
Abstract
Background
Thalassemias are common inherited blood disorders that have been extensively studied in Asia. Thus far, data on mutations of the HBB gene in Vietnamese patients with β-thalassemia are limited to small studies.
Methods
We recruited 696 β–thalassemia patients and carriers in southern Vietnam and analyzed for the HBB gene mutations using Sanger sequencing technology.
Results
We documented 27 types of known mutations and 10 types of novel variants on 737 alleles out of 1392 surveyed alleles. The three most common mutations, which account for more than ¾ of all mutant alleles, were c.79G > A (HbE), c.124_127delTTCT, and c.52A > T. The novel variants were mainly located in 5′ untranslated region (c.-92delC and c.-67A > G) and 3′ untranslated region (c.*4C > T, c.*116_*117insA, c.*142 T > C, c.*156G > C, c.*176_*177insA, and c.*247 T > C), except for one in intron 2 (c.316-99 T > G) and one in exon 3 (c.385delG).
Conclusion
We provide here a comprehensive mutation spectrum of the HBB gene in Southern Vietnam, which is crucial for carrier screening and prenatal diagnosis in the future.
Similar content being viewed by others
Avoid common mistakes on your manuscript.
Introduction
Thalassemias are the most common hereditary anemia disorders which cause serious health problem in developing countries, especially in Mediterranean areas, Africa, Middle East, Indian areas, and Southeast Asia. The disease is classified into two most common types according to the mutations of α-globin chain (α-thalassemia) and β-globin chain (β-thalassemia).The mutations result in an absence or reduction of α-globin or β-globin protein synthesis, leading to varied clinical aspects of thalassemia from blood transfusion-dependent to no symptom. The majority of mutations causing α-thalassemia are large deletions of one or two α-globin genes [1], while the nucleotide substitutions, frameshifts or minor deletions of the HBB gene causing β-thalassemia are frequently reported [2,3,4,5]. In transfusion-dependent cases, the clinical symptoms are fatigue, pale skin, weakness, jaundice, splenomegaly, bone deformity, iron loading, and growth retardation which require costly treatment [6]. Genetic counseling and prenatal diagnosis are principle methods to reduce the burden of health care problems in severe thalassemia cases.
Vietnam is located in the Southeast Asia where both β–thalassemia and hemoglobin (Hb) E are common. There were two community-based surveys of thalassemia and hemoglobinopathies on large individuals carrying out in the southern area and central, which showed that the frequency of HbE was markedly variation, ranging from 1 to 36% [7, 8]. The frequency of β–thalassemia carrier varied from 1.5 to 25% depending on different ethnic groups [5]. Several molecular based studies of the HBB gene mutations have been carried out in Vietnamese patients with β–thalassemia [4, 5, 9,10,11]. Most of these studies were performed on small number of patients, ranging from 22 to 50 patients and their families. In fact, Vo et al. analyzed on a large number of 244 patients in Northern Vietnam, but they screened for only 4 commonly known mutations [4]. Thus far, from all the above studies, totally 10 types of HBB mutations have been documented in Vietnamese, namely c.-78A > G, c.45_46insG, c.52A > T, c.79G > A, c.79G > T, c.92 + 1G > T, c.126_129delCTTT, c.216_217insA, c.287_288insA, and c.316-197C > T. We lack a large cohort study of HBB gene mutations in β-thalassemia patients and carriers in the Vietnamese. Given that more than 943 mutations in the HBB gene have been reported worldwide, we conducted the present study to uncover a comprehensive spectrum of the HBB gene mutations from 696 β–thalassemia patients and carriers in Southern Vietnam.
Materials and methods
Subjects
This study includes a total of 696 unrelated subjects in Southern Vietnam who were referred to the Blood Transfusion and Hematology Hospital at Ho Chi Minh City for HBB gene mutational analyses and had positive results, from February 2012 to April 2020. Subjects were suspected to have β–thalassemia disorder based on a mean corpuscular volume (MCV) < 80.0 fL and/or mean corpuscular hemoglobin (MCH) < 27 pg and HbA2 > 3.4%, after ruling out iron deficiency and other causes of microcytic hypochromic anemia. Because most specimens were from outside hospitals, we did not have access to detailed clinical data and treatment information. Before entry into the study, written informed consents were given by all the participants or their guardians. The protocol for this study was approved by the Ethics Committee of Blood Transfusion and Hematology Hospital at Ho Chi Minh City, Vietnam (approval number 0968/TMHH-HĐĐĐ).
Sample collection and DNA extraction
From each patient, we collected 2 mL of peripheral blood with EDTA anticoagulant. DNA was extracted using the GeneJET Genomic DNA Purification Kit (Thermo Scientific, USA) and stored at 2–8 °C until use. DNA quantity and purity were assessed using the NanoDrop™ 1000 (Thermo Scientific). DNA concentration was measured and DNA purity was calculated via the standard A260/280 and A260/230 ratios.
Mutation analysis
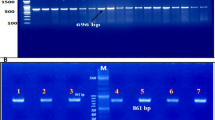
HBB gene mutational analysis was performed at the Center for Molecular Biomedicine, University of Medicine and Pharmacy at Ho Chi Minh City or at Department of Molecular Cytogenetics, Ho Chi Minh City Blood Transfusion and Hematology Hospital. The complete HBB gene was amplified with polymerase chain reaction (PCR) using Takara TaqHotStart Polymerase (Takara Bio, Shiga, Japan). PCR was done at an annealing temperature of 60 °C, in separate 25 µL reactions consisting of 1 × PCR Buffer, 1.5 mM MgCl2, 200 µM each dNTP, 0.5U Taq Hot Start Polymerase, 0.1 µM each forward and reverse primers, and 25–50 ng of genomic DNA. The forward primer (5′-GGCTGAGGGTTTGAAGTCCA -3′) and reverse primer (5′-GCAGCCTCACCTTCTTTCAT-3′), which covered the β–globin gene region from c.-233 to c.*279, were designed based on referenced genomic and coding sequences of the HBB gene (NG_059281.1 and NM_000518.5). The 1936-bp PCR products after amplification were purified with the Illustra™ GFX™ PCR DNA and Gel Band Purification Kit (GE Healthcare, USA) or with the ExoSAP-IT reagent (Thermo Scientific, USA) and were sequenced and analyzed in both sense and antisense directions using a Bigdye Terminator v3.1 cycle sequencing kit (Thermo Scientific, USA). The sequencing products were run on an ABI 3500 Genetic Analyzer (Applied Biosystems, USA). Mutations were analyzed using SeqScape Software version 2.6 (Thermo Fisher, Scientific, Waltham, MA, U.S.A) and compared with the reference sequence of HBB.
Results
This study reported on 696 subjects who had mutation of the HBB gene from detailed genetic analysis. The majority of subjects were β–thalassemia carriers who had asymptomatic or mild anemia with MCV < 80.0 fL and/or MCH < 27 pg. In many cases, β–thalassemia status was suspected through an examination for other diseases or a medical checkup. Regarding ethnicity, in this cohort study, Kinh people accounted for 89.51% (623 subjects), followed by Chinese (7.61%, 53 subjects), Khmer and others ethnic groups (2.88%, 20 subjects).
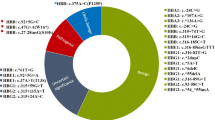
We detected 37 types of HBB mutations and novel variants on 737 alleles out of 1392 surveyed alleles (696 subjects). Figure 1 and Table 1 show the distribution and frequencies of these mutations and variants. In addition to 27 mutation types that have been reported in the literature, there were 10 novel variants in Vietnamese probands. The three most common mutations were c.79G > A (HbE), c.124_127delTTCT, and c.52A > T which account for 33.65%, 23.47%, and 19.95%, respectively. Other recurrent mutations with less commonness were c.-78A > G (3.39%), c.287_288insA (2.85%), c.92 + 1G > T (2.85%), c.216_217insA (2.17%), c.-100G > A (1.90%), c.*129 T > A (1.76%), and c.316-197C > T (1.49%), accounting in total for 16.41%. The 17 remaining types of reported mutation were found in 4.61% (34/737) of mutated alleles. Among these rare mutations, each of 10 mutations (c.-81A > G, c.-11_-8delAAAC, c.48G > A, c.67G > A, c.251G > A, c.364G > C, c.410G > A, c.92 + 5G > C, c.*110 T > C, and c.*132C > T) was detected in single cases.

Distribution of HBB gene mutations and variants detected in the study
We also detected 10 novel variants of HBB gene in 15 subjects. Among them, only one subject was at compound heterozygous status, harboring the well-known mutant allele c.79G > A in combination with the novel allele c.*116_*117insA. The other 14 subjects solely carried novel monoallelic variants. The novel variants were mainly located in 5′UTR (c.-92delC and c.-67A > G) and 3'UTR after stop codon (c.*4C > T, c.*116_*117insA, c.*142 T > C, c.*156G > C, c.*176_*177insA, and c.*247 T > C). There were one novel variant in intron 2 (c.316-99 T > G) and one in exon 3 (c.385delG).
Among 696 subjects with HBB gene mutation, 681 people solely carried known mutations (97.84%). We could classify the 681 carriers of reported mutations into two groups: 640 with monoallelic mutations and 41 with biallelic mutations (Table 2), accounting for 91.95% and 5.89% of the total cohort, respectively. More specifically, Table 2 shows that among 41 subjects with biallelic mutations, there were 28 who harbored at least one c.79G > A (HbE) allele.
Discussion
Thus far, reports on HBB gene mutations in Vietnamese are limited in terms of sample size and technique used. The present work is the largest study on 696 Vietnamese β–thalassemia patients and carriers who were confirmed to harbor HBB gene mutations. Sequencing analysis using Sanger method uncovered 27 types of well-known mutations and 10 novel variants. Among them, the three most common mutations which account for more than ¾ of all mutant alleles were c.79G > A (HbE), c.124_127delTTCT, and c.52A > T. The c.79G > A (HbE) mutation is the most common in Vietnamese, consistent with previous studies [4, 5, 9, 10, 12]. The initial study on 23 β–thalassemia patients in Northern Vietnam using sequence analysis also mainly detected c.79G > A, c.124_127delTTCT, and c.52A > T in addition to an indigenous mutation to Vietnam, c.287_288insA [12]. Recently, a small report from 22 β–thalassemia patients in Central Vietnam showed that the three mutations c.79G > A, c.52A > T, and c.124_127delTTCT accounted for 29.2%, 25.0%, and 18.8% of mutant alleles, respectively [9]. However, it should be noted that in Myanmar [13], Thailand [14], and China [15], the most common mutation was c.124_127delTTCT, not c.79G > A like our population.
Other mutations were dispersed throughout the full length of the HBB gene, from the promoter to 3'UTR after the stop codon. For this broad spectrum, sequencing technique might be most suitable for detecting all mutations or variants of the HBB gene, while other PCR-based methods only detect common mutations [5, 7, 10].
Gene sequencing was also able to detect 10 novel HBB variants, of which 7 variants were found only once in single individuals. Although the pathogenicity of each novel variant needs to be validated in functional study, we provide here evidence suggesting that some variants might be pathogenic. Among these novel variants, only the c.385delG was located in coding region of the HBB gene, resulting in a frameshift after codon 129 on exon 3 (p.Ala129fsGln). Within this location, the c.383_385delAGG mutation has been recurrently reported in Japanese patients [16,17,18], supporting that the c.385delG variant detected in our patient might be a pathogenic mutation causative of β–thalassemia.
There were 2 novel variants located in the 5'UTR region, namely c.-92delC and c.-67A > G. At c.-92 site, the c.-92C > G mutation has been reported in compound heterozygous status with the c.92G > C on exon 1, which led to anemia [19,20,21]. Moosa et al. further showed that the c.-92C > G could create a putative binding site for transcription factor Egr1, which proves the crucial role of this site in 5′UTR region [21].
The other 6 novel variants located in 3′UTR region after stop codon, including c.*4C > T, c.*116_*117insA, c.*142 T > C, c.*156G > C, c.*176_*177insA, and c.*247 T > C. Stability of HBB mRNA was heavily determined by the 3′UTR region, which was associated with 3′ end processing, polyadenylation, and mRNA capping [22]. The novel variant c.*4C > T was found in a 27-year-old male patient with mild anemia, without any additional HBB or HBA gene mutations (data not shown). Next to this site, there were 2 known mutations, namely c.*5G > A and c.*6C > G [15, 23]. Moreover, the c.*6C > G mutation has been shown to decrease stability of HBB mRNA which explained for a patient with β–thalassemia intermedia [23]. Therefore, the c.*4C > T is more likely to be a causative mutation explaining for our patient's mild anemia.
The variant c.*116_117insA is located between the polyadenylation A signal and the polyadenylation A cleavage sites, suggesting that this variant might interfere with mRNA maturation. Within this location, the c.*118A > G mutation in combination with the c.118C > T mutation was reported to be causative of β-thal intermedia in 2 children who were born to a β-thalassemia trait mother with monoallelic c.118C > T mutation [22]. To the best of our knowledge, mutation related to the positions of c.-67, c.316–99 and the others in 3′UTR in this study have not been documented in literature.
It should also be noted that among “well-known” mutations found in this study, a thalassemic behavior for some mutations such as c.251G > A (Hb Pyrgos), c.410G > A (Hb Hope), and c.364G > C (Hb D Punjab) has been still argued. Therefore, clinical interpretation for these mutations requires great precaution.
Limitations of this study are the lack of α-thalassemia mutational information and detailed clinical data, which make a genotype–phenotype analysis impossible in our cohort population. More works are needed to overcome this challenge. Moreover, Sanger sequencing is unable to detect a large deletion of HBB gene, which may account for 10% of β-thalassemia trait.
In conclusion, this study provides a comprehensive mutation spectrum of the HBB gene in Southern Vietnam. We believe that this is an important reference for further studies to see if the mutation spectrum is distinct for different regions of Vietnam, which is crucial for carrier screening and prenatal diagnosis in the future.
References
Guida V, Colosimo A, Fiorito M et al (2004) Denaturing HPLC-based assay for molecular screening of nondeletional mutations causing α-thalassemias. Clin Chem 50:1242–1245
Hassan S, Ahmad R, Zakaria Z et al (2013) Detection of β-globin gene mutations among β-thalassaemia carriers and patients in Malaysia: application of multiplex amplification refractory mutation system–polymerase chain reaction. Malays J Med Sci MJMS 20:13
Chan OTM, Westover KD, Dietz L et al (2010) Comprehensive and efficient HBB mutation analysis for detection of β-hemoglobinopathies in a pan-ethnic population. Am J Clin Pathol 133:700–707
Vo LTT, Nguyen TT, Le HX et al (2018) Analysis of common β-thalassemia mutations in north Vietnam. Hemoglobin 42:16–22
Svasti MLS, Hieu TM, Munkongdee T et al (2002) Molecular analysis of β-thalassemia in South Vietnam. Am J Hematol 71:85–88
Weatherall DJ, Clegg JB (1996) Thalassemia—a global public health problem. Nat Med 2:847–849
Nguyen HV, Sanchaisuriya K, Nguyen D et al (2013) Thalassemia and hemoglobinopathies in Thua Thien Hue province, central Vietnam. Hemoglobin 37:333–342
O’Riordan S, Hien TT, Miles K et al (2010) Large scale screening for haemoglobin disorders in southern Vietnam: implications for avoidance and management. Br J Haematol 150:359–364
Doro MG, Casu G, Frogheri L et al (2017) Molecular characterization of β-thalassemia mutations in Central Vietnam. Hemoglobin 41:96–99
Le TH, Pissard S, Pham HV et al (2001) Molecular analysis of β-thalassemia in South Vietnam. Hemoglobin 25:305–309
Anh TM, Sanchaisuriya K, Kieu GN et al (2000) Thalassemia and hemoglobinopathies in an ethnic minority group in Northern Vietnam. Hemoglobin 43:249–253
Filon D, Oppenheim A, Rachmilewitz EA et al (2000) Molecular analysis of β-thalassemia in Vietnam. Hemoglobin 24:99–104
Win N, Harano T, Harano K et al (2002) A wider molecular spectrum of beta-thalassaemia in Myanmar. Br J Haematol 117:988–992
Panichchob P, Iamdeelert P, Wongsariya P et al (2021) Molecular spectrum of β-Thalassemia mutations in Central to Eastern Thailand. Hemoglobin 45:97–102
Zhao J, Li J, Lai Q et al (2020) Combined use of gap-PCR and next-generation sequencing improves Thalassaemia carrier screening among premarital adults in China. J Clin Pathol 73:488–492
Ohba Y, Hattori Y, Harano T et al (1997) Beta-thalassemia mutations in Japanese and Koreans. Hemoglobin 21:191–200
Fucharoen S, Fucharoen G, Fukumaki Y et al (1990) Three-base deletion in exon 3 of the beta-globin gene produced a novel variant (beta gunma) with a thalassemia-like phenotype. Blood 76:1894–1896
Hattori Y, Yamane A, Yamashiro Y et al (1989) Characterization of beta-thalassemia mutations among the Japanese. Hemoglobin 13:657–670
Nagar R, Sinha S, Raman R (2015) Genotype-phenotype correlation and report of novel mutations in β-globin gene in thalassemia patients. Blood Cells Mol Dis 55:10–14
Yasmeen H, Toma S, Killeen N et al (2016) The molecular characterization of Beta globin gene in thalassemia patients reveals rare and a novel mutations in Pakistani population. Eur J Med Genet 59:355–362
Moosa MM, Ayub MI, Bashar AE et al (2011) Combination of two rare mutations causes β-thalassaemia in a Bangladeshi patient. Genet Mol Biol 34:406–409
Herrera MA, de La Fuente-Gonzalo F, González FA et al (2015) Identification of a novel mutation in the β-globin gene 3’ untranslated region (HBB: c.*+118A > G) in Spain. Hemoglobin 39:30–35
Jankovic L, Dimovski AJ, Kollia P et al (1991) A C—G mutation at nt position 6 3’ to the terminating codon may be the cause of a silent beta-thalassemia. Int J Hematol 54:289–293
Acknowledgements
The authors would like to thank the people of Southern Vietnam for participating in this study.
Funding
No.
Author information
Authors and Affiliations
Contributions
PTX, HQC, and HAV designed the study and wrote the manuscript. PTX, HDBT, LVHT, NTHH, HN, NTB, and PCD recruited the patients. PTX, HQC, NTTH, CVD, and HAV designed and performed the experiments. All authors read and approved the manuscript.
Corresponding author
Ethics declarations
Conflict of interest
The authors have no competing interests.
Ethical approval
This study was approved by the Ethics Committee of Blood Transfusion and Hematology Hospital at Ho Chi Minh City, Vietnam (approval number 0968/TMHH-HĐĐĐ).
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
About this article

Cite this article
Xinh, P.T., Chuong, H.Q., Ha, N.T.T. et al. Spectrum of HBB gene mutations among 696 β–thalassemia patients and carriers in Southern Vietnam. Mol Biol Rep 49, 2601–2606 (2022). https://doi.org/10.1007/s11033-021-07062-w
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s11033-021-07062-w