
Abstract
Purpose
The constitutive activation of STAT3 through receptor tyrosine kinases triggered breast cancer cell growth and invasion-metastasis. Atiprimod impacts anti-proliferative, anti-carcinogenic effects in hepatocellular carcinoma, lymphoma, multiple myeloma via hindering the biological activity of STAT3. Dose-dependent atiprimod evokes first autophagy as a survival mechanism and then apoptosis due to prolonged ER stress in pituitary adenoma cells. The therapeutic efficiency and mechanistic action of atiprimod in breast cancer cells have not been investigated yet. Thus, we aimed to modulate the pivotal role of ER stress in atiprimod-triggered apoptosis in MDA-MB-231 and MDA-MB-468 breast cancer cells.
Results
Dose- and time-dependent atiprimod treatment inhibits cell viability and colony formation in MDA-MB-468 and MDA-MB-231 breast cancer cells. A moderate dose of atiprimod (2 μM) inhibited STAT3 phosphorylation at Tyr705 residue and also suppressed the total expression level of p65. In addition, nuclear localization of STAT1, 3, and NF-κB was prevented by atiprimod exposure in MDA-MB-231 and MDA-MB-468 cells. Atiprimod evokes PERK, BiP, ATF-4, CHOP upregulation, and PERK (Thr980), eIF2α (Ser51) phosphorylation’s. However, atiprimod suppressed IRE1α-mediated Atg-3, 5, 7, 12 protein expressions and no alteration was observed on Beclin-1, p62 expression levels. PERK/eIF2α/ATF4/CHOP axis pivotal role in atiprimod-mediated G1/S arrest and apoptosis via Bak, Bax, Bim, and PUMA upregulation in MDA-MB-468 cells. Moreover, atiprimod renders MDA-MB-231 more vulnerable to type I programmed cell death by plasmid-mediated increased STAT3 expression.
Conclusion
Atiprimod induced prolonged ER stress-mediated apoptosis via both activating PERK/eIF2α/ATF4/CHOP axis and suppressing STAT3/NF-κB transcription factors nuclear migration in TBNC cells.
Similar content being viewed by others
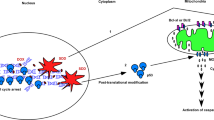


Avoid common mistakes on your manuscript.
Introduction
Breast cancer (BC) is the second cause of cancer-related death among females worldwide. Age, gender, family history, BRCA1, and BRCA2 gene mutations are known as major risk factors for BCs [1]. The underlying molecular mechanism of BC development is associated with various hormones such as Epidermal growth factor (EGF), Vascular endothelial growth factor (VEGF), progesterone, and estrogen. Due to growth triggering signals, the extracellular receptor profile of BC is correlated with aggressiveness and determines the response of BC against drug regimens [2]. Among BCs approximately 65–70% cases have Estrogen receptor positive (ER +) expression profile and give therapeutic achievement against adjuvant anti-estrogen therapy. However, 10–20% of BC cases are ER- and give no significant response against hormonal or targeted therapy. Although surgery, radiotherapy, and chemotherapy (olaparib, Talzenna, atezolizumab) combinations preferred for ER- BCs treatment, still new therapeutic agents needed to develop for triple-negative breast cancer (TNBCs) therapy [3].
Platelet-derived growth factors (PDGF) and EGF, and IL-6 family members induced cell proliferation, development, survival, angiogenesis, metastasis, and invasion through activation one of the key transcription factor, signal transducer and activator of transcriptions-3 (STAT3) [4]. Upstream kinases such as Janus kinases (JAKs) and c-Src kinase activation lead phosphorylation of STAT3 at Tyr 705 residue. Following homodimerization, STAT3 nuclear localization leading transcription of target gene expressions. Various target genes related to cellular processes such as cell cycle (Cyclin D1), angiogenesis (VEGF and HIF-1α), cell migration (MMP-2/9), and anti-apoptotic proteins (survivin, Bcl-xL, Bcl-2) expressions are under the control of phosphorylated STAT-3 homodimers nuclear localization [5]. Constitutively active STAT3 is demonstrated in an ample number of tumor types such as multiple myeloma, hepatocellular carcinoma, leukemia, lymphoma, ovarian, breast, and prostate cancers [6]. Moreover, STAT3/pSTAT3 expression was higher in breast cancer tissue samples compared to normal tissues [7]. Due to the essential role of STAT3 activation in carcinogenesis, different inhibitors targeting STAT3 are being designed, produced and developed as cancer therapeutics [8].
Midostaurin, lestaurtinib, K252a, CIMO, azaspirane, and atiprimod are well-known azaspirane-based tyrosine kinase inhibitors and their therapeutic efficiency has been reported in various clinical trials [9]. Atiprimod, known as an effective JAK2/JAK3 inhibitor, has anti-inflammatory, anti-proliferative, and anti-cancerogenic effects [10]. According to preclinical studies, dose-dependent atiprimod or CIMP inhibited cell proliferation, triggered apoptosis via activating caspase-3, -9 in various cancer cells such as myeloid lymphoma, hepatocellular carcinoma, respectively [11, 12]. The molecular mechanism underlying the atiprimod-induced apoptotic effect on hepatocellular carcinoma has been reported to have a suppressive effect on the STAT-3 molecule [13]. Although FDA approved atiprimod for the treatment of myeloid leukemia and mantle lymphoma, atiprimod therapeutic effect has been increased by combined treatment such as bortezomib [14, 15]. Recently, we reported that atiprimod triggered mild-ER stress and autophagy as an early response and finalized apoptosis via activating caspase-3 and -9 in GH3 cells [16].
One of the major obstacles in cancer therapy is the potential effect of drugs on endoplasmic reticulum (ER) stress-mediated activation of survival mechanism before type II programmed cell death decision has been taken. Due to drug dose- and time-dependent manner, ER stress performed through mild or severe conditions and leads to apoptotic cell death decision or autophagy as a cell survival mechanism [17]. To overcome unfolded protein response (UPR), ER receptors; PERK, IRE1α, and ATF6 inhibit protein translation, activate UPR target genes expression and induced translation of ER chaperon (BiP) [18]. The cross-talk between ER stress and autophagy has been modulated in IRE1α deficient or chemically inhibited JNK activation within cells as ER-stress-mediated autophagy has been inhibited. However, autophagy induced following ER stress has been reported in knock-down of ATF6 or PERK deficient cells [19]. Autophagy is a conserved self-degradation process of cells against pathogen infection, serum deprivation, organelle damage and ER stress [20]. Although autophagy is assumed as a protective mechanism of cancer cells against cancer therapeutic agents’ apoptotic efficiency due to studies with autophagy inhibitors, hyperactivation of autophagy can lead to cell death [21]. However, the pivotal role of mild or severe ER stress embarks on apoptotic cell death through either Ca2+ mediated caspase-12 activation or triggering PERK/eIF2α/ATF4/CHOP axis. We aimed in this study to modulate the role of ER stress in atiprimod-triggered apoptosis in MDA-MB-231, MDA-MB-468 breast cancer cells.
Material and methods
Cell culture, and drug
MDA-MB-231 (ATCC HTB-26) and MDA-MB-468 (ATCC CVCL_0419) human breast cancer cells were incubated in DMEM F12 medium (GIBCO-Life Technologies, Carlsbad, CA, USA) including Fetal bovine serum (15%), and penicillin/streptomycin (1%). The potential chemotherapeutic agent; N,N-diethyl-8,8-dipropyl-2-azaspiro[4.5]decane-2-propanamine dihydrochloride was obtained from Tocris (Bristol, UK).
Cell viability assay
The impact of atiprimod (0–5 µM) on BC cell lines viabilities were determined by MTT colorimetric assay. Each cell line was seeded at a density of 1 × 104 cells per well in 96-well plates and treated with drug exposure for 24 h. Then, 10 μl of MTT dye (5 mg/ml in PBS) was applied to each well for 4 h. Due to mitochondrial enzyme activation, formazan crystals converted from MTT dye were solubilized by 100 µl dimethyl sulfoxide (DMSO). The absorbance of the suspension was measured at 570 nm by a Bio-Rad microplate reader.
Cell growth and proliferation assay
The time-dependent manner impact of atiprimod on cell growth and colony diameter was determined by trypan blue, hanging drop assays that were described previously [22].
DiOC6, PI, DAPI, AO staining
Atiprimod-mediated mitochondrial membrane potential loss, cell death, nuclear condensation, and autophagosome vacuole formation in MDA-MB-468 and MDA-MB-231 cells were determined by DiOC6, PI, DAPI, and AO staining, respectively through protocol performed in our previous study [22].
GFP-LC-3II and CHOP activation via plasmid transfection and FACS flow analysis
BCs were seeded in 12 well-plate at a density of 5 × 104, 0.5 µg GFP-LC3 (addgene) or pmCherry-1 CHOP promoter (− 649/ + 136) (addgene #36,035) plasmids transfected by using Fugene HD reagent. Following 48 h transfection, 24 h drug (2 μM) applied to the culture medium. The fluorescence of green fluorescence and mCherry were detected by the BD Accuri cell flow cytometer via FL-1 and FL-3 channels, respectively. 1 × 104 cells/samples were analyzed for each condition.
Cell cycle analysis
5 × 104 cells/well were seeded in 12-well plates and treated with 0–2.5 µM atiprimod for 24 h. Following trypsinization, the cells were fixed with 70% Ethanol and incubated at -20 °C. Discarding ethanol by centrifugation, cells were stained by PI/RNase staining buffer for 30 min and 1 × 104 cells were analyzed by flow cytometer (BD Accuri, Becton Dickinson USA) via using C6 plus software.
Apoptotic death determination by annexin V/ PI staining
1 × 105 cells/Petri MDA-MB-231 and MDA-MB-468 cells were seeded into 60 mm Petri dishes and treated with a selected dose of atiprimod for 24 h. Before drug exposure, cells were trypsinized, pellets were applied with FITC Annexin V Apoptosis Detection Kit, and flow cytometric analysis was performed with cells were analyzed by flow cytometer (BD Accuri, Becton Dickinson USA) via using C6 plus software.
Electrophoretic mobility shift assay (EMSA) and immunoblotting
The effect of atiprimod on STAT3, NF-κB DNA binding activity was determined by EMSA Assay that was previously described. In addition, alteration following atiprimod exposure on Atg-3, Atg-5, Atg-12, Atg-16, Beclin-1, LC-3, p62, STAT-1, pSTAT-1, PERK, Calnexin, STAT-3, pSTAT-3, PDI, CHOP, XBP, Ire-1α, ATF6, BiP, NF-κB, STAT-5, pSTAT-5, PARP, Bax, Bad, Bid, Bim, Bcl-2, Puma protein levels were determined by immunoblotting according to our previous study [22].
DCFDH2 staining
Cells were seeded in 96-well plates (1 × 104 cells/well) and then treated with atiprimod, the media was carefully discarded, and the cells were stained with DCFH-DA (0.5 µM). Drug-induced ROS generation in treated samples and untreated control samples was visualized using fluorescence microscopy Ex./Em.: 495 nm/529 nm (Magnification: 20 ×).
STAT3 plasmid transfection
Overexpression of STAT3 was performed by transfecting 1 µg STAT3 pcDNA3 (Addgene, #8706) for 48 h. After a 24 h transfection period, 2 µM atiprimod applied to the cell medium for 24 h. The potential incline on atiprimod-mediated cell viability loss, growth inhibition, cell death induction was determined in MDA-MB-231 cells by appropriate assays, respectively.
Data analysis
Statistical analysis of the MTT, hanging drop, PI, Annexin V/PI assays were performed by using GraphPad Prism 8 (https://www.graphpad.com). Statistically significant results were performed by two-way ANOVA analysis (where indicated). Due to at least triple replications of each experiment, the mean difference was calculated and represented as standard deviation (SD). Statistical significance was assumed when the p-value was determined below 0.05. The alterations on protein expression profiles in MDA-MB-231 and MDA-MB-468 cells following atiprimod exposure were determined by using the Image J program through analyzing band intensities [23].
Results
STAT3 and NF-κB molecule’s expression profile has a pivotal role in atiprimod-mediated TNBC cell proliferation
Increasing dose of atiprimod exposure (0–5 µM) declined the cell viability of MDA-MB-231 and MDA-MB-468 BC cells as compared to untreated cells. Although MDA-MB-468 cells were more sensitive against atiprimod exposure, prolonged drug treatment overcame atiprimod resistance in MDA-MB-231 BC cells (Fig. 1a). We selected 2 µM atiprimod exposure as its IC50 level for MDA-MB-468 breast cancer cells. A moderate dose of atiprimod (2 μM) hinders the MDA-MB-468 and MDA-MB-231 cell growth, colony formation and cell migration (Fig. 1b, d, e, f). 2 µM atiprimod causes cytostatic effects on MDA-MB-231 cells but the cytotoxic effect was observed in MDA-MB-468 cells after 48 h atiprimod exposure by growth assay (Fig. 1b, e). Similarly, the evident impact of atiprimod on MDA-MB-231 and MDA-MB-468 mitochondrial membrane potential loss (Δψm), cell death and nuclear condensation was determined in a dose-dependent manner (Fig. 1c). Time-dependent atiprimod (2 μM) exposure decreased the colony formation and prevented cell migration in each BC cells by colony formation and wound healing assay, respectively (Fig. 1d-f). Due to the hanging drop assay, 2 μM atiprimod decreased colony diameter 352 μm to 280 μm in MDA-MB-231, and 366 μm to 259 μm in MDA-MB-468 cells after 4 days drug exposure (Fig. 1e). Following drug exposure, downregulation of STAT-3, p65 expression, dephosphorylation of STAT3 (Fig. 2a), and also nuclear localization of pSTAT3, p65 and p50 were inhibited in each estrogen receptor-negative BCs (Fig. 2b). According to the EMSA assay, atiprimod prevented DNA binding affinity of both STAT1/3 and p65 in each BC cell line (Fig. 2c, d).



Dose-dependent atiprimod exposure inhibits cell viability, growth in MDA-MB-231 and MDA-MB-468 breast cancer cells in a dose- and time-dependent manner. Dose-dependent atiprimod (0–5 µM) mediated cell viability loss determined in a time-dependent manner (24 and 48 h) by MTT assay in a MDA-MB-231 (left) and MDA-MB-468 (right). 2 µM atiprimod treatment inhibited the proliferation of b MDA-MB-231 (left) and MDA-MB-468 (right) cells were demonstrated by the Trypan blue dye exclusion assay. The impact of atiprimod (0–3 μM) on cell death mechanism was demonstrated in c MDA-MB-231 and MDA-MB-468 breast cancer cells by DiOC6, DAPI, PI staining, respectively. Atiprimod (0–2 μM) mediated MMP loss, nuclear condensation and cell death were determined by appropriate staining and cells were visualized by fluorescence microscopy (400 × magnification). d The effect of atiprimod (2 μM) on colony formation was determined by colony formation assay in MDA-MB-231 and MDA-MB-468 cells. E Inhibitive effect of atiprimod on MDA-MB-231 and MDA-MB-468 breast cancer cells 3D spheroid formation was observed in a time-dependent manner by hanging drop assay. Three independent experiments were analyzed and presented as the Mean ± Std Dev. p values were determined by comparing 2 μM atiprimod treated MDA-MB-231 and MDA-MB-468 cells with untreated control cells within 4 days. F The suppressive effect of atiprimod (2 μM) exposure on MDA-MB-231 and MDA-MB-468 cells migration was checked by wound healing assay for 48 h. MDA-MB-231 cells; atiprimod treated vs untreated cells for 24 h; p: 0.00213, for 48 h,; p: 0.00115 and MDA-MB-468 cells, atiprimod vs untreated cells for 24 h; p:0.00157, for 48 h; p: 0.0289 (n:3; Magnification 40 ×. Scale bar 100 µm). Barr graphic represents the average measurement of three independent experiments

Suppressive effect of atiprimod on STAT signaling axis and NF-κB activation in MDA-MB-231 and MDA-MB-468 breast cancer cells. a Alteration on STAT-1, STAT-3, pSTAT-3 (Ty705), p65 expression levels following atiprimod application was detected by western blotting in MDA-MB-231 and MDA-MB468 cells. b The effect of atiprimod on STAT1, STAT3, p65 and p100 transcription factors expression profile was determined by nuclear and cytoplasmic lysates of MDA-MB-231 and MDA-MB-468 cells. c The inhibitive effect of atiprimod on the biological activity of nuclear-migrated STAT1/3 and p65 was confirmed by EMSA assay. H3 is used as a nuclear control, and β-tubulin was used as a cytoplasmic loading control. Each blot was repeated twice
Atiprimod induced PERK/eIF2α/ATF4/CHOP axis in MDA-MB-231 and MDA-MB-468 breast cancer cells
CHOP activation was determined following 2 μM atiprimod exposure in each BC cell line by fluorescence microscopy (Fig. 3a). Similarly, atiprimod triggered ER stress-mediated CHOP activation determined in both MDA-MB-231 and MDA-MB468 cells by FACS flow analysis following the CHOP promoter with ATF4 binding mCherry plasmid transfection (Fig. 3b). A significant atiprimod-triggered PERK, BiP, ATF4 and CHOP expressions were detected following drug treatment in each BC cell line. Moreover, phosphorylation of PERK (Thr980) and eIF2α (Ser51) were upregulated by atiprimod exposure in MDA-MB-231 and MDA-MB-468 cells. However, atiprimod significantly downregulates IRE1α expression in each BC cell line. Concomitantly, atiprimod induced a significant increase in PDI, Calnexin protein expressions. Although Ero1- expression slightly upregulated in MDA-MB-231 cells, no significant effect was observed in MDA-MB-468 cells following atiprimod treatment (Fig. 3c). Following drug exposure, CHOP and ATF6 nuclear localization were observed in each BC cell (Fig. 3d).

PERK/ATF4/CHOP axis triggered by atiprimod exposure in MDA-MB-231 and MDA-MB-468 cells. a ATF4, BiP, PERK, pPERK (Thr980), peIF2α (Ser51), CHOP, Calnexin, PDI upregulation and IRE1α downregulation was determined after atiprimod exposure in MDA-MB-231 and MDA-MB-468 cells. CHOP activation was determined by b fluorescence microscopy and flow cytometry in atiprimod treated BCs before mCherry-CHOP (− 649/ + 136) plasmid transfection. Tunicamycin (1 μM) was used as an ER stress inducer agent. c Nuclear localization of CHOP, ATF6α were demonstrated by western blotting. β-tubulin-1 was used as a cytoplasmic, Histon-3 was selected as a nuclear loading control. Blotting images are the representative figure of two independent experiments
The autophagosome formation following atiprimod treatment was not altered
In our previous work, although we demonstrated atiprimod triggered autophagy in pituitary adenoma cells, we couldn’t visualize exact autophagy vacuole formation following drug exposure in each BC cell line (Fig. 4a). When we checked the expression profiles of autophagosome key players, atiprimod downregulated Atgs-3, 5, 12 expressions, and no significant alteration on Beclin-1 expression was detected (Fig. 4b). Although a slight degradation of p62 was determined following drug exposure in MDA-MB-231 cells, no significant effect on LC3-II cleavage was detected. However, the cleavage of LC3II was illustrated in MDA-MB-468 cells by immunoblotting and GFP FACS analysis (Fig. 4c).

Inhibitive effect of atiprimod on autophagosome formation in each breast cancer. a No significant acidic vacuole formation was determined following atiprimod (2 μM) exposure in MDA-MB-231 and MDA-MB468 cells by AO staining. Cells were visualized by fluorescence microscopy (Magnification 40× ×, scale bar 100 μm). b Atiprimod inhibited autophagosome formation via downregulating Atg-3, Atg-5, Atg-12, Beclin-1 expression levels and noteworthy alteration on p62 protein expression levels was determined in each breast cancer cells. β-actin was used as a loading control. c No alteration on LC-3II cleavage after atiprimod was measured by flow cytometry in the FL1 channel via GFP-LC3 plasmid transfection. Serum starvation (%0.5 FBS) was used as a positive control for autophagy induction
Atiprimod triggered G1 arrest and apoptosis in MDA-MB-468 cells
A negative impact of atiprimod on cell viability was observed in each breast cancer cell and drug-triggered ROS-generation was determined by DCFH-DA staining (Fig. 5a). Due to cell cycle analysis, atiprimod induced 2.2 and 4.3% subG1 population accumulation in MDA-MB-231 and MDA-MB-468 cells, respectively. In addition, drug-induced cell cycle arrest at the S phase in MDA-MB-231 and G1 phase in MDA-MB-468 cells, respectively (Fig. 5b). Similar to PI analysis, 2 µM atiprimod treatment induced apoptotic cell death by 2.6% in MDA-MB-231, and 7.5% in MDA-MB-468 cells (Fig. 5b). Moreover, atiprimod-mediated upregulation in pro-apoptotic proteins (Bad, Bax, Bid, Bim, PUMA) and Bcl-2 downregulation was determined in each BC cell line. However, this effect was more sharply in BCs with high STAT3 expressing (MDA-MB-468) cells as compared to low STAT3 expressing BCs (MDA-MB-231) (Fig. 5c). Thus, we forced STAT3 expression via transfection of pcDNA3.1 plasmid with the STAT3 gene inserted in MDA-MB-231 BC cells and demonstrate the alteration of atiprimod (2 μM) effect on cell viability loss, cell growth, and death by MTT, trypan blue exclusion, colony assays and PI staining. Atiprimod-mediated cell viability loss and growth inhibition were increased in MDA-MB-231 parental cells as compared to STAT3 overexpressing BC cells. A similar effect was observed by PI staining (Fig. 6a-d).

Atiprimod triggered cell cycle arrest and ROS-generation-mediated apoptosis in each breast cancer cells. A Atiprimod mediated cell viability loss was determined by Mitotracker/DAPI dual staining in MDA-MB-231 and MDA-MB-468 cells. Atiprimod-induced ROS generation was observed by DCFH-DA staining. Given microscopy images were representative figures of at least two replicate experiments. Magnification: 20 ×, scale bar is 100 μm. b Atiprimod induced G1 arrest in MDA-MB-231 and S phase arrest in MDA-MB-468 cells were determined by PI staining. The apoptotic analysis was preceded with Annexin V/PI staining under flow cytometry after atiprimod exposure in each BC cell. c Atiprimod-induced apoptotic cell death via downregulation of Bcl2, fPARP, and upregulation of Bad, Bax, Bid, PUMA, Bim upregulation especially in MDA-MB-468 cells. GAPDH was used as a loading control

The apoptotic effect of atiprimod increased by STAT-3 overexpression in MDA-MB-231 cells. a pcDNA STAT3 transfection increase the atiprimod-mediated cell growth inhibition in MDA-MB-231 cells as compared to parental cells by trypan blue exclusion assay. b STAT3 overexpression inclines the atiprimod triggered cell viability loss in MDA-MB-231 cells. c Atiprimod-induced cell death increased in MDA-MB-231 STAT3 + cells as compared to MDA-MB-231 wt cells. d Plasmid-mediated STAT3 upregulation decline atiprimod mediated colony dimeter in MDA-MB-231 breast cancer cells (Magnification: 400 ×, Scale: 100 μm)
Discussion
Atiprimod, an FDA-approved JAK2 inhibitor, has anti-inflammatory, anti-proliferative, and anti-carcinogenic effects [11]. The apoptotic effect of atiprimod was demonstrated in various cancer cells such as multiple myeloma [24], hepatocellular carcinoma [13], myeloma [11], mantle lymphoma [8], acute myeloid leukemia [25], and pituitary adenoma [16]. Although the effect of one of the azaspirane derivatives, CIMO, on ER- and ER + breast cancer was evaluated in a dose- and time-dependent manner [12], there is not any study exhibiting the effect of atiprimod on BC cells. By this study, we demonstrate the dose- and time-dependent atiprimod effect on MDA-MB-231 and MDA-MB-468 ER- breast cancer cells. First of all, we revealed atiprimod inhibited cell viability, proliferation and induce Δψm in each ER- breast cancer cells. However, MDA-MB-468 cells were shown more sensitive against atiprimod as compared to MDA-MB-231 breast cancer cells. Similar to our results, atiprimod inhibited cell proliferation and colony formation in HepG2 hepatocellular carcinoma [13], OPM1 and INA-6 myeloma cells [11]. In our previous study, we demonstrated that pituitary adenoma cell proliferation was prevented by atiprimod in a dose-dependent manner [16]. In addition, dose-dependent atiprimod (0–4 μM) inhibited colony formation in OCIM-2, K562, OCI-3, HL-60 and KG-1 acute myeloid leukemia cell lines were indicated [25].
Constitutively active STAT3 expression was displayed in an ample number of solid tumors such as leukemia, lymphoma, hepatocellular carcinoma, prostate, breast, and ovarian cancers [6]. The essential effect of active STAT3 on cancer metastasis due to its targeting genes' role in cellular proliferation, invasion, metastasis, and angiogenesis [26]. As STAT3 has a major role in cancer progression, various inhibitors targeting STAT3 were designed, investigated for their therapeutic effect on different solid tumors [9]. Inhibition of JAK2 and STAT3, 5 expression profile and decline in dephosphorylation of STAT3, 5 molecules expression was revealed in a dose-dependent manner in K562 cells following atiprimod exposure [25]. In addition, dephosphorylation of STAT3 at Tyr705 residue inhibited STAT3 nuclear localization without any effect on Ser727 phosphorylation in hepatocellular carcinoma cells following CIMO treatment [9]. Similarly, CIMO treatment prevents the expression of pSTAT3 (Y705), and represses the expression of STAT-3 regulated genes such as CDK2, CDK4 in MDA-MB-231 breast cancer cells [12]. Similarly, in our previous study, we indicated that dose-dependent atiprimod exposure prevented STAT3 phosphorylation in pituitary adenoma cells [16]. Besides STAT3, NF-κB is also known as an important transcription factor for the regulation of tumors malignancy, angiogenesis, and invasiveness, resistance against various chemotherapeutic agents [27]. Dose- and time-dependent exposure of atiprimod prevented the DNA binding of NF-κB in U266-B1 myeloma cells [24]. Our recent work results illustrated that atiprimod prevented nuclear localization of p65, p50/52 and inhibited DNA binding of p65 in MDA-MB-231 and MDA-MB-468 breast cancer cells (Fig. 2b, d). Thus, atiprimod inhibited cell proliferation, colony formation, and invasion through negatively regulating pSTAT3, p65 nuclear localization and DNA binding in each ER- breast cancer cells. Due to MTT, growth assay, hanging drop results, MDA-MB-231 cells showed a resistant profile against atiprimod treatment as compared to MDA-MB-468 cells (Fig. 1a-e). This might be due to basal cytoplasmic STAT3 and nuclear pSTAT3 levels as its expression is higher in MDA-MB-468 cells than in MDA-MB-231 breast cancer cells.
The molecular machinery underlying drug-mediated apoptotic cell death in preclinical studies was essential to understand the therapeutic efficiency of drugs. The apoptotic effect of atiprimod has been reported in hepatocellular carcinoma, myeloid leukemia and pituitary adenoma cells [11, 13, 16]. However, molecular machinery underlying atiprimod-triggered apoptosis has not been investigated in breast cancer cells, yet. There was only our previous work demonstrating the dose-dependent effect of atiprimod on apoptotic cell death via ER stress activation in the GH3 rat pituitary adenoma cell line [16]. In our recent study, we tried to evaluate the apoptotic effect of atiprimod on two TNBC breast cancer cells regarding ER stress. Anti-carcinogenic agents trigger cellular stress that leads to chemo-resistance through one of the stress-response pathways; Unfolded Protein Response (UPR) [28]. Although protein synthesis is an essential process for cancer cell differentiation and, invasion-metastasis, translation of protein attenuation increased protein clearance and elevated ER-resident chaperone expression maintenance because of the accumulation of unfolded proteins [29]. Protein kinase RNA-activated-like ER kinase (PERK), Inositol requiring enzyme-α (IRE1-α), Activating transcription factor 6 α (ATF-6 α) are well-known three UPR sensors that activate during UPR stress [30]. During unstressed conditions, UPR sensors are inactivated by binding of ER chaperon GRP78/BiP. However, UPR-induced BiP activation triggers PERK, IRE1-α and ATF6-α activation. By our previous and recent study, we demonstrated atiprimod-induced ER stress via BiP upregulation in GH3 [16] and TBNC, respectively. Increased PERK, ATF-6 and IRE-1α expression in the histochemical analysis of breast, brain, liver, pancreatic cancer cells are evidence for the essential role of UPR on tumor growth and chemoresistance [31]. In addition, UPR can be assumed as a touchstone in the cellular discussion through activation of pro-survival events or pro-death pathways. While acute UPR can activate cell survival pathways such as autophagy, chronic ER stress can trigger autophagic and/or apoptotic cell death [32]. PERK/eIF2α /ATF4 activation induces the expression of autophagy-related gene expression and also IRE1α/XBPs activate the expression of Beclin-1 to initiate autophagosome formation. In our study, although we demonstrated that atiprimod (2 μM) induced ER stress through increased protein expression and phosphorylation of PERK, a sharp downregulation of IRE-1α expression and no alteration of XBP expression levels was revealed following drug exposure in both MDA-MB-231 and MDA-MB-468 breast cancer cells. Although we determined atiprimod-triggered PERK activation, the suppressive effect of drug exposure on autophagy key players expression profile such as Atg-3, Atg-5, Atg-12 was determined in each breast cancer cell. Autophagy, an evolutionary conserved process, is generally activated during nutrient deprivation, pathogen infection, accumulation of damaged proteins or organelles. Last decade, chemotherapeutic agents enabled to trigger autophagy as cell death or survival mechanism in various cancer cells [33]. Although the activation of Atg family members following dose-dependent atiprimod was displayed in GH3 cells [16], no significant activation was illustrated following 2 μM atiprimod in MDA-MB-231 and MDA-MB-468 cells. Thus, we evaluate that 2 μM atiprimod might be an effective apoptotic dose for TNBC without activating autophagy as a survival mechanism via suppressing IRE-1α/XBP/Beclin-1 axis. The spliced form of XBP1 induced the ER-associated protein degradation (ERAD) that is activated under the accumulation of damaged or improperly folded proteins. Calnexin and calreticulin control the maturation of glycoproteins, protein desulphated isomerases (PDIs) catalysis the folding of proteins via desulphated bond formation. Each key molecule triggered during UPR activated conditions and activates the ERAD system to overwhelm unfolded/misfolded protein accumulation [34]. In our experimental system, we demonstrated atiprimod exposure triggered UPR mediated-ERAD in each breast cancer cell via Calnexin and PDIs upregulation. Another URP sensor receptor; ATF6, is activated through cleavage of S1P, S2P in Golgi and cATF6 nuclear localization acts as a transcription factor for XBPs, ER chaperon’s expression [34]. Nuclear translocation of both cATF6 and CHOP was determined following atiprimod exposure in each breast cancer cells (Fig. 3c). However, this effect was detected especially in high STAT3 expressing TNBC cells. IRE-1α is demonstrated as an essential target molecule that cross-talk between UPR and inflammatory response. Although the exact mechanism of controlling of IRE-1α on IKK activity regulation has not been identified yet, NF-κB nuclear localization through IKB degradation subsequent activation of IRE-1α forms a complex with TRAF2 [35]. Thus, the preventive effect of atiprimod on IRE-1α expression might directly inhibit the pro-survival pathway; autophagy and also indirectly inhibited NF-κB activation in BCs especially MDA-MB-468 cells.
Prolonged and uncontrolled UPR activation in various cancers (lung, hepatocellular, pancreatic, and breast) triggers tumor development and resistance against chemotherapy. Generally, the UPR axis might be assumed as a protective process for cancer cells to escape from apoptotic cell death. However, moderate or severe UPR evokes apoptosis through PERK/eIF2α/ATF4/CHOP axis [36]. Activation of PERK via phosphorylation subsequently inactivates the global protein translation initiation key target; eukaryotic Initiation factor 2 alpha (eIF2α) by phosphorylation at Ser51 residue. Increased peIF2α Ser51 accumulation has a pivotal role in cancer cells through the attenuation of global gene translation, but increased in some selective gene expressions such as activating transcription factor 4 (ATF4)[37]. PERK-mediated eIF2α phosphorylation and ATF4 activation has an impact on the apoptotic pathway via binding on the CHOP promoter region. PERK/eIF2α/ATF4-dependent CHOP activation evokes the apoptotic pathway acting as a suppressor on anti-apoptotic proteins (Bcl2) and enhancer for the pro-apoptotic proteins that have BH3 domain (Bid, Bad, Bim, Noxa, Puma) [36]. Various studies demonstrated the pivotal role of dose- and time-period of ER stress determines the activation of cell survival or cell death in tumor cells. Consequently, severe or prolonged ER stress due to chemotherapeutic agents may evoke apoptotic cell death in tumor cells [38]. Moreover, ER stress-mediated G1/S arrest implicated in downregulation of cyclinD1 via ATF4-dependent CHOP activation through PERK-triggered eIF2α phosphorylation [39]. In our previous study, we demonstrated although the low dose of atiprimod (1 μM) induced mild ER stress via IRE1α -mediated autophagy induction as a survival mechanism, a higher dose of atiprimod (3 μM) triggered severe ER stress-mediated apoptotic cell death in pituitary adenoma cells [16]. In our recent study, IC50 dose of atiprimod (2 μM) evoke G1/S arrest and apoptosis in MDA-MB-468 cells through activation of PERK/eIF2α /ATF4/CHOP axis and upregulate pro-apoptotic proteins such as Bad, Bid, Bim (Fig. 5b-c). Although drug-induced apoptosis was revealed in each TNBC, MDA-MB-468 cells illustrated sensitivity against atiprimod exposure. This might be due to the different genomic expression profiles of each TBNC cell. As atiprimod act as a STAT3 inhibitor, we investigated the potential incline in atiprimod-mediated apoptosis when increasing STAT3 expression in MDA-MB231 cells. Concomitantly, the plasmid-derived STAT3 overexpression overcome resistance against atiprimod in MDA-MB-231 breast cancer cells (Fig. 5d–g). Besides STAT3, MDA-MB-468 cells have high Ki-67 levels. Similarly, the positive correlation between high pSTAT3, Ki-67, survivin levels, and high-risk breast cancer patients’ primary tissues was illustrated [40]. Thus, the sensitive profile of MDA-MB-468 cells against atiprimod might be also due to both high pSTAT3 and Ki-67 levels.
Conclusion
This study evaluates the apoptotic impact of atiprimod for the first time in MDA-MB-231 and MDA-MB-468 TNBC cells via inhibiting STAT3 and NF-κB signaling pathways. Molecular mechanistic effect of atiprimod on the cell death decision of TBNC cells through inhibiting STAT3 Tyr705 phosphorylation and preventing the DNA binding affinity of STAT1/3 and NF-κB p65 in MDA-MB-231 and MDA-MB-468 breast cancer cells. Moreover, autophagy as a survival mechanism against chemotherapeutic drugs was not activated following atiprimod exposure in breast cancer cells. In contrast, drug-triggered ER stress via activated PERK/eIF2α/ATF4/CHOP axis in MDA-MB-231 and MDA-MB468 breast cancer cells. Moreover, forced STAT3 expression overcomes atiprimod resistance in MDA-MB-231 breast cancer cells. Further in vitro and in vivo studies are needed to highlight the exact mechanism underlying the therapeutic impact of atiprimod in breast cancer cells, especially with aggressive types like TNBCs.
Data availability
Data transparency.
Abbreviations
- ATF6:
-
Activating transcription factor 6
- AO:
-
Acridine orange
- CHOP:
-
C/EBP-homologous protein
- DiOC6:
-
3,3′-Dihexyloxacarbocyanineiodide
- EDTA:
-
Ethylenediaminetetraaceticacid
- eIF-2 α:
-
Eukaryotic initiation factor 2
- HRP:
-
Horseradish peroxidase
- IRE-1α:
-
Inositol-requiring protein-1α,
- ψ:
-
Mitochondrial membrane potential
- PERK:
-
Protein kinase RNA- like endoplasmic reticulum kinase
- PI3K:
-
Phosphatidylinositol-3-kinase
- PBS:
-
Phosphate-buffered saline
- PI:
-
Propidium iodide
- PVDF:
-
Polyvinyldifluoride
- SAPK/JNK:
-
Stress-activated protein kinase/ Jun amino-terminal kinase
- ROS:
-
Reactive oxygen species
- STAT:
-
Signal transducer and activator of transcriptions
- TNBC:
-
Triple negative breast cancer
- UPREs:
-
Unfolded protein response
References
Kamińska M, Ciszewski T, Łopacka-Szatan K, Miotła P, Starosławska E (2015) Breast cancer risk factors. Przeglad Menopauzalny. https://doi.org/10.5114/pm.2015.54346
Lehmann BD, Pietenpol JA (2014) Identification and use of biomarkers in treatment strategies for triple-negative breast cancer subtypes. J Pathol. https://doi.org/10.1002/path.4280
Khosravi-Shahi P, Cabezón-Gutiérrez L, Aparicio Salcedo MI (2019) State of art of advanced triple negative breast cancer. Breast J 25(5):967–970. https://doi.org/10.1111/tbj.13369
McMurray JS (2006) A new small-molecule Stat3 inhibitor. Chem Biol. https://doi.org/10.1016/j.chembiol.2006.11.001
Huang S (2007) Regulation of metastases by signal transducer and activator of transcription 3 signaling pathway: clinical implications. Clin Cancer Res. https://doi.org/10.1158/1078-0432.CCR-06-2313
Bromberg J (2002) Stat proteins and oncogenesis. J Clin Invest 109(9):1139–1142. https://doi.org/10.1172/JCI200215617
Li Y et al (2019) Clinicopathological and prognostic role of STAT3/p-STAT3 in breast cancer patients in China: a meta-analysis. Sci Rep. https://doi.org/10.1038/s41598-019-47556-z
Wang X, Crowe PJ, Goldstein D, Yang JL (2012) STAT3 inhibition, a novel approach to enhancing targeted therapy in human cancers (Review). Int J Oncol. https://doi.org/10.3892/ijo.2012.1568
Mohan CD et al (2014) Development of a novel azaspirane that targets the Janus Kinase-signal transducer and activator of transcription (STAT) pathway in hepatocellular carcinoma in vitro and in vivo. J Biol Chem. https://doi.org/10.1074/jbc.M114.601104
Badger AM et al (1999) Atiprimod (SKandF 106615), a novel macrophage targeting agent, enhances alveolar macrophage candidacidal activity and is not immunosuppresive in candida-infected mice. Int J Immunopharmacol 21(3):161–176. https://doi.org/10.1016/S0192-0561(98)00076-9
Neri P et al (2007) Biological pathways and in vivo antitumor activity induced by Atiprimod in myeloma. Leukemia. https://doi.org/10.1038/sj.leu.2404912
Sulaiman NBS et al (2016) An azaspirane derivative suppresses growth and induces apoptosis of ER-positive and ER-negative breast cancer cells through the modulation of JAK2/STAT3 signaling pathway. Int J Oncol 49(3):1221–1229. https://doi.org/10.3892/ijo.2016.3615
Choudhari SR et al (2007) Deactivation of Akt and STAT3 signaling promotes apoptosis, inhibits proliferation, and enhances the sensitivity of hepatocellular carcinoma cells to an anticancer agent, Atiprimod. Mol Cancer Ther 6(1):112–121. https://doi.org/10.1158/1535-7163.MCT-06-0561
O’Connor OA et al (2005) Phase II clinical experience with the novel proteasome inhibitor bortezomib in patients with indolent non-Hodgkin’s lymphoma and mantle cell lymphoma. J Clin Oncol. https://doi.org/10.1200/JCO.2005.02.050
Sun L et al (2012) Combination of atiprimod and the proteasome inhibitor bortezomib induces apoptosis of mantle cell lymphoma in vitro and in vivo. Leuk Res. https://doi.org/10.1016/j.leukres.2011.09.014
Coker-Gurkan A, Ayhan-Sahin B, Keceloglu G, Obakan-Yerlikaya P, Arisan E, Palavan-Unsal N (2019) Atiprimod induce apoptosis in pituitary adenoma: endoplasmic reticulum stress and autophagy pathways. J Cell Biochem 120(12):19749–19763. https://doi.org/10.1002/jcb.29281
Holohan C, Van Schaeybroeck S, Longley DB, Johnston PG (2013) Cancer drug resistance: an evolving paradigm. Nat Rev Cancer. https://doi.org/10.1038/nrc3599
Chevet E, Hetz C, Samali A (2015) Endoplasmic reticulum stress-activated cell reprogramming in oncogenesis. Cancer Discov 5(6):586–597. https://doi.org/10.1158/2159-8290.CD-14-1490
Storniolo A, Alfano V, Carbotta S, Ferretti E, Di Renzo L (2018) IRE1α deficiency promotes tumor cell death and eIF2α degradation through PERK dipendent autophagy. Cell Death Discov. https://doi.org/10.1038/s41420-017-0002-9
Paglin S et al (2001) A novel response of cancer cells to radiation involves autophagy and formation of acidic vesicles. Cancer Res 61(2):439–444
Galluzzi L et al (2018) Molecular mechanisms of cell death: recommendations of the nomenclature committee on cell death 2018. Cell Death Differ. https://doi.org/10.1038/s41418-017-0012-4
Coker-Gurkan A et al (2018) Curcumin inhibits autocrine growth hormone-mediated invasion and metastasis by targeting NF-κB signaling and polyamine metabolism in breast cancer cells. Amino Acids 50(8):1045–1069. https://doi.org/10.1007/s00726-018-2581-z
Rueden CT et al (2017) ImageJ2: ImageJ for the next generation of scientific image data. BMC Bioinform. https://doi.org/10.1186/s12859-017-1934-z
Amit-Vazina M et al (2005) Atiprimod blocks STAT3 phosphorylation and induces apoptosis in multiple myeloma cells. Br J Cancer 93(1):70–80. https://doi.org/10.1038/sj.bjc.6602637
Faderl S, Ferrajoli A, Harris D, Van Q, Kantarjian HM, Estrov Z (2007) Atiprimod blocks phosphorylation of JAK-STAT and inhibits proliferation of acute myeloid leukemia (AML) cells. Leuk Res. https://doi.org/10.1016/j.leukres.2006.05.027
Bhattacharya S, Ray RM, Johnson LR (2005) STAT3-mediated transcription of Bcl-2, Mcl-1 and c-IAP2 prevents apoptosis in polyamine-depleted cells. Biochem J 392(2):335–344. https://doi.org/10.1042/BJ20050465
Fan Y, Mao R, Yang J (2013) NF-κB and STAT3 signaling pathways collaboratively link inflammation to cancer. Protein Cell. https://doi.org/10.1007/s13238-013-2084-3
Tsuruo T et al (2003) Molecular targeting therapy of cancer: drug resistance, apoptosis and survival signal. Cancer Sci. https://doi.org/10.1111/j.1349-7006.2003.tb01345.x
Dejeans N, Barroso K, Fernandez-Zapico ME, Samali A, Chevet E (2015) Novel roles of the unfolded protein response in the control of tumor development and aggressiveness. Semin Cancer Biol. https://doi.org/10.1016/j.semcancer.2015.04.007
Harding HP, Zhang Y, Ron D (1999) Protein translation and folding are coupled by an endoplasmic- reticulum-resident kinase. Nature. https://doi.org/10.1038/16729
Epple LM et al (2013) Induction of the unfolded protein response drives enhanced metabolism and chemoresistance in glioma cells. PLoS ONE. https://doi.org/10.1371/journal.pone.0073267
Avril T, Vauléon E, Chevet E (2017) Endoplasmic reticulum stress signaling and chemotherapy resistance in solid cancers. Oncogenesis 6(8):e373–e373. https://doi.org/10.1038/oncsis.2017.72
Farooqi AA et al (2015) Anticancer drugs for the modulation of endoplasmic reticulum stress and oxidative stress. Tumor Biol. https://doi.org/10.1007/s13277-015-3797-0
Hoseki J, Ushioda R, Nagata K (2010) Mechanism and components of endoplasmic reticulum-associated degradation. J Biochem. https://doi.org/10.1093/jb/mvp194
Urano F et al (2000) Coupling of stress in the ER to activation of JNK protein kinases by transmembrane protein kinase IRE1. Science 287(5453):664–666. https://doi.org/10.1126/science.287.5453.664
Puthalakath H et al (2007) ER stress triggers apoptosis by activating BH3-only protein bim. Cell. https://doi.org/10.1016/j.cell.2007.04.027
Rozpedek W, Pytel D, Mucha B, Leszczynska H, Diehl JA, Majsterek I (2016) The role of the PERK/eIF2α/ATF4/CHOP signaling pathway in tumor progression during endoplasmic reticulum stress. Curr Mol Med. https://doi.org/10.2174/1566524016666160523143937
Schönthal AH (2012) Endoplasmic reticulum stress: its role in disease and novel prospects for therapy. Scientifica (Cairo). https://doi.org/10.6064/2012/857516
Raven JF et al (2008) PKR and PKR-like endoplasmic reticulum kinase induce the proteasome-dependent degradation of cyclin D1 via a mechanism requiring eukaryotic initiation factor 2α phosphorylation. J Biol Chem. https://doi.org/10.1074/jbc.M709677200
Diaz N et al (2006) Activation of Stat3 in primary tumors from high-risk breast cancer patients is associated with elevated levels of activated Src and survivin expression. Clin Cancer Res. https://doi.org/10.1158/1078-0432.CCR-04-1749
Acknowledgements
Authors would thank to TUBITAK 2209A Undergraduate Research Project for Grant (1919B011701280).
Funding
The project was supported by TUBITAK 2209A Undergraduate Research Project Grant (1919B011701280).
Author information
Authors and Affiliations
Contributions
AC: Conceptualization, project administration, supervision and roles/writing—original draft. PO and EA: Validation, writing—review & editing. EC and SS: methodology. The authors have no conflict of interest to declare.
Corresponding author
Ethics declarations
Conflict of interest
The authors declare that they have no conflict of interest.
Ethical approval
All the experiments were performed by using commercially available cell line from ATCC company. Neither human’s nor animal’s alive body material was used in this research.
Disclosure summary
The authors have nothing to disclosure.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
About this article

Cite this article
Coker-Gurkan, A., Can, E., Sahin, S. et al. Atiprimod triggered apoptotic cell death via acting on PERK/eIF2α/ATF4/CHOP and STAT3/NF-ΚB axis in MDA-MB-231 and MDA-MB-468 breast cancer cells. Mol Biol Rep 48, 5233–5247 (2021). https://doi.org/10.1007/s11033-021-06528-1
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s11033-021-06528-1