNA expression of S100A12 in a concentration-dependent manner. The mRNA expression increased by 2-folds at a concentration of 100 ng/mL compared to untreated cells (control) which was consistent up to 500 ng/mL of LPS (Fig. 4a). A slight increase in the mRNA expression of S100A12 was observed with oxLDL at concentrations of 50 and 100 µg/mL. The mRNA expression of RAGE in VSMCs with the treatment of LPS slightly increases with 100 ng/mL but decreases with an increase in the concentration of LPS (Fig. 4b). This decrease in mRNA expression may be due to the use of higher concentration of LPS, the effect is well known as the Hook effect [32] which explains the fact that at a high enough concentration the amount of formed product decreases. The oxLDL treatment induced concentration-dependent effect and produced significant enhancement by 3.5-fold compared to untreated cells in the mRNA expression of RAGE (Fig. 4b).

mRNA expression of the genes S100A12 (a) and RAGE (b) in smooth muscle cells treated with different concentrations of LPS and oxLDL. *p < 0.05, **p < 0.01, ***p < 0.001 and ****p < 0.0001. mRNA messenger ribonucleic acid, RAGE receptor for advanced glycation end products, LPS lipopolysaccharide, oxLDL oxidized low-density lipoprotein
Discussion
In the present study, we found increased protein expression of S100A12 and RAGE in the carotid arteries from the swine group with angioplasty and treated with LPS compared to oxLDL. The higher expression of S100A12 and RAGE in the carotid arteries with LPS treatment suggests that this lipid antigen may be the most critical factor contributing to the progression of atherogenic inflammation in CVDs [33]. It is well known that inflammation is intimately associated with atherosclerosis and LPS has been identified to be a potent pathogen-associated molecular pattern (PAMP) inflammatory molecule aggravating the pathology [34]. Particularly, previous studies showed that the in vivo administration of LPS increased the level of S100A12 in the plasma in healthy volunteers [35]. These findings were also supported by another study in Takayasu arteries in healthy volunteers. The mRNA expression of S100A12 was also found significantly increased after the stimulation by LPS in healthy volunteers [36]. S100A12 is the ligand for RAGE, therefore increased expression of RAGE in the current study probably points towards the preferential engagement of RAGE with S100A12 in atherosclerotic inflammation [37]. Various studies have documented that RAGE activation upon binding with S100A12 associates with various inflammatory disease conditions including CVD, peripheral arterial disease, and diabetes [20, 34, 38, 39]. These studies suggest the potential association of the LPS-S100A12-RAGE axis in chronic inflammation in atherosclerosis and progression and plaque development. Further, denudation of endothelium by balloon angioplasty induces atherosclerosis in arteries [40, 41]. This suggests that treating arteries with LPS after balloon angioplasty will have synergistic pathological effects. In this study, the histological findings of narrowed lumen of carotid arteries in angioplasty + LPS group compared to angioplasty or LPS alone support the notion of a synergistic effect of angioplasty and LPS. This might be due to increased lipid accumulation in adventitia and inflammation due to LPS in a denuded artery with endothelial injury and dysfunction [41,42,43].
In this study, we induced endothelial injury with balloon angioplasty first followed by LPS administration and increased lesion in such arteries compared to LPS or angioplasty alone support the notion that LPS administration after angioplasty increases atherosclerotic plaque formation probably due to increased inflammation caused by S100A12-RAGE axis activation. These results are further supported by the findings of light necrotic core observed in the angioplasty + LPS group carotid arteries on H&E staining probably due to the lipid accumulation (Fig. 1d). Lipid accumulation in these arteries is due to the hypercholesterolemic diet given to microswine [44]. Lipid accumulation and LPS-mediated chronic inflammation induce more detrimental effects than angioplasty or LPS alone (Fig. 1d). LPS aggravates the inflammatory process through S100A12- RAGE axis by producing pro-inflammatory cytokines like IL-1β, IL-6, and TNF-α [45]. Angioplasty procedure directly damages the arteries. The acute response to this damage or injury induced by angioplasty initiates vascular sepsis and restenosis consisting of thrombus formation and inflammation in the intimal layer of carotid arteries. The simultaneous injection of LPS or oxLDL further aggravates the inflammatory response to induce the formation of plaque inside the carotid arteries [46]. Therefore, the treatment of LPS or oxLDL alone without any injury at the carotid arteries cannot induce the formation of plaque inside the arteries as much as of angioplasty with LPS or oxLDL. In this study, the aim was to delineate the effect of LPS on S100A12 and RAGE expression. However, the effect on downstream signaling pathway is yet to be explored. Since lipid accumulation underlies the pathogenesis of atherosclerotic plaque formation [47], we investigated the effect of minimally oxidized LDL (oxLDL) on the expression of S100A12 and RAGE. The results of this study revealed increased expression of S100A12 and RAGE in oxLDL-treated arteries with angioplasty but oxLDL alone showed a similar change in the expression levels as that of the LPS alone.
Overall, the increased level in the expression of S100A12 and RAGE in different treatment groups were found with the following trend: Angioplasty with LPS > angioplasty with oxLDL > angioplasty ≥ LPS alone ≈ oxLDL. Therefore, our study generated another hypothesis that LPS potentially can induce the protein expression of both S100A12 and RAGE in carotid arteries in the swine group with angioplasty. Gene expression of S100A12 and RAGE evaluated in VSMCs isolated from control carotid arteries and treated with LPS (100 and 500 ng/mL) and oxLDL (50 and 100 µg/mL) further corroborated the IHC results. The mRNA expression of S100A12 was found significantly increased with LPS and oxLDL compared to the untreated cells while the expression of RAGE was increased with 100 ng/mL LPS and oxLDL. Increased expression of S100A12 and RAGE in cultured cells with LPS and oxLDL support the findings of immunostaining. Altogether, these findings suggest S100A12 and its receptor RAGE as potential therapeutic targets to attenuate inflammation and progression of atherosclerosis [20, 48].
Conclusion
S100A12 and RAGE play a critical role in atherosclerotic inflammation which is the major cause of cardiovascular diseases. This study revealed that S100A12 and RAGE expression was increased in the arteries with the vascular injury and treated with LPS more than the arteries with angioplasty and treated with oxLDL. Lipid antigens (LPS) perform their function to further accelerate the inflammatory process by increasing the expression of these proteins. The findings of our study signify that LPS is the potential inducer of the protein and gene expression of S100A12 and RAGE in atherosclerotic inflammation in the carotid arteries. Therefore, S100A12 and RAGE could be potential novel targets to inhibit chronic inflammatory cascade and thus can attenuate the progression of atherosclerosis and plaque instability in carotid arteries.
References
Moran AE, Roth GA, Narula J, Mensah GA (2014) 1990–2010 global cardiovascular disease atlas. Glob Heart 9:3–16. https://doi.org/10.1016/j.gheart.2014.03.1220
Lozano R, Naghavi M, Foreman K, Lim S, Shibuya K, Aboyans V et al (2012) Global and regional mortality from 235 causes of death for 20 age groups in 1990 and 2010: a systematic analysis for the Global Burden of Disease Study 2010. Lancet 380:2095–2128. https://doi.org/10.1016/S0140-6736(12)61728-0
https://www.who.int/news-room/fact-sheets/detail/cardiovascular-diseases-(cvds). Accessed 18 July 2021
Kubo T, Imanishi T, Takarada S, Kuroi A, Ueno S, Yamano T, Tanimoto T, Matsuo Y, Masho T, Kitabata H, Tsuda K, Tomobuchi Y, Akasaka T (2007) Assessment of culprit lesion morphology in acute myocardial infarction: ability of optical coherence tomography compared with intravascular ultrasound and coronary angioscopy. J Am Coll Cardiol 50:933–939. https://doi.org/10.1016/j.jacc.2007.04.082
Mozaffarian D, Benjamin EJ, Go AS, Arnett DK, Blaha MJ, Cushman M et al (2016) Executive summary: heart disease and stroke statistics-2016 update a report from the American Heart Association. Circulation 133:447–454. https://doi.org/10.1161/Cir.0000000000000366
Rai V, Rao VH, Zhifei Shao, Agrawal DK (2016) Dendritic cells expressing triggering receptor expressed on myeloid cells-1 correlate with plaque stability in symptomatic and asymptomatic patients with carotid stenosis. PLoS One 11:e0154802. https://doi.org/10.1371/journal.pone.0154802
Hanck AB (1973) Plasma cholesterol level in healthy subjects and its modification by large doses of L(+)-ascorbic acid. Z Ernahrungswiss 12:152–158. https://doi.org/10.1007/BF02023914
Jonasson L, Holm J, Skalli O, Bondjers G, Hansson GK (1986) Regional accumulations of T-cells, macrophages, and smooth-muscle cells in the human atherosclerotic plaque. Arteriosclerosis 6:131–138. https://doi.org/10.1161/01.Atv.6.2.131
Jonasson L, Holm J, Skalli O, Gabbiani G, Hansson GK (1985) Expression of class II transplantation antigen on vascular smooth muscle cells in human atherosclerosis. J Clin Investig 76:125–131. https://doi.org/10.1172/JCI111934
Hansson GK, Jonasson L (2009) The discovery of cellular immunity in the atherosclerotic plaque. Arterioscler Thromb Vasc Biol 29:1714–1717. https://doi.org/10.1161/Atvbaha.108.179713
Xu QB, Oberhuber G, Gruschwitz M, Wick G (1990) Immunology of atherosclerosis: cellular composition and major histocompatibility complex class II antigen expression in aortic intima, fatty streaks, and atherosclerotic plaques in young and aged human specimens. Clin Immunol Immunopathol 56:344–359. https://doi.org/10.1016/0090-1229(90)90155-j
Binder CJ, Horkko S, Dewan A, Chang MK, Kieu EP, Goodyear CS, Shaw PX, Palinski W, Witztum JL, Silverman GJ (2003) Pneumococcal vaccination decreases atherosclerotic lesion formation: molecular mimicry between Streptococcus pneumoniae and oxidized LDL. Nat Med 9:736–743. https://doi.org/10.1038/nm876
Stemme S, Faber B, Holm J, Wiklund O, Witztum JL, Hansson GK (1995) T lymphocytes from human atherosclerotic plaques recognize oxidized low density lipoprotein. Proc Natl Acad Sci USA 92:3893–3897. https://doi.org/10.1073/pnas.92.9.3893
Perez Avila J (1989) Assistance to patients with AIDS. Rev Cubana Med Trop 41:319–320
Libby P (2012) Inflammation in atherosclerosis. Arterioscler Thromb Vasc Biol 32:2045–2051. https://doi.org/10.1161/ATVBAHA.108.179705
Nagareddy PR, Noothi SK, Flynn MC, Murphy AJ (2018) It’s reticulated: the liver at the heart of atherosclerosis. J Endocrinol 238:R1–R11. https://doi.org/10.1530/JOE-18-0082
Husain K, Hernandez W, Ansari RA, Ferder L (2015) Inflammation, oxidative stress and renin angiotensis system in atherosclerosis. World J Biol Chem 3:209–217. https://doi.org/10.4331/wjbc.v6.i3.209
Xiao X, Yang C, Qu SL, Shao YD, Zhou CY, Chao R, Huang L, Zhang C (2019) S100 proteins in atherosclerosis. Clin Chim Acta 502:293–304. https://doi.org/10.1016/j.cca.2019.11.019
Camoretti-Mercado B, Karrar E, Nunez L, Bowman MAH (2012) S100A12 and the airway smooth muscle: beyond inflammation and constriction. J Allergy Ther 3(Suppl 1):S1-S007. https://doi.org/10.4172/2155-6121.S1-007
Oesterle A, Bowman MA (2015) S100A12 and the S100/calgranulins: emerging biomarkers for atherosclerosis and possibly therapeutic targets. Arterioscler Thromb Vasc Biol 35:2496–2507. https://doi.org/10.1161/ATVBAHA.115.302072
Chiou JW, Fu B, Chou RH, Yu C (2016) Blocking the interactions between calcium-bound S100A12 protein and the V domain of RAGE using tranilast. Plos One. https://doi.org/10.1371/journal.pone.0162000
Hofmann MA, Drury S, Fu CF, Qu W, Taguchi A, Lu Y, Avila C, Kambham N, Bierhaus A, Nawroth P, Neurath MF, Slattery T, Beach D, McClary J, Nagashima M, Morser J, Stern D, Schmidt AM (1999) RAGE mediates a novel proinflammatory axis: a central cell surface receptor for S100/calgranulin polypeptides. Cell 97:889–901. https://doi.org/10.1016/S0092-8674(00)80801-6
Yan L, Per Bjork, Butu R, Gawdzik J, Earley J, Kim G, Bowman MAH (2013) Beneficial effects of quinoline-3-carboxamide (ABR-215757) on atherosclerotic plaque morphology in S100A12 transgenic ApoE null mice. Atherosclerosis 228:69–79. https://doi.org/10.1016/j.atherosclerosis.2013.02.023
Zhao P, Wu M, Yu H, Huang Y, Wang Y, Wang W, Yin W (2013) Serum S100A12 levels are correlated with the presence and severity of coronary artery disease in patients with type 2 diabetes mellitus. J Investig Med 61:861–866. https://doi.org/10.2310/JIM.0b013e318292fb1e
Realegeno S, Kelly-Scumpia KM, Dang AT, Lu J, Teles R, Liu PT, Schenk M, Lee EY, Schmidt NW, Wong GCL, Sarno EN, Rea TH, Ochoa MT, Pellegrini M, Modlin RL (2016) S100A12 is part of the antimicrobial network against Mycobacterium leprae in Human macrophages. Plos Pathog. https://doi.org/10.1371/journal.ppat.1005705
Gottsch JD, Eisinger SW, Liu SH, Scott AL (1999) Calgranulin C has filariacidal and filariastatic activity. Infect Immun 67:6631–6636
Schmidt AM, Du Yan S, Yan SF, Stern DM (2001) The multiligand receptor RAGE as a progression factor amplifying immune and inflammatory responses. J Clin Investig 108:949–955. https://doi.org/10.1172/Jci200114002
Lira-Junior R, Holmstrom SB, Clark R, Zwicker S, Majster M, Johannsen G, Axtelius B, Akerman S, Svensson M, Klinge B, Bostrom EA (2020) S100A12 expression is modulated during monocyte differentiation and reflects periodontitis severity. Front Immunol 11:86. https://doi.org/10.3389/fimmu.2020.00086
Sur S, Swier VJ, Radwan MM, Agrawal DK (2016) Increased expression of phosphorylated polo-like kinase 1 and histone in bypass vein graft and coronary arteries following angioplasty. PLoS ONE 11:e0147937. https://doi.org/10.1371/journal.pone.0147937
Holzinger D, Kessel C, Omenetti A, Gattorno M (2015) From bench to bedside and back again: translational research in autoinflammation. Nat Rev Rheumatol 11:573–585. https://doi.org/10.1038/nrrheum.2015.79
Perera C, McNeil HP, Geczy CL (2010) S100 calgranulins in inflammatory arthritis. Immunol Cell Biol 88:41–49. https://doi.org/10.1038/icb.2009.88
Calabrese EJ (2013) Biphasic dose responses in biology, toxicology and medicine: accounting for their generalizability and quantitative features. Environ Pollut 182:452–460. https://doi.org/10.1016/j.envpol.2013.07.046
Bowman JD, Surani S, Horseman MA (2017) Endotoxin, toll-like receptor-4, and atherosclerotic heart disease. Curr Cardiol Rev 13:86–93. https://doi.org/10.2174/1573403X12666160901145313
Rai V, Agrawal DK (2017) The role of damage- and pathogen-associated molecular patterns in inflammation-mediated vulnerability of atherosclerotic plaques. Can J Physiol Pharmacol 95:1245–1253. https://doi.org/10.1139/cjpp-2016-0664
Brooks D, Barr LC, Wiscombe S, McAuley DF, Simpson AJ, Rostron AJ (2020) Human lipopolysaccharide models provide mechanistic and therapeutic insights into systemic and pulmonary inflammation. Eur Respir J 56:1901298. https://doi.org/10.1183/13993003.01298-2019
Foell D, Wittkowski H, Kessel C, Luken A, Weinhage T, Varga G, Vogl T, Wirth T, Viemann D, Bjork P, van Zoelen MA, Gohar F, Srikrishna G, Kraft M, Roth J (2013) Proinflammatory S100A12 can activate human monocytes via toll-like receptor 4. Am J Respir Crit Care Med 187:1324–1334. https://doi.org/10.1164/rccm.201209-1602OC
Chellan B, Sutton NR, Bowman MAH (2018) S100/RAGE-mediated inflammation and modified cholesterol lipoproteins as mediators of osteoblastic differentiation of vascular smooth muscle cells. Front Cardiovasc Med 5:163. https://doi.org/10.3389/fcvm.2018.00163
Egana-Gorrono E, Lopez-Diez R, Yepuri G, Ramirez LS, Reverdatto S, Gugger PF, Shekhtman A, Ramasamy R, Schmidt AM (2020) Receptor for advanced glycation end products (RAGE) and mechanisms and therapeutic opportunities in diabetes and cardiovascular disease: insights from human subjects and animal models. Front Cardiovasc Med 7:37. https://doi.org/10.3389/fcvm.2020.00037
Malmstedt J, Karvestedt L, Swedenborg J, Brismar K (2015) The receptor for advanced glycation end products and risk of peripheral arterial disease, amputation or death in type 2 diabetes: a population-based cohort study. Cardiovasc Diabetol 14:93. https://doi.org/10.1186/s12933-015-0257-5
Smet BJ, Pasterkamp G, Helm YJ, Borst C, Post MJ (1998) The relation between de novo atherosclerosis remodeling and angioplasty-induced remodeling in an atherosclerotic Yucatan micropig model. Arterioscler Thromb Vasc Biol 18:702–707. https://doi.org/10.1161/01.atv.18.5.702
Rai V, Agrawal DK (2021) Immunomodulation of IL-33 and IL-37 with vitamin D in the neointima of coronary artery: a comparative study between balloon angioplasty and stent in hyperlipidemic microswine. Int J Mol Sci 22:8824. https://doi.org/10.3390/ijms22168824
Wang J, Si Y, Wu C, Sun L, Ma Y, Ge A, Li B (2012) Lipopolysaccharide promotes lipid accumulation in human adventitial fibroblasts via TLR4-NF-κB pathway. Lipids Health Dis 11:139. https://doi.org/10.1186/1476-511X-11-139
Autar A, Taha A, Duin RV, Krabbendam-Peters I, Duncker DJ, Zijlstra F, Beusekom HMM (2020) Endovascular procedures cause transient endothelial injury but do not disrupt mature neointima in drug eluting stents. Sci Rep 10:2173. https://doi.org/10.1038/s41598-020-58938-z
Yin K, You K, Swier V, Tang L, Radwan MM, Pandya AN, Agrawal DK (2015) Vitamin D protects against atherosclerosis via regulation of cholesterol efflux and macrophage polarization in hypercholesterolemic swine. Arterioscler Thromb Vasc Biol 35:2432–2442. https://doi.org/10.1161/ATVBAHA.115.306132
Gupta GK, Agrawal T, Core MGD, Hunter WJ, Agrawal DK (2012) Decreased expression of vitamin D receptors in neointimal lesions following coronary artery angioplasty in atherosclerotic swine. PLoS One 7:e42789. https://doi.org/10.1371/journal.pone.0042789
Wilensky RL, March KL, Gradus-Pizlo I, Sandusky G, Fineberg N, Hathaway DR (1995) Vascular injury, repair, and restenosis after percutaneous transluminal angioplasty in the atherosclerotic rabbit. Circulation 92:2995–3005. https://doi.org/10.1161/01.CIR.92.10.2995
Pirillo A, Norata GD, Catapano AL (2013) LOX-1, OxLDL, and atherosclerosis. Mediat Inflamm 2013:152786. https://doi.org/10.1155/2013/152786
Singh H, Rai V, Agrawal DK (2021) Discerning the promising binding sites of S100/calgranulins and their therapeutic potential in atherosclerosis. Expert Opin Ther Pat 10:1–13. https://doi.org/10.1080/13543776.2021.1937122
Acknowledgements
Carotid artery tissues in this study were obtained from another ongoing study in Yucatan microswine where Dr. Mohamed M. Radwan performed the surgery and radiology of the carotid artery in pigs. Dr. Sunil K. Nooti helped in the preparation of cDNA of the cultured smooth muscle cells.
Funding
The research work of DK Agrawal is supported by research Grant R01 HL144125 and R01 HL147662 from the National Heart, Lung, and Blood Institute, National Institutes of Health, USA. The content of this article is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health.
Author information
Authors and Affiliations
Contributions
HS: conceptualization, methodology, experimentation, data curation, writing—original draft preparation. VR and DKA: data curation, writing-reviewing and editing, investigation, formal analysis. DKA: conceptualization, supervision, investigation, writing-reviewing and editing. All authors read and approved the final manuscript.
Corresponding author
Ethics declarations
Conflict of interest
All authors have read the journal’s policy on disclosure of potential conflicts of interest. Author DKA has received research grants from the National Institutes of Health, USA. Other authors have no relevant affiliations or financial or non-financial involvement with any organization or entity with financial or non-financial interest or conflict with the subject matter or materials discussed in the manuscript apart from those disclosed. As the corresponding author, I declare that this manuscript is original; that the article does not infringe upon any copyright or other proprietary rights of any third party; that neither the text nor the data have been reported or published previously. All authors have read the journal’s policy on disclosure of potential conflicts of interest and declare that they have no conflict of interest.
Consent to participate
This article does not contain any studies with human participants performed by any of the authors.
Consent to publish
As the corresponding author, I verify that all authors significantly contributed to various aspect of the study, have read the manuscript, and consented to submit for publication in Molecular Biology Reports.
Animal studies
The animal studies were conducted with prior approval (R19IACUC026) from the Institutional Animal Care and Use Committee of Western University of Health Sciences, Pomona, California, USA, and the NIH and institutional guidelines for the care and use of animals were followed.
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
About this article

Cite this article
Singh, H., Rai, V. & Agrawal, D.K. LPS and oxLDL-induced S100A12 and RAGE expression in carotid arteries of atherosclerotic Yucatan microswine. Mol Biol Rep 49, 8663–8672 (2022). https://doi.org/10.1007/s11033-022-07703-8
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s11033-022-07703-8