
Abstract
Background
Familial adenomatous polyposis (known also as classical or severe FAP) is a rare autosomal dominant colorectal cancer predisposition syndrome, characterized by the presence of hundreds to thousands of adenomatous polyps in the colon and rectum from an early age. In the absence of prophylactic surgery, colorectal cancer (CRC) is the inevitable consequence of FAP. The vast majority of FAP is caused by germline mutations in the adenomatous polyposis coli (APC) tumor suppressor gene (5q21). To date, most of the germline mutations in classical FAP result in truncation of the APC protein and 60% are mainly located within exon 15.
Material and methods
In this first nationwide study, we investigated the clinical and genetic features of 52 unrelated Algerian FAP families. We screened by PCR-direct sequencing the entire exon 15 of APC gene in 50 families and two families have been analyzed by NGS using a cancer panel of 30 hereditary cancer genes.
Results
Among 52 FAP index cases, 36 had 100 or more than 100 polyps, 37 had strong family history of FAP, 5 developed desmoids tumors, 15 had extra colonic manifestations and 21 had colorectal cancer. We detected 13 distinct germline mutations in 17 FAP families. Interestingly, 4 novel APC germline pathogenic variants never described before have been identified in our study.
Conclusions
The accumulating knowledge about the prevalence and nature of APC variants in Algerian population will contribute in the near future to the implementation of genetic testing and counseling for FAP patients.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Familial adenomatous polyposis (FAP, OMIM + 175,100) is a rare autosomal dominant colorectal cancer predisposition syndrome, characterized by the presence of hundreds to thousands of adenomatous polyps in the colon and rectum from an early age, with over 90% risk of development of carcinoma in one or more of the polyps [1, 2]. The incidence of FAP is 3–10/100000, it is the most common adenomatous polyposis syndrome and accounting for less than 1% of all CRC cases [2]. CRC arise in FAP patients if untreated, patients develop colorectal cancer in a mean age of 40 years [1].
To date, various studies have described two FAP phenotypes: the classical form (CFAP), defined as > 100 colorectal polyps and early onset (polyp formation in the second decade of life); the attenuated FAP (AFAP) which is a milder form of classic FAP with less polyps (< 100) and a later age of polyp/cancer onset [2]
Prophylactic surgery (total abdominal colectomy), usually by age 40, is the gold-standard therapy to mitigate this risk [2]. Although surgery is the mainstay of treatment for FAP, some medical treatments have been used for prevention of adenoma formation like sulindac and celecoxib, that have been shown to reduce the number and size of rectal polyps [2]. Other gastrointestinal features (duodenal adenomas, fundic gland cysts) and extra gastrointestinal manifestations including congenital hypertrophy of the retinal pigment epithelium (CHRPE), dental abnormalities, osteomas, soft tissue tumors like epidermoid cysts, desmoid tumors are frequently described in FAP patients [3]. In addition, cancers of the thyroid, brain and biliary tracts are found to be associated with FAP [3].
FAP is caused by germline mutations in the APC gene located at 5q21-q22. The most frequent transcript is 8532 bp long with exon 15 accounting for 77% of the coding region. The APC gene encodes a tumor suppressor protein (comprising 2843 amino acids) that acts as an antagonist of the Wnt signaling pathway. It is also involved in other processes including cell migration and adhesion, transcriptional activation, and apoptosis. APC protein plays a central role in the Wnt-signalling pathway, especially in regards to the degradation of β-catenin within the cell cytoplasm. If APC is mutated, the β- catenin-Tcf complex is not suppressed and leads to constitutive activation of several genes and oncogenes controlling cell growth and division. Mutations in APC affect the ability of the cell to maintain normal growth and function, which results in cell overgrowth/adenoma formation and cancer development [4]
The 5’ coding region of exon 15 includes a mutation-cluster region and mutations contributing to classical FAP occur between exon 5 and the 5’ portion of exon 15 [5,6,7]. Since the identification of the APC gene, 1801 unique variants of the APC gene have been reported in the InSiGHT database (http://insight-database.org/).
Since the cloning of the APC gene [8, 9], several studies in populations and ethnic groups from America, Asia, Europe have reported large APC mutations spectrum, mutational hotspots and various genotype phenotype correlations [3, 4, 8,9,10,11,12,13,14,15,16,17,18]. To date, clinical and genetics features of FAP are largely unknown in Algeria and in the other North African countries. To our best knowledge, only three APC genotype–phenotype association studies have been reported in one Algerian FAP family and in small set of Tunisian FAP patients, respectively [19,20,21]. The present study is the first nationwide FAP cohort being reported in Algeria with molecular analysis of germline mutations in the APC gene.
Patients and methods
Study population
This nationwide study investigated index cases and relatives from 52 unrelated FAP families selected between March 2012 and May 2019. Patients and relatives were referred through gastroenterology service of Mustapha Bacha University Hospital (Algiers), academic general surgery services of public hospital of Kouba (Algiers) and academic medical oncology services and academic general surgery services of public hospital of Rouiba (Eastern suburb of Algiers). Clinical and pathological information was extracted from medical records of the patients with particular attention to the age at diagnosis, number and location of adenomas, the presence of colorectal cancer or extracolonic manifestations. Family histories of FAP were obtained from interviews, pedigrees and chart review of the index cases. Cancer registries of the three public hospitals covered an area of 20 provinces among 48 of Algeria (Fig. 1). All patients, relatives and parents/legally authorized representatives of the minor subjects provided an informed consent before blood sampling and molecular analysis of the APC gene.

Map showing the 20 Algerian provinces (indicated by red circle symbol) covered by the cancer registries of the three Public hospitals where the FAP patients included in our study were diagnosed and treated
DNA isolation
Genomic DNA was extracted from peripheral blood lymphocytes using Promega Wizard Genomic DNA Purification Kit (Promega, Madison, MI, USA) (Cat. # A1120) and in accordance with the manufacturer’s protocols.
Mutation analysis
The entire exon 15 of the APC gene (6.5 Kb) has been screened by PCR- direct sequencing in a cohort of 52 FAP index cases and 9 relatives, respectively. PCR and Sanger sequencing were performed as described elsewhere [22]. Primers used to screen exon 15 of the APC gene are available in Supplementary file1 (See Supporting Table 1).Identified pathogenic DNA sequence variants were confirmed by sequencing both DNA strands on at least two independent PCR products.
NGS analysis
Two patients were analyzed by Color Genomics using a cancer panel of 30 hereditary cancer genes test (Color genomics, Burlingame, San Francisco, USA, https://www.color.com). Pathogenic variant identified in the exon 12 of APC gene by NGS has been confirmed by PCR-Sanger sequencing and performed as described elsewhere [22]. Primers used to screen exon 12 of the APC gene are available in Supplementary file1 (See Supporting Table 1).
Nomenclature and variant analysis
All nucleotide numbers refer to the wild-type cDNA human sequence of APC gene (NM_000038.6) as reported in the GenBank database. The description of nucleotide sequence variants is in accordance with HGVS (Human Genome Variation Society) nomenclature rules (www.hgvs.org/mutnomen). The HGVS approved systematic nomenclature follows the rule where the nucleotide + 1 is the A of the ATG translation initiation codon. The pathogenic variants identified in our study were screened for clinical significance and if it were reported or novel in InSiGHT database, UMD APC-mutations database, ClinVar database, gnomAD genomes and gnomAD exomes database. Novel variants not described in the InSiGHT database or ClinVar database were classified according to ACMG classification.
Results
Patient characteristics
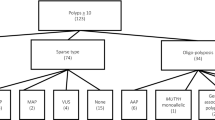
In this nationwide study, there were 29 women and 23 men diagnosed with severe FAP (Tables 1, 2, 3 and Figs. 1, 2, 3 summarize the results). The mean age at diagnosis was 32.92 years (range 12–54 years), 76.91% cases (40/52) have been diagnosed at age under 40 years and 69.23% cases (36/52) had 100 or more than 100 polyps (Table 1). Among 52 FAP index cases, 37 had strong family history of FAP, 5 developed desmoids tumors, 15 had extracolonic manifestations, 21 had colorectal cancer. In addition, there were 220 FAP affected individuals among the 52 FAP families included in our study (Table 2).


a–l Pedigrees of 12 FAP families with deleterious mutations in the APC gene

Sequence forward electropherogram of germline pathogenic variants identified in the APC gene of FAP index patients: a index patient F1610 carrier of the novel germline pathogenic variant c.1605dupT; b index patient F1506 carrier of germline pathogenic variant c.3927_3931delAAAGA; c mother of index patient F1506 negative for the germline mutation c.3927_3931delAAAGA; d index patient F1606 carrier of the germline pathogenic variant c.2544dupA
Analysis of the exon 15 of the APC gene using PCR-sanger-sequencing
The germline mutations analysis using PCR-Sanger sequencing of the entire exon 15 of the APC gene detected the presence of pathogenic sequence variant in 21 individuals (Table 3). Among them, 16 FAP index cases (32%) and five relatives from 4 FAP families, respectively (Table 3 and Fig. 2). The mutation spectrum consists of twelve point mutations. All these mutations have been detected in the 5′ coding region of the exon 15 of the APC gene. Of these 12 point mutations, 10 were frameshift and 2 were non sense substitutions. Four of these point mutations are novel. Interestingly, the most occurring mutations c.3927_3931delAAAGA and c.4728dupA were identified in four and two index cases in 6 unrelated FAP families, respectively (Table 3 and Fig. 3).
NGS analysis
The analysis of two patients using a cancer panel of 30 hereditary cancer genes (Color Genomics) revealed a novel pathogenic mutation in the exon 12 of the APC gene, a frameshit sequence variant c.1605dupT in the proband F1610 diagnosed at age 28 years with severe FAP (Table 2 and Fig. 2, pedigree a). This patient has a strong family history of FAP and 16 individuals among his relatives are FAP affected along 3 generations.
Out of 16 relatives, five were deceased and only 4 first relatives (3 sisters and one brother) were available for the screening for the new candidate mutation c.1605dupT. Unfortunately, healthy individuals of the family F1610 were not available for the screening of the new candidate mutation c.1605dupT. Subsequently genetic testing was performed using PCR- Sanger sequencing in the four relatives affected with FAP (3 sisters and one brother) of our index case F1610. The four relatives were APC mutation positive for the new candidate mutation c.1605dupT. (Table 3 and Figs. 2–3, pedigree a).
The new frameshift sequence variant c.1605dupT co-segregated with FAP syndrome in 4 first relatives of our index case F1610, we can classify it as Class 5 variant with the status: “Pathogenic” according to ACMG classification.
Genotype–phenotype correlations in FAP patients with APC mutation
We noticed that the mean age for 17 FAP patients with APC mutation was 27 years and five developed a colorectal cancer, two had desmoid tumors and 3 had extracolonic manifestations (Table 2). Sixteen FAP index cases were found to carry a germline mutation in the 5′ coding region of the exon 15 of the APC gene and the mutations are located between codons 843–1577 (Tables 2, 3).
Discussion
To date, there is a limited information in the mutational spectrum of the APC gene in Algerian population [19]. In our present study, we used PCR-Sanger sequencing for the analysis of the exon 15 of the APC gene and we successfully detected germline pathogenic variants in 16 of 50 FAP families. In addition, NGS analysis of two patients using a panel of 30 hereditary cancer genes revealed a novel point germline mutation in the exon 12 of the APC gene in a young FAP patient.
Thirteen distinct APC germline pathogenic variants were found in 17 unrelated FAP families confirming mutation heterogeneity in APC gene as already reported in various populations and ethnic groups [3, 4, 8,9,10,11,12,13,14,15,16,17,18].The detected mutations in the 5′ coding region of exon 15 scattered between codon 843 and codon 1577. Five mutations are located inside the mutation cluster region (MCR). Here, we report for the first time in Algerian population in the APC gene the 5 bp canonical deletion c.3927_3931delAAAGA located in the mutational hot spot at codon 1309 and it was detected in 7.69% unrelated FAP index cases (4/52) of our 52 FAP families. To date, this mutation has been reported at moderate frequency in South American populations and at high frequency in several European populations and Asian populations [5, 23,24,25,26,27,28]. Three patients’ carrier of this mutation showed severe early onset polyposis among them one patient developed a CRC at early age, respectively. In addition, among them a young female patient has been diagnosed with severe FAP at age 12 years (patient F1506), her parents were not affected and she went under colectomy at the age of 16 years. As the pathogenic germline variant c.3927_3931delAAAGA occurs at high frequency de novo [29], we noticed that out of 4 index cases carriers of this mutation, three of them (patients F1210, F1409 and F1506) have not reported a family history of FAP. Interestingly, subsequently genetic testing of the mother of patient F1506 showed the absence of the mutation c.3927_3931delAAAGA. Although the healthy father of the patient F1506 and the healthy parents of the two other index cases (Patients F1210, F1409) were not tested, we could not rule out that the mutation c.3927_3931delAAAGA had occurred de novo in our three FAP index cases.
We also detected a second germline mutation c.3925G > T in the mutational hot spot at codon 1309 in the young FAP patient F1418 with strong family history of FAP along three generations. He was diagnosed with severe FAP with more than 100 colonic adenomas, duodenal adenomas and anastomotic stenosis. To date, the pathogenic germline variant c.3925 G > T has been reported in two German patients who showed somatic APC mosaicism [30], one Dutch patient (Carli Tops, unpublished result reported in InSIGHT database, http://insight-database.org/genes/APC) and one French patient, respectively [23].
Interestingly, in our study, we also detected the rare germline pathogenic variant c.3905delT in a young female FAP index case (Patient F1201) with strong family history of FAP along four generations. The rare germline mutation c.3905delT has been reported one time in FAP patient from Italy [31]. Our patient F1201 was diagnosed with an early profuse colorectal polyposis and presented with more than 100 colonic adenomas. Her mother and one maternal uncle were diagnosed with FAP and CRC and deceased at age 43 and 40 years, respectively.
We did not detect a mutation in the second mutational hot spot at codon 1061 in our FAP families, but we identified the common germline pathogenic variant c.3202_3205delTCAA at codon 1068 in a young female FAP patient and her brother. She presented 100 colonic adenomas, duodenal adenomas and developed CRC at age 40 years. Interestingly, these clinical manifestations have been already reported in two Japanese FAP patients’ carriers of the germline pathogenic variant c.3202_3205delTCAA [32].
We detected the rare germline pathogenic variant c.2527_2530delAGTT in a patient without a family history of FAP but with a familial CRC history. He was diagnosed with FAP at age 40 years and developed less than 100 colorectal polyps, adenomas all over the duodenum. He developed a rectal cancer at age 46 years and he deceased. This patient showed some differences in genotype–phenotype correlations reported in Polish and Australian FAP patients, respectively, carriers of the germline mutation c.2527_2530delAGTT y [33, 34].
We also identified for the first time in Algerian FAP patients, the germline pathogenic variant c.2544dupA in a young female index FAP patient diagnosed at age 18 years and her affected sister. She had a strong family history of FAP along three generations and 7 of her relatives are FAP affected. She showed a profuse polyposis with several hundred of colorectal polyps and extracolonic manifestations.
In our study, we detected the common germline pathogenic variant c.2805C > A in a female patient who developed a severe FAP with more than 100 colorectal polyps. She had strong family history of FAP along three generations and 9 of her relatives are FAP affected (5 of them developed CCR). In addition, her father developed a thyroid cancer. Interestingly, the pathogenic variant c.2805C > A has been already identified in one large Algerian FAP family [19]. The variant has been detected in the index case and 12 FAP affected relatives along 4 generations [19]. To date, the germline pathogenic variant c.2805C > A has also been reported in Korean, Singapore and Portuguese FAP families, respectively, with thyroid tumor as extracolonic manifestation [35,36,37].
The common germline pathogenic variant c.3471_3474delGAGA at codon 1157 has been detected in our current study in a FAP patient diagnosed at late age of 44 years and developed less than 100 colonic polyps, duodenal polyps and desmoid tumors. He had a family history of FAP and six of his relatives were FAP affected (4 of them had colon surgery). Interestingly, the two extracolonic manifestations of this patient have been reported in a familial CRC patient carrier of the pathogenic variant c.3471_3474delGAGA and a germline mutation in MSH2 gene, respectively [38].
We identified a novel germline mutation in the 5′ coding region of the exon 15 in the APC gene c.3784delT at codon 1262 in a young female FAP patient and her two FAP affected sisters (ISF131301 and ISF131302). She had strong family history of FAP (5 relatives developed a severe FAP), in addition, she was diagnosed with a CRC at early age 24 years and her sister ISF131301 had developed desmoid tumors at age 37 years.
Interestingly, our 2 Algerian FAP patients with mutations in codons 1157 and 1262, respectively, who developed desmoid tumors, showed different genotype–phenotype correlations reported in previous studies in Caucasian patients that associated desmoid tumors beyond codon 1444 [39,40,41].
The rare germline pathogenic variant c.4384_4385delAA at codon 1462 reported two times in two French FAP families [23], was detected in our study in young female FAP index patient who developed fewer than 50 colorectal polyps, duodenal polyposis, desmoid tumors and mandibular and dental abnormalities. These extracolonic manifestations have been reported in previous studies in FAP patients’ carriers of germline mutations in the APC gene located between codons 1444–1578 and 767–1578, respectively [42].
We detected a novel germline mutation in the exon 15 of the APC gene, c.4559dupA/p.Thr1487Asnf*27 in a young female FAP patient diagnosed with profuse colorectal polyposis at early age of 14 years. She developed duodenal polyps and desmoid tumors at age 21 years. She had a strong family history of FAP along 4 generations and five of her relatives are FAP affected.
Interestingly, in our current study, we identified for the first time in two unrelated Algerian FAP families, the very rare germline mutation c.4728dupA/Glu1577Argfs*14 reported only one time in InSIGHT database in one Japanese FAP family back in the year 1994 [43]. In the first FAP family (F1407) the index patient carrier of this mutation was diagnosed with FAP at age 37 years and developed less than 100 colonic polyps. In the second FAP family, the index case (F1704) and his sister (F1703) were found to carry the pathogenic variant c.4728dupA. He developed less than 50 colonic polyps and osteomas. His sister has developed desmoid tumors. Thirteen relatives of this second family were FAP affected. As the codon 1577 is the most 3’ end mutation in our current study, our two FAP families’ carriers of the germline pathogenic variant c.4728dupA could be affected with AFAP.
As the germline pathogenic variant c.4728dupA has been identified in two unrelated FAP families, the codon 1577 could be a new mutational hot spot in Algerian FAP families.
For the first time, NGS analysis using a panel of 30 hereditary cancer genes revealed a novel germline pathogenic variant in the exon 12 of the APC gene c.1605dupT/p.(Glu536*) in a young FAP index patient diagnosed with a profuse colorectal polyposis at age 28 year. He developed a colorectal cancer at age 31 years. His family had strong history of FAP and 16 of his relatives were FAP affected along 3 generations. Subsequently genetic testing was performed in four relatives of our index case (3 sisters and one brother affected with FAP syndrome). All the four relatives were found to carry the new candidate mutation c.1605dupT. Previous studies have reported germline mutations in the exon 12 of the APC gene in patients with classical FAP [23, 44, 45]. In addition, our results are in agreement with previous studies [16, 46,47,48] that showed that NGS technology and the screening of cancer panel of hereditary cancer genes are powerful tools to identify new APC candidate mutations in large FAP families.
There are some limitations in our present study, which should be considered. We studied a small set of FAP families and we only screened the exon 15 of the APC gene. A limitation of our study is that several exons of the APC gene have not been analyzed in our patients. We are also aware that LGR have not been screened in the APC gene using NGS or MLPA. Screening of all coding exons of the APC gene including flanking regions in a large cohort of FAP families, and using NGS analysis of cancer panel of hereditary colorectal cancer genes will let us to know about the mutational profile of APC gene. We also should include in our screening the four other polyposis associated genes MUTYH, NHTL1, POLD1 and POLE in order to assess their prevalence in Algerian population.
A key strength of the present study is that we performed the first genetic testing of APC germline mutations in Algerian FAP cohort using PCR-direct sequencing and NGS analysis. We also reported for the first time the genotype phenotype correlations in FAP patients. The mutational spectrum knowledge of polyposis-associated genes in FAP families will contribute to the implementation of cost-effective strategies for the prevention and the treatment of familial adenomatous polyposis.
Conclusions
Our present study provides for the first time insight in the clinical and genetic features of FAP syndrome in Algerian population. Our data confirmed the mutation heterogeneity in the APC gene in FAP patients. The screening of the APC gene in large cohort of FAP patients will help to implement affordable genetic testing and to improve the clinical management and risk assessment of FAP syndrome.
Data availability
Data that support the conclusions of this article are included within the article.
Abbreviations
- APC :
-
Adenomatous polyposis coli gene
- CRC:
-
Colorectal cancer
- CHRPE:
-
Congenital hypertrophy of the retinal pigment epithelium
- FAP:
-
Familial adenomatous polyposis syndrome
- LGR:
-
Large genomic rearrangement
- MLPA:
-
Multiplex ligation probe amplification
- NGS:
-
Next generation sequencing
References
Bülow S (1991) Diagnosis of familial adenomatous polyposis. World J Surg 15(1):41–46
Byrne RM, Tsikitis VL (2018) Colorectal polyposis and inherited colorectal cancer syndromes. Ann Gastroenterol 31(1):24–34
Friedl W, Aretz S (2005) Familial adenomatous polyposis: experience from a study of 1164 unrelated german polyposis patients. Hered Cancer Clin Pract 3(3):95–114
Talseth-Palmer BA (2017) The genetic basis of colonic adenomatous polyposis syndromes. Hereditary cancer in clinical practice 15:5. https://doi.org/10.1186/s13053-017-0065-x
Nallamilli B, Hegde M (2017) Detecting APC gene mutations in familial adenomatous polyposis (FAP). Curr Protoc Hum Genet, 92, 10.8.1–10.8.16.
Fodde R, Smits R, Clevers H (2001) APC, signal transduction and genetic instability in colorectal cancer. Nat Rev Cancer 1(1):55–67
Kerr SE, Thomas CB, Thibodeau SN, Ferber MJ, Halling KC (2013) APC germline mutations in individuals being evaluated for familial adenomatous polyposis: a review of the Mayo Clinic experience with 1591 consecutive tests. J Mol Diagnostics 15(1):31–43
Groden J, Thliveris A, Samowitz W, Carlson M, Gelbert L, Albertsen H, Joslyn G, Stevens J, Spirio L, Robertson M (1991) Identification and characterization of the familial adenomatous polyposis coli gene. Cell 66(3):589–600
Kinzler KW, Nilbert MC, Su LK, Vogelstein B, Bryan TM, Levy DB, Smith KJ, Preisinger AC, Hedge P, McKechnie D (1991) Identification of FAP locus genes from chromosome 5q21. Science 253(5020):661–665
Hutter P, Rey-Berthod C, Chappuis PO, Couturier A, Membrez V, Murphy A, Joris F, Schorderet DF, Delozier-Blanchet C, Soravia C (2001) Molecular and clinical characteristics in 32 families affected with familial adenomatous polyposis. Hum Mutat 18(6):550
Andresen PA, Heimdal K, Aaberg K, Eklo K, Ariansen S, Silye A, Fausa O, Aabakken L, Aretz S, Eide TJ, Gedde-Dahl T Jr (2009) APC mutation spectrum of Norwegian familial adenomatous polyposis families: high ratio of novel mutations. J Cancer Re Clin Oncol 135(10):1463–1470
Papp J, Kovacs ME, Matrai Z, Orosz E, Kásler M, Børresen-Dale AL, Olah E (2016) Contribution of APC and MUTYH mutations to familial adenomatous polyposis susceptibility in Hungary. Fam Cancer 15(1):85–97
Miyoshi Y, Ando H, Nagase H, Nishisho I, Horii A, Miki Y, Mori T, Utsunomiya J, Baba S, Petersen G (1992) Germ-line mutations of the APC gene in 53 familial adenomatous polyposis patients. Proc Natl Acad Sci USA 89(10):4452–4456
Khan N, Lipsa A, Arunachal G, Ramadwar M, Sarin R (2017) Novel mutations and phenotypic associations identified through APC, MUTYH, NTHL1, POLD1, POLE gene analysis in indian familial adenomatous polyposis cohort. Sci Rep 7(1):2214
Li N, Kang Q, Yang L, Zhao XJ, Xue LJ, Wang X, Li AQ, Li CG, Sheng JQ (2019) Clinical characterization and mutation spectrum in patients with familial adenomatous polyposis in China. J Gastroenterol Hepatol 34(9):1497–1503
Giang H, Nguyen VT, Nguyen SD, Nguyen HP, Vo BT, Nguyen TM, Nguyen NH, Truong KD, Do TT, Phan MD, Nguyen HN (2018) Detection of a heterozygous germline APC mutation in a three-generation family with familial adenomatous polyposis using targeted massive parallel sequencing in Vietnam. BMC Med Genet 19(1):188
Araujo LF, Molfetta GA, Vincenzi OC, Huber J, Teixeira LA, Ferraz VE, Silva WA Jr (2019) Molecular basis of familial adenomatous polyposis in the southeast of Brazil: identification of six novel mutations. Int J Biol Markers 34(1):80–89
Terlouw D, Suerink M, Singh SS, Gille H, Hes FJ, Langers A, Morreau H, Vasen H, Vos YJ, van Wezel T, Tops CM, Ten Broeke SW, Nielsen M (2020) Declining detection rates for APC and biallelic MUTYH variants in polyposis patients, implications for DNA testing policy. Eur J Hum Genet, EJHG 28(2):222–230
Mehenni H, Friedl W, Nelen MR, Hutter P, Brundler MA (2005) An unexpected Cowden syndrome case found among members of a large familial adenomatous polyposis kindred. Euro J Gastroenterol Hepatol 17(12):1407–1412
Bougatef K, Marrakchi R, Moussa A, Blondeau-Lahely Y, Najjar T, Coulet F, Colas C, Ben Ayed F, Elgaaied BAA, Soubrier F (2008) First genetic analysis in Tunisian familial adenomatous polyposis probands. Oncol Rep 19(5):1213–1218
Miladi-Abdennadher I, Amouri A, Ayadi L, Khabir A, Ellouze S, Tahri N, Frikha M, Sellami-Boudawara T, Mokdad-Gargouri R (2011) A novel pathogenic germline mutation in the adenomatous polyposis coli gene in a Tunisian family with FAP. Fam Cancer 10(3):567–571
Development of a resequencing workflow for variant analysis in the APC gene (2011) http://tools.thermofisher.com/content/sfs/brochures/cms_095915.pdf (www.lifetechnologies.com)
Lagarde A, Rouleau E, Ferrari A, Noguchi T, Qiu J, Briaux A, Bourdon V, Rémy V, Gaildrat P, Adélaïde J, Birnbaum D, Lidereau R, Sobol H, Olschwang S (2010) Germline APC mutation spectrum derived from 863 genomic variations identified through a 15-year medical genetics service to French patients with FAP. J Med Genet 47(10):721–722
Bertario L, Russo A, Sala P, Varesco L, Giarola M, Mondini P, Pierotti M, Spinelli P, Radice P, Registry HCT (2003) Multiple approach to the exploration of genotype-phenotype correlations in familial adenomatous polyposis. J Clin Oncol 21(9):1698–1707
Plawski A, Lubiński J, Banasiewicz T, Paszkowski J, Lipinski D, Strembalska A, Kurzawski G, Byrski T, Zajaczek S, Hodorowicz-Zaniewska D, Gach T, Brozek I, Nowakowska D, Czkwaniec E, Krokowicz P, Drews M, Zeyland J, Juzwa W, Słomski R (2004) Novel germline mutations in the adenomatous polyposis coli gene in Polish families with familial adenomatous polyposis. J Med Genet 41(1):e11
Kim DW, Kim IJ, Kang HC, Park HW, Shin Y, Park JH, Jang SG, Yoo BC, Lee MR, Hong CW, Park KJ, Oh NG, Kim NK, Sung MK, Lee BW, Kim YJ, Lee H, Park JG (2005) Mutation spectrum of the APC gene in 83 Korean FAP families. Hum Mut 26(3):281
Cao X, Hong Y, Eu KW, Loi C, Cheah PY (2006) Singapore familial adenomatous polyposis (FAP) patients with classical adenomatous polyposis but undetectable APC mutations have accelerated cancer progression. Am J Gastroenterol 101(12):2810–2817
Cruz-Correa M, Pérez-Mayoral J, Dutil J, Echenique M, Mosquera R, Rivera-Román K, Umpierre S, Rodriguez-Quilichini S, Gonzalez-Pons M, Olivera MI, Pardo S, Consortia PRCCG (2017) Hereditary cancer syndromes in Latino populations: genetic characterization and surveillance guidelines. Hered Cancer Clin Pract 15:3
Aretz S, Uhlhaas S, Caspari R, Mangold E, Pagenstecher C, Propping P, Friedl W (2004) Frequency and parental origin of de novo APC mutations in familial adenomatous polyposis. Eur J Hum Genet: EJHG 12(1):52–58
Aretz S, Stienen D, Friedrichs N, Stemmler S, Uhlhaas S, Rahner N, Propping P, Friedl W (2007) Somatic APC mosaicism: a frequent cause of familial adenomatous polyposis (FAP). Hum Mut 28(10):985–992
Gismondi V, Bafico A, Biticchi R, Pedemonte S, Molina F, Heouaine A, Sala P, Bertario L, Presciuttini S, Strigini P, Groden J, Varesco L (1997) Characterization of 19 novel and six recurring APC mutations in Italian adenomatous polyposis patients, using two different mutation detection techniques. Hum Mut 9(4):370–373
Enomoto M, Konishi M, Iwama T, Utsunomiya J, Sugihara KI, Miyaki M (2000) The relationship between frequencies of extracolonic manifestations and the position of APC germline mutation in patients with familial adenomatous polyposis. Jpn J Clin Oncol 30(2):82–88
Plawski A, Slomski R (2008) APC gene mutations causing familial adenomatous polyposis in Polish patients. J Appl Genet 49(4):407–414
Chen CS, Phillips KD, Grist S, Bennet G, Craig JE, Muecke JS, Suthers GK (2006) Congenital hypertrophy of the retinal pigment epithelium (CHRPE) in familial colorectal cancer. Fam Cancer 5(4):397–404
Won Y-J, Park KJ, Kwon H-J, Lee J-H, Kim J-H, Kim YJ, Chun SH, Han H-J, Park J-G (1999) Germline mutations of the APC gene in Korean familial adenomatous polyposis patients. J Hum Genet 44:103–108
Cao X, Eu KW, Seow-Choen F, Zao Y, Cheah PY (2000) APC mutation and phenotypic spectrum of Singapore familial adenomatous polyposis patients. Eur J Hum Genet 8:42
Filipe B, Baltazar C, Albuquerque C, Fragoso S, Lage P, Vitoriano I, Mão de Ferro S, Claro I, Rodrigues P, Fidalgo P (2009) APC or MUTYH mutations account for the majority of clinically well-characterized families with FAP and AFAP phenotype and patients with more than 30 adenomas. Clin Genet 76:242–255
Soravia C, DeLozier CD, Dobbie Z, Berthod CR, Arrigoni E, Bründler MA, Blouin JL, Foulkes WD, Hutter P (2005) Double frameshift mutations in APC and MSH2 in the same individual. Int J of Colorectal Dis 20(5):466–470
Caspari R, Olschwang S, Friedl W, Mandl M, Boisson C, Böker T, Augustin A, Kadmon M, Möslein G, Thomas G (1995) Familial adenomatous polyposis: desmoid tumours and lack of ophthalmic lesions (CHRPE) associated with APC mutations beyond codon 1444. Hum Mol Genet 4(3):337–340
Giardiello FM, Petersen GM, Piantadosi S, Gruber SB, Traboulsi EI, Offerhaus GJ, Muro K, Krush AJ, Booker SV, Luce MC, Laken SJ, Kinzler KW, Vogelstein B, Hamilton SR (1997) APC gene mutations and extraintestinal phenotype of familial adenomatous polyposis. Gut 40(4):521–525
Disciglio V, Fasano C, Cariola F, Forte G, Grossi V, Sanese P, Lepore Signorile M, Resta N, Lotesoriere C, Stella A, Lolli I, Simone C (2020) Gastric polyposis and desmoid tumours as a new familial adenomatous polyposis clinical variant associated with APC mutation at the extreme 3’-end. J Med Genet 57(5):356–360
Groen EJ, Roos A, Muntinghe FL, Enting RH, de Vries J, Kleibeuker JH, Witjes MJ, Links TP, van Beek AP (2008) Extra-intestinal manifestations of familial adenomatous polyposis. Ann Surg Oncol 15(9):2439–2450
Miyaki M, Konishi M, Kikuchi-Yanoshita R, Enomoto M, Igari T, Tanaka K, Muraoka M, Takahashi H, Amada Y, Fukayama M (1994) Characteristics of somatic mutation of the adenomatous polyposis coli gene in colorectal tumors. Cancer Res 54(11):3011–3020
Gómez-Fernández N, Castellví-Bel S, Fernández-Rozadilla C, Balaguer F, Muñoz J, Madrigal I, Milà M, Graña B, Vega A, Castells A, Carracedo A, Ruiz-Ponte C (2009) Molecular analysis of the APC and MUTYH genes in Galician and Catalonian FAP families: a different spectrum of mutations?. BMC Med Genet 10:57
Vandrovcová J, Stekrová J, Kebrdlová V, Kohoutová M (2004) Molecular analysis of the APC and MYH genes in Czech families affected by FAP or multiple adenomas: 13 novel mutations. Hum Mutat 23(4):397
Zhan Q, Wang L, Xu X, Sun Y, Li L, Qi X, Chen F, Wei X, Raff ML, Yu P, Jin F (2020) An APC Mutation in a Large Chinese Kindred With Familial Adenomatous Polyposis Was Identified Using Both Next Generation Sequencing and Simple STR Marker Haplotypes. Front Genet 11:191
Zhang Z, Liang S, Wang D, Liang S, Li Y, Wang B, Jiang T, Zhao G, Zhang X, Banerjee S (2017) A novel pathogenic single nucleotide germline deletion in APC gene in a four generation Chinese family with familial adenomatous polyposis. Sci Rep 7(1):12357. https://doi.org/10.1038/s41598-017-10395-x
Olkinuora AP, Peltomäki PT, Aaltonen LA, Rajamäki K (2021) From APC to the genetics of hereditary and familial colon cancer syndromes. Hum Mol Genet 30(R2):R206–R224. https://doi.org/10.1093/hmg/ddab208
Acknowledgements
We deeply thank the patients and their families for their participation. This study was supported by the Algerian National Research Program FNRSDT (D01N01UN160420130007) and PRFU (Project N° D01N01UN160420180005). Farid Cherbal would like to deeply and warmly thank Dr Alicia Zhou and Color Genomics (Burlingame, California, United States, https://www.color.com) for performing the NGS analysis in our Algerian FAP patients using Hereditary Cancer Test. Farid Cherbal would like to thank Imene Tabet, Sihem Hamine, Fatma Narimane Nouredine, Sarah Sabri and Adam Walid Damache for their big help with the FAP patients and cancer data. Farid Cherbal would like to thank Dr Philippe Maillet (Ornex, France), Prof. Kada Boualga (Anti Cancer Center, Blida, Algeria), Daoud Cherbal (Paris, France) and Romaissa Cherbal (Paris, France) for their permanent support to the research program on hereditary cancers in Algerian population.
Funding
The funded was provided by FNRSDT, (Grant No: D01N01UN160420130007). PRFU, (Grant No D01N01UN160420180005).
Author information
Authors and Affiliations
Contributions
FK collected and selected the FAP families, carried out the molecular genetics studies, analysis and interpretation of data, funding acquisition (Doctoral fellowship). FC prepared the study concept and design, supervised the study, participated in FAP families’ recruitment, carried out the molecular genetics studies, did the sequence alignment, data analysis and interpretation, funding acquisition, writing, drafting, reviewing and revising the manuscript. ALB participated in FAP families’ recruitment, analysis and interpretation of data. KL, HM, FZ and MM contributed to data collection and clinical data analysis. All authors read and approved the final manuscript.
Corresponding author
Ethics declarations
Conflict of interest
The authors declare non conflict of interest.
Ethical approval
All patients, relatives and parents/legally authorized representatives of the minor subjects tested for APC germline mutations and screened by PCR-direct sequencing and NGS analysis, respectively, signed written informed consent. The study was approved by the institutional review boards and ethical approval was obtained from appropriate institutions (USTHB, EPH Mohamed El Kolli, EPH Bachir Mentouri, University Hospital Mustapha Bacha, FNRSDT D01N01UN160420130007 and PRFU D01N01UN160420180005, 61 participants, start date: 3/1/2012, end date: 5/8/2019).
Informed consent
Informed consent was obtained from all individual participants and from parents/legally authorized representatives of the minor subjects included in the study.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
This study has been presented (in part) in a poster session at the 68th Annual Meeting of the American Society of Human Genetics (ASHG), 18 October 2018 in San Diego, California, USA.
Supplementary Information
Below is the link to the electronic supplementary material.
Rights and permissions
About this article

Cite this article
Khider, F., Cherbal, F., Boumehdi, AL. et al. Germline mutations of the adenomatous polyposis coli (APC) gene in Algerian familial adenomatous polyposis cohort: first report. Mol Biol Rep 49, 3823–3837 (2022). https://doi.org/10.1007/s11033-022-07228-0
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s11033-022-07228-0