
Abstract
The Indian antelope or blackbuck (Antilope cervicapra) is endemic to the Indian subcontinent, inhabiting scrublands and dry grasslands. Most of the blackbuck populations are small, isolated, and threatened by habitat fragmentation and degradation. Management of such disjunct populations requires genetic characterization, which is critical for assessing hazards of stochastic events and inbreeding. Addressing the scarcity of such information on the blackbuck, we describe a novel panel of microsatellite markers that could be used to monitor blackbuck demography and population genetic parameters using non-invasive faecal sampling. We screened microsatellites (n = 40) that had been reported to amplify in bovid and cervid species using faecal samples of the blackbuck collected from Kaimoor Wildlife Sanctuary, Uttar Pradesh, India and its vicinities. We selected 12 markers for amplification using faecal DNA extracts (n = 140) in three multiplex reactions. We observed a mean amplification success rate of 72.4% across loci (92.1–25.7%) with high allele diversity (mean number of alleles/locus = 8.67 ± 1.03). Mean genotyping error rates across the markers were low to moderate (allelic drop-out rate = 0.09; false allele rate = 0.11). The proportions of first- and second-order relatives in the study population were 0.69% and 6.21%, respectively. Based on amplification success, genotyping error rates and the probability of identity (PID), we suggest (i) a panel of five microsatellite markers (cumulative PID = 1.24 × 10–5) for individual identification and population monitoring and (ii) seven additional markers for conservation genetics studies. This study provides essential tools capable of augmenting blackbuck conservation strategies at the landscape level, integral to protecting the scrubland-grassland ecosystem.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
The primary causative factors driving extinction events in the Anthropocene have been human interference, introduction of invasive species, habitat fragmentation, degradation and inadequacy of natural habitat. Protection of the focal species does not always guarantee species survival due to extrinsic factors such as the influence of predators, pathogens, and natural catastrophes as well as intrinsic factors such as species demography, inclusive fitness and genetic variability [1]. Most of these factors induce departure from panmixia, subsequently changing the genetic characteristics at different scales [2]. Therefore, informing conservation strategies with genetic characteristics in hopes of preventing genetic erosion and establishing connectivity for a healthy population has been encouraged.
The Indian antelope or blackbuck, one of the indicator species of the scrub-grassland ecosystem, used to be the most abundant wild animal across the Indian subcontinent [3] and was once widespread across a vast range starting from the Indus River in the west and spanning across the Indo-Gangetic Plain and from the Deccan plateau down to Point Calimere of the eastern coastal plains. Due to indiscriminate hunting during the 20th century, a sharp decline of the blackbuck population caused local extinctions at the range extremities, i.e. Pakistan and Bangladesh, causing the populations to be restricted to the scrublands and dry grasslands of India and Nepal in the form of fragmented populations along with a small population in Pakistan reintroduced from Texas, USA [4,5,6]. Additionally, land-use changes have caused a 31% loss of scrub-grassland habitat in India from 2005 to 2015 [7], affecting several species. Introduction of the Wild Life (Protection) Act, 1972 of India helped to curb blackbuck poaching to some extent. However, still at issue are villagers who frequent the habitat-agricultural land interfaces and consider the blackbuck to be an agricultural pest in various high-density locations.
The viability of small, isolated populations is known to be under constant threat from stochastic destabilizing effects such as inbreeding, demographic changes, and susceptibility to diseases [8]. Moreover, the management of smaller populations with low effective population sizes (Ne) is necessary to avoid the extinction vortex for the species, whereas loss of metapopulation dynamics hinders the long-term probability of survival. Information on fine-scale population status is essential in the drafting of such management strategies.
To obtain demographic information on wild species, identifying unique individuals using non-invasive genetic samples, e.g. excrements, hair, etc., has been one of the most widely used tools, especially for unmarked species [9]. Resultant information unveils parameters (e.g. relative abundance, habitat use patterns, etc.) essential for management of species. On the other hand, understanding genetic variation is fundamental to understanding biological diversity, which affects the variability in a species at both the population and ecosystem levels [10]. Additionally, abiotic extrinsic variables can modify demographic and genetic traits in the form of selection pressure, thereby influencing the species’ persistence. The most versatile and cost-effective tool for addressing ecological questions like gene-flow, migration rates, effective population size, recent and past bottlenecks and relatedness is the use of neutral nuclear markers such as microsatellites [11, 12]. However, developing novel microsatellite markers for numerous species of conservation importance is resource- and time-intensive. Cross-amplification of microsatellite markers developed for closely related species is a popular alternative for resource-intensive development of species-specific markers. However, mutation-driven processes often give rise to homoplasy of allele sizes, amplification of non-orthologues, and ascertainment bias [13,14,15]. In spite of these limitations, cross-amplification of markers developed for closely related species have widely been used to characterize population genetic parameters [16,17,18,19].
The majority of the blackbuck populations in India are small and fragmented, requiring insights into their population genetic status to plan effective conservation measures. In the absence of species-specific markers, such information on the blackbuck could be obtained from potentially cross-amplifying microsatellites developed for other ungulate species. The available literature indicates successful amplification of bovid microsatellites in cervid species and vice-versa [20, 21]. Therefore, we document for the first time a thorough screening of bovid and cervid cross-amplifying microsatellites and propose a panel of markers for the identification of unique blackbuck individuals and for further investigation into their population genetic parameters across their patchy distribution along the scrublands and dry grasslands of India.
Material and methods
Study area
The study was conducted in Kaimoor Wildlife Sanctuary (KWLS), which is situated across the Mirzapur and Sonbhadra districts of the state of Uttar Pradesh (UP), India, and adjoining blackbuck habitats in the neighbouring areas. We performed extensive sampling in the KWLS and the territorial forest ranges of the Robertsganj, Ghurma, Ghorawal and Halia lying between 24° 27′ 36.93″ N, 83° 09′ 37.44″ E and 24° 54′ 25.46″ N, 82° 15′ 54.23″ E (Fig. 1). The forest type in the area is mixed and dry deciduous, with primary tree vegetation consisting of Bakli (Anogeissus latifolia), Mahua (Madhuca longifolia), and Dhaak (Butea monosperma). The mammalian wildlife comprises the blackbuck (Antilope cervicapra), chinkara (Gazella bennettii), nilgai (Boselaphus tragocamelus), sloth bear (Melursus ursinus), leopard (Panthera pardus), golden jackal (Canis aureus), and Bengal fox (Vulpes bengalensis).

Collection of blackbuck faecal pellets
During the study period (October to December 2018), we conducted an intensive search for blackbuck pellets in the scrublands and adjoining agricultural fields throughout the study area by vehicle survey and on foot. We collected blackbuck faecal pellets, identified by characteristic morphology that differs from the other ungulates commonly found in the study area (chinkara, domestic sheep and goat), from spatially segregated pellet groups. We stored the pellets in labelled resealable pouches in the field before transferring to 50 ml sterile containers with silica gel as a desiccant for preservation at room temperature until further processing.
DNA extraction
We scraped the outer surfaces of 5–6 pellets from each pellet group containing sloughed off intestinal epithelial cells into 2 ml polypropylene centrifuge tubes using sterile blades. We isolated genomic DNA from the sampled pellet groups (n = 140) using QIAamp Fast DNA Stool Mini Kits employing a silica membrane column-based purification of faecal DNA according to the manufacturer specified protocol. We used a dedicated faecal DNA isolation facility while having negative controls (i.e. blanks) for each DNA extraction batch to account for possible contamination. We stored the DNA eluates at – 20 °C for later analysis.
Confirmation of species identity
To confirm whether the samples collected belonged to blackbuck, we amplified a partial fragment of the Cytochrome b gene of the mtDNA genome using the primer pair H15149-L14841 [22] from a subset (n = 19) of the faecal extracts. Amplification was carried out with Maxima Hot Start Green 2X PCR Master Mix (Thermo Scientific); 15 µg bovine serum albumin (BSA); 3 pmol each of forward and reverse primers; 2.0 µl genomic DNA (gDNA) of variable concentrations; and nuclease-free water to bring the total reaction volume to 15 µl. A negative control and a positive control were also set up along with the reaction mixture to cross-check for contamination. The cycling conditions for the Cytochrome b fragment amplification were: initial denaturation at 94 °C for 5 min; 35 cycles of denaturation at 94 °C for 40 s; annealing temperature of 53 °C for 45 s; and extension at 72 °C for 40 s with a final extension step at 72 °C for 10 min. Amplicons were visualized under a UV transilluminator after being run through 2% agarose gels stained with ethidium bromide (EtBr). We performed enzymatic clean-up of the amplicons to remove unincorporated primers and dNTPs using Exonuclease I and Shrimp Alkaline Phosphatase (Thermo Scientific Inc.) following a cycling condition of 37 °C and 85 °C each for 15 min. Purified products were then sequenced from both directions using BigDye Terminator v3.1 Cycle Sequencing Kit (Applied Biosystems, CA). The fragments were then dissolved in Hi-Di Formamide (Applied Biosystems, CA) and directly electrophoresed on an ABI 3500XL Genetic Analyzer. The sequences generated were examined manually to eliminate ambiguity in base-calling. We compared the sequences with blackbuck mtDNA Cytochrome b data based on the internal database of the Wildlife Institute of India and the public domain (National Center for Biotechnology Information—GenBank) to ascertain the species from the faecal DNA samples under study.
Screening of microsatellite markers
As there is no published literature on the use of microsatellites in the blackbuck, we decided to screen a subset of markers known to amplify in bovids (n = 33) and cervids (n = 7) (Supplementary Table S1) [21, 23,24,25]. Despite having access to reference tissue samples, we chose to use confirmed faecal DNA of the blackbuck for standardization as we aimed to generate good quality data from low concentration gDNA templates. Initially, we used four faecal DNA samples, confirmed to be of blackbuck through sequence similarity, in a temperature gradient of 51 to 61 °C with each of the 40 microsatellites tested. For the amplification, we used Qiagen Multiplex PCR Master Mix (2X), 10 µg BSA, 0.25 µM each of forward and reverse primers, 2 µl of gDNA having variable DNA quantity, and nuclease-free water to bring the volume up to 10 µl. The thermocycler conditions included an initial denaturation step at 95 °C for 15 min followed by 40 cycles of denaturation at 95 °C for 30 s, annealing along the temperature gradient for 60 s, and extension at 72 °C for 40 s. The final extension step was at 60 °C for 30 min, followed by a hold at 4 °C. We ran the products on an agarose gel with a 100 bp-sized marker, and EtBr intercalated products were visualized using a UV transilluminator. We identified the optimal temperatures for individual markers in case there were visible bands and confirmed the annealing conditions by running a second experiment with specific temperatures along with negative and positive controls.
Optimization of multiplex reaction
Once the annealing temperatures were standardized, we optimized primers, indicating positive results for co-amplification in groups of four markers. We considered annealing temperatures, amplicon sizes and dye labels (G5 dye set; Applied Biosystems, CA) to design such multiplex reactions. We employed a similar reaction composition and thermal profile used during the microsatellite screening, except that we used a total pool of 1 µM of primers constituting the multiplex. We amplified three such multiplex panels (Table 1) with blackbuck faecal DNA (n = 140). We amplified a subset (n = 16) of the faecal DNA extracts for five additional replicates each across the three multiplex panels to compute genotyping error rates using a multi-tube genotyping approach [26]. All amplicons were run for fragment analysis in an ABI 3500XL Genetic Analyzer with GeneScan 500 LIZ (Applied Biosystems, CA) as a size standard.
Microsatellite data analysis
We used GENEMAPPER v5.0 (Applied Biosystems) to manually verify and score alleles from the fragment analysis electropherograms. In order to characterize the profiles of each marker for consistent allele calling, we also recorded intensities of ‘stutter’ [27] and ‘+ A’ [28] peaks in terms of relative fluorescence unit (RFU), caused by slipped strand mispairing and addition of nucleotide residues during PCR, respectively [29]. We used the AUTOBIN v0.9 macro [30] for binning the raw allele scores into fragment sizes. We used GenAlEx v6.5.0.1 [31] for calculation of observed heterozygosity (Ho), expected heterozygosity (He), probability of misidentifying two unrelated individuals as a single individual (PID) and the probability of misidentifying two siblings as a single individual (PIDsibs). Null allele frequencies were calculated using the software FREENA [32]. We used GIMLET v1.3.3 [33] to compute allelic drop-out (ADO) and false allele (FA) rates per heterozygote and homozygote genotypes, respectively, based on six multi-tube repeat genotypes each for a subset of the DNA extracts (n = 16). We report the mean rates of ADO and FA along with the standard error across four different thresholds (two, three, four and five recurrences of an allele) to construct consensus genotype following Hansen et al. [34]. We computed the estimates of kinship amongst the genotypes using a full likelihood approach and accounting for genotyping errors [35]. We used the software COLONY v2.0.6.6 [36] to determine kinship with assumptions of polygamy and the possibility of inbreeding while implementing a weak sibship prior and three independent medium-length runs. A matrix of pairwise kinship was constructed based on the full-likelihood computations. Additionally, we calculated the Cavalli-Sforza and Edwards measure of pairwise genetic distances (Dc) [37], which can accommodate null alleles [32, 38], between the samples to construct a neighbour-joining dendrogram using the software POPULATIONS v1.2.32 [39] and subsequently visualized using FigTree v1.4.2 [40].
Results
Confirmation of species identity
Based on the Cytochrome b sequence data generated (~ 330 bp fragments), 100% (n = 19) of the samples were assigned to the blackbuck, based on matches with sequences from the GenBank database as well as our sequence repository at the species level. Hence, we presume all other samples collected, having similar pellet morphology to those confirmed by the sequence data, were of blackbuck origin.
Screening and optimization of microsatellite amplification
We found positive amplification visible on agarose gel in 12 out of the 40 markers tested (Supplementary Table S1). The optimal annealing temperature range for these markers varied from 51 to 57 °C (Table 1, Supplementary Fig. S1). We did not observe any amplification of the six cervid markers with blackbuck faecal DNA [21]. Markers yielding positive amplification (n = 12) contained directly dye-labelled forward primers (n = 9) and tailed forward primers with unique M13-analogue sequences corresponding to dye-labelled oligonucleotides (n = 3, Table 1) [41, 42]. The electropherograms of all 12 markers produced discrete peaks with low to moderate stuttering (Supplementary Fig. S2), facilitating unambiguous allele calling, a pre-requisite of high-quality data with minimal scoring errors. The mean ratio of the peak heights of the − 2 bp ‘stutter’ (S1) and the first allele (A1) varied between 0.06 ± 0.01 (MAF209) and 0.49 ± 0.12 (INRA005) while the second (A2) to first allele (A1) ratio was between 0.38 ± 0.03 (BM415) and 0.88 ± 0.03 (TGLA122) for heterozygous genotypes (Table 2). We did not observe any significant occurrence of ‘+ A’ peaks in any of the loci. The mean amplification success rate was 72.4%, varying between 92.1% in TGLA122 and 25.7% in MAF209 (Table 2).
Microsatellite marker characteristics
The genotyping error rates varied considerably across loci. We did not find any ADO or FA at the loci BM8125 or OarCP34, whereas the ADO rate was minimal (≤ 0.05) for MAF65, INRA005 and MAF209 (Table 2). Low FA rates (≤ 0.5) were observed at OarFCB226, SPS115 and BM415, while ETH10 had high error rates (ADO = 0.18 ± 0.03, FA = 0.17 ± 0.03). Null allele frequencies varied between 0.09 and 0.38 for the loci with a mean value of Hnull = 0.18 ± 0.03.
The number of alleles was lowest in OarCP34 (n = 4) and highest in BM415 (n = 16). Observed heterozygosity (Ho) varied between 0.02 (SPS115) and 0.65 (ETH10) (Table 2). Expected heterozygosity (He) ranged from 0.14 (BM8125) to 0.83 (INRA005) (Table 2). We found low observed heterozygosity (Ho = 0.34 ± 0.07) across the markers for the study population. Cumulative PID and PIDsib for the 12 markers were 1.67 × 10–10 and 1.53 × 10–4, respectively, corresponding to a < 0.001% probability of two individuals being considered as one and a 0.015% chance of full siblings misidentified as a single individual.
Kinship and genetic distance among genotypes
We used a subset of the samples (n = 112) with a 10.2% gap in the genotype data to compute kinship and pairwise genetic distances. Out of the possible 6216 pairs of samples, we identified 43 (0.69%) dyads of first-order and 386 (6.21%) dyads of second-order kin (Supplementary Fig. S3). The NJ dendrogram depicting pairwise genetic distances (Dc) across the 112 samples revealed the presence of three major genetic clusters, two major and one minor in terms of numbers, in the study area (Supplementary Figure S4). Two sub-clusters each were observed within both of the major genetic clusters.
Panel for individual identification
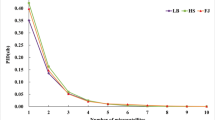
We identified five markers (BM415, MAF65, OarFCB226, SRCRSP06 and TGLA122) that had low to moderate rates of ADO and FA as well as low cumulative misidentification rates of one in > 80,000 individuals (PID = 1.24 × 10–5; PIDsib = 1.14 × 10–2) (Figs. 2 and 3) as a panel to identify blackbuck individuals.

Comparison of genotyping error rates and probability of identity (PID) for microsatellite loci (n = 12) amplified from faecal DNA of the blackbuck. The diameters of the bubbles are proportional to the amplification success rate

Cumulative probability of identity of unrelated individuals (PID) and sibling probability of identity (PIDsib) for increasing combination of loci (n = 5) suggested for identifying blackbuck individuals
Discussion
This study is the first to test cross-amplified microsatellite markers of cervid and bovid origin for amplification success, variability, and error rates for identifying and characterizing the blackbuck population in India. The resulting set of markers prescribed in this study would support the use of a non-invasive, cost-effective strategy for genetic analysis of this species, augmenting conservation policies.
Available literature indicates that the majority of the microsatellite markers screened during the course of this study were polymorphic in wild bovid and cervid species, having up to 13 alleles (mean 4.3 alleles per locus) (Supplementary Table S1). However, non-amplification of a large proportion (70%) of the screened markers in the blackbuck indicated that the primer binding flanking sequences for the microsatellites were not conserved and might have undergone mutation in the species. Hindrance of proper annealing due to the low amount of template DNA as well as the presence of PCR inhibitors could not be ruled out. Therefore, we suggest a similar screening of a large number of markers before the inception of a microsatellite-based study on any species that does not have a successful panel described for non-invasive samples by any earlier study.
Successful amplification of microsatellites from excremental DNA are often governed by climatic conditions, method of preservation and time elapsed between deposition and collection of samples [43]. Our rate of successful amplification (72.4%) was comparable to the observed rates (58–100%) in other tropical wild bovid species using dry storage over silica gel at ambient temperature [44]. Accurate and reproducible allele scoring from faecal samples has been a significant limitation in getting reliable data using non-invasive samples [26]. Therefore, characterizing each observed allele across loci using stutter-to-peak and first-to-second allele ratios would aid consistent allele scoring.
The low Ho observed in this study could either be driven by the characteristic erosion of small isolated populations or by the presence of null alleles. It is possible to overestimate the null allele frequencies due to the effects of inbreeding [45]. Therefore, range-wide genetic characterization of similar populations is required to determine the factors driving low heterozygosity conclusively. However, the presence of only 0.69% first-order relatives in the study population indicates that severe inbreeding might be unlikely. In addition, the uniform branch distribution in the genetic distance dendrogram (Supplementary Fig. S4) indicates a long-term stable effective population size as postulated by Spong et al. [46].
Genotyping errors are inherent properties of microsatellite markers, often incurring positive bias, directly proportional to the number of markers used, while enumerating unique individuals from non-invasive DNA sources [47]. With a speculated abundance of > 50,000 blackbucks across India [48] and most of the populations having < 7000 individuals [6], the panel of five microsatellites (BM415, MAF65, OarFCB226, SRCRSP06 and TGLA122) provided sufficient resolution while incurring low ADO and FA. The effect of genotyping errors on population genetics parameters and diversity statistics based on allele frequencies are lower than those errors on individual identification [49]. Therefore, we recommend using data for an additional seven markers (BM8125, ETH10, ETH152, INRA005, OarCP34, SPS115 and MAF209) in conjunction with the individual identification panel for population and landscape genetic characterization of the blackbuck. A suggested workflow for the projects related to conservation genetics of the blackbuck is provided in Supplementary Fig. S5.
Conclusion
With the waning of scrublands and dry grasslands across India to provide for the increasing human population and agro-economic development encompassing agriculture and plantation forestry, the conservation of species specialized for such habitat has become a necessity. The blackbuck's remaining distribution across India is in several isolated habitat patches, demanding immediate intervention as a flagship species of the scrub-grassland ecosystem. Most of the blackbuck populations are severely understudied, and our data indicate low observed heterozygosity with high allele diversity for one such population. Therefore, there is a need to initiate a pan-India study for understanding genetic variability, inbreeding status and functional connectivity of the blackbuck populations. We present markers with low to moderate error rates, low PID and higher success rates, which can produce reliable genotypes from blackbuck faecal DNA samples using the fewest possible loci for individual identification with sufficient resolution. Hence, our panel of standardized microsatellites could supplement future studies on population and landscape genetics of the blackbuck, ideally leading to the conservation of the scrub-grassland ecosystem.
Data availability
Raw genotypes used in this study will be made available upon reasonable request. All other data have been included in the form of tables in the manuscript and supplementary materials.
References
Balkenhol N, Cushman SA, Storfer AT, Waits LP (2016) Landscape genetics: concepts, methods, applications. Wiley-Blackwell, Oxford, UK
Barton N, Clark A (1990) Population structure and processes in evolution. In: Wöhrmann K, Jain SK (eds) Population biology. Springer-Verlag, Berlin, Germany, pp 115–173
Rahmani AR (1991) Present distribution of the blackbuck Antilope cervicapra Linn in India, with special emphasis on the lesser known populations. J Bombay Nat Hist Soc 88:35–46
Khanal L, Chalise MK (2010) Population status of blackbuck (Antilope cervicapra) at Khairarpur, Bardiya. Nepal J Nat Hist Museum 25:266–275
Mirza ZB, Waiz A (1973) Food availability for blackbuck (Antilope cervicapra) at Lal Suhanra Sanctuary, Pakistan. Biol Conserv 5:119–122. https://doi.org/10.1016/0006-3207(73)90091-8
Meena R, Saran RP (2018) Distribution, ecology and conservation status of blackbuck (Antilope cervicapra): an update. Int J Biol Res 3:79–86
MoEFCC (2018) Report from India. Ministry of environment, forest and climate change, government of India - United Nations Convention to Combat Desertification. New Delhi, India
Lacy RC (2000) Considering threats to the viability of small populations using individual-based models. Ecol Bull 39-51
Lamb CT, Ford AT, Proctor MF, Royle JA, Mowat G, Boutin S (2019) Genetic tagging in the anthropocene: scaling ecology from alleles to ecosystems. Ecol Appl 29:e01876. https://doi.org/10.1002/eap.1876
Primack RB (2010) Essentials of conservation biology. Sunderland, MA, USA
Selkoe KA, Toonen RJ (2006) Microsatellites for ecologists: a practical guide to using and evaluating microsatellite markers. Ecol Lett 9:615–629. https://doi.org/10.1111/j.1461-0248.2006.00889.x
Carroll EL, Bruford MW, DeWoody JA, Leroy G, Strand A, Waits L, Wang J (2018) Genetic and genomic monitoring with minimally invasive sampling methods. Evol Appl 11:1094–1119. https://doi.org/10.1111/eva.12600
Estoup A, Jarne P, Cornuet J-M (2002) Homoplasy and mutation model at microsatellite loci and their consequences for population genetics analysis. Mol Ecol 11:1591–1604
Yue G-H, Balazs K, Laszlo O (2010) A new problem with cross-species amplification of microsatellites: generation of non-homologous products. Zool Res 31:131–140. https://doi.org/10.3724/SP.J.1141.2010.02131
Li B, Kimmel M (2013) Factors influencing ascertainment bias of microsatellite allele sizes: impact on estimates of mutation rates. Genetics 195:563–572. https://doi.org/10.1534/genetics.113.154161
Kim KS, Min MS, An JH, Lee H (2004) Cross-species amplification of bovidae microsatellites and low diversity of the endangered Korean goral. J Hered 95:521–525. https://doi.org/10.1093/jhered/esh082
Maudet C, Luikart G, Taberlet P (2001) Development of microsatellite multiplexes for wild goats using primers designed from domestic bovidae. Genet Sel Evol 33:S193–S203. https://doi.org/10.1186/BF03500880
Miller SM, Clarke AB, Bloomer P, Guthrie AJ, Harper CK (2016) Evaluation of microsatellites for common ungulates in the South African wildlife industry. Conserv Genet Resour 8:329–341. https://doi.org/10.1007/s12686-016-0554-7
Osmers B, Petersen BS, Hartl GB, Grobler JP, Kotze A, Van Aswegen E, Zachos FE (2012) Genetic analysis of southern African gemsbok (Oryx gazella) reveals high variability, distinct lineages and strong divergence from the East African Oryx beisa. Mamm Biol 77:60–66. https://doi.org/10.1016/j.mambio.2011.08.003
Røed KH (1998) Microsatellite variation in scandinavian cervidae using primers derived from bovidae. Hereditas 129:19–25. https://doi.org/10.1111/j.1601-5223.1998.00019.x
Gaur A, Singh A, Arunabala V, Umapathy G, Shailaja K, Singh L (2003) Development and characterization of 10 novel microsatellite markers from chital deer (Cervus axis) and their cross-amplification in other related species. Mol Ecol Notes 3:607–609. https://doi.org/10.1046/j.1471-8286.2003.00528.x
Kocher TD, Thomas WK, Meyer A, Edwards SV, Paabo S, Villablanca FX, Wilson AC (1989) Dynamics of mitochondrial DNA evolution in animals: amplification and sequencing with conserved primers. Proc Natl Acad Sci USA 86:6196–6200. https://doi.org/10.1073/pnas.86.16.6196
FAO (2011) Molecular genetic characterization of animal genetic resources. commission on genetic resources for food and agriculture food and agriculture organization. Rome, Italy
Kappes SM, Keele JW, Stone RT, Mcgraw RA, Sonstegard TS, Smith TPL, Lopez-Corrales NL, Beattie CW (1997) A second-generation linkage map of the bovine genome. Genome Res 7:235–249
Vial L, Maudet C, Luikart G (2003) Thirty-four polymorphic microsatellites for European roe deer. Mol Ecol Notes 3:523–527. https://doi.org/10.1046/j.1471-8286.2003.00499.x
Taberlet P, Griffin S, Goossens B, Questiau S, Manceau V, Escaravage N, Waits LP, Bouvet J (1996) Reliable genotyping of samples with very low DNA quantities using PCR. Nucleic Acids Res 24:3189–3194. https://doi.org/10.1093/nar/24.16.3189
Hauge XY, Litt M (1993) A study of the origin of ‘shadow bands’ seen when typing dinucleotide repeat polymorphisms by the PCR. Hum Mol Genet 2:411–415
Clark JM (1988) Novel non-templated nucleotide addition reactions catalyzed by procaryotic and eucaryotic DNA polymerases. Nucleic Acids Res 16:9677–9686. https://doi.org/10.1093/nar/16.20.9677
Matsumoto T, Yukawa W, Nozaki Y et al (2004) Novel algorithm for automated genotyping of microsatellites. Nucleic Acids Res 32:6069–6077. https://doi.org/10.1093/nar/gkh946
Salin F (2010) Autobin v0.9. http://www4.bordeaux-aquitaine.inra.fr/biogeco/Resources/Logiciels/Autobin. Accessed 26 April 2016.
Peakall R, Smouse PE (2012) GenALEx 6.5: genetic analysis in Excel. Population genetic software for teaching and research - an update. Bioinformatics 28:2537–2539. https://doi.org/10.1093/bioinformatics/bts460
Chapuis MP, Estoup A (2007) Microsatellite null alleles and estimation of population differentiation. Mol Biol Evol 24:621–631. https://doi.org/10.1093/molbev/msl191
Valière N (2002) GIMLET: a computer program for analysing genetic individual identification data. Mol Ecol Notes 2:377–379
Hansen H, Ben-David M, McDonald DB (2008) Effects of genotyping protocols on success and errors in identifying individual river otters (Lontra canadensis) from their faeces. Mol Ecol Resour 8:282–289. https://doi.org/10.1111/j.1471-8286.2007.01992.x
Wang J (2004) Sibship reconstruction from genetic data with typing errors. Genetics 166:1963–1979. https://doi.org/10.1534/genetics.166.4.1963
Jones OR, Wang J (2010) COLONY: a program for parentage and sibship inference from multilocus genotype data. Mol Ecol Resour 10:551–555. https://doi.org/10.1111/j.1755-0998.2009.02787.x
Cavalli-Sforza LL, Edwards AW (1967) Phylogenetic analysis: models and estimation procedures. Am J Hum Genet 19:233–257. https://doi.org/10.2307/2406616
Séré M, Thévenon S, Belem AMG, De Meeûs T (2017) Comparison of different genetic distances to test isolation by distance between populations. Heredity 119:55–63. https://doi.org/10.1038/hdy.2017.26
Langella O (2002) POPULATIONS, a free population genetics software. https://bioinformatics.org/populations. Accessed 31 March 2019.
Rambaut A (2014) FigTree v1.4.2: tree figure drawing tool. http://tree.bio.ed.ac.uk/software/figtree. Accessed 17 Jan 2018.
Schuelke M (2000) An economic method for the fluorescent labeling of PCR fragments. Nat Biotechnol 18:233–234. https://doi.org/10.1038/72708
Forgiarini C, Curto M, Stiehl-Alves EM, Bräuchler C, Kollmann J, Meimberg H, de Souza-Chies TT (2017) Fifteen microsatellite markers for Herbertia zebrina (Iridaceae): an endangered species from South American grasslands. Appl Plant Sci 5:1–4. https://doi.org/10.3732/apps.1700035
Rivière-Dobigny T, Doan LP, Le QN, Maillard JC, Michaux J (2009) Species identification, molecular sexing and genotyping using non-invasive approaches in two wild bovids species: Bos gaurus and Bos javanicus. Zoo Biol 28:127–136. https://doi.org/10.1002/zoo.20211
Soto-CalderÓn ID, Ntie S, Mickala P, Maisels F, Wickings EJ, Anthony NM (2009) Effects of storage type and time on DNA amplification success in tropical ungulate faeces: technical advances. Mol Ecol Resour 9:471–479. https://doi.org/10.1111/j.1755-0998.2008.02462.x
Chybicki IJ, Burczyk J (2009) Simultaneous estimation of null alleles and inbreeding coefficients. J Hered 100:106–113. https://doi.org/10.1093/jhered/esn088
Spong G, Johansson M, Björklund M (2000) High genetic variation in leopards indicates large and long-term stable effective population size. Mol Ecol 9:1773–1782. https://doi.org/10.1046/j.1365-294X.2000.01067.x
Creel S, Spong G, Sands JL, Rotella J, Zeigle J, Joe L, Murphy KM, Smith D (2003) Population size estimation in yellowstone wolves with error-prone noninvasive microsatellite genotypes. Mol Ecol 12:2003–2009. https://doi.org/10.1046/j.1365-294X.2003.01868.x
Rahmani AR (2001) India. In: Mallon DP, Kingswood SC (Eds) Antelopes: global survey and regional action plans. Part 4: North Africa, the Middle East and Asia. Gland, Switzerland, pp 178–187
Pompanon F, Bonin A, Bellemain E, Taberlet P (2005) Genotyping errors: causes, consequences and solutions. Nat Rev Genet 6:847–859. https://doi.org/10.1038/nrg1707
Shukla MA, Joshi BD, Kumar VP, Mehta AK, Goyal SP (2019) Investigating the genetic diversity and presence of forensically informative nucleotide sequences in Indian antelope (Antilope cervicapra) using multiple genes of the mitochondrial genome. Mol Biol Rep 46:6187–6195. https://doi.org/10.1007/s11033-019-05054-5
Acknowledgements
The authors thank the Director, the Dean, Faculty of Wildlife Science, the Research Coordinator and the Nodal Officer, Wildlife Forensic and Conservation Genetics Cell, Wildlife Institute of India, Dehradun, Uttarakhand, India, for facilitating the study. We appreciate Mr. A. Madhanraj for logistic support. We also thank the Uttar Pradesh State Forest Department for providing permission for the fieldwork. The study was financed by M/s Welspun Energy UP Pvt Ltd.
Funding
The financial support for this study was granted by M/s Welspun Energy UP Pvt Ltd. The funding agency had no role in the design or execution of the experiments.
Author information
Authors and Affiliations
Contributions
Concept: SPG, BH, RD; Fieldwork: RD, KA, KAK, HK; Sample processing: RD, VK, KA, KAK, HK, NK; Performed the experiments: RD, VK, NK; Analysed the data: RD, SPG, BH; Authored the original manuscript: RD; Reviewed and commented on the draft manuscript: SPG, BH; approved the initial and revised drafts: RD, VK, KA, KAK, HK, NK, SPG, BH.
Corresponding authors
Ethics declarations
Conflict of interest
The authors declare no conflicts of interest.
Ethical approval
This study did not involve handling of animals. We did not use any tissue samples. We received permission to collect non-invasive faecal samples from Uttar Pradesh State Forest Department vide letter no. 456 dated 20th August 2018.
Consent for publication
Not applicable as this study does not contain data from any individual person. All authors approved and consented to the submission of the final draft of the manuscript.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Below is the link to the electronic supplementary material.
Rights and permissions
About this article

Cite this article
De, R., Kumar, V., Ankit, K. et al. Cross-amplification of ungulate microsatellite markers in the endemic Indian antelope or blackbuck (Antilope cervicapra) for population monitoring and conservation genetics studies in south Asia. Mol Biol Rep 48, 5151–5160 (2021). https://doi.org/10.1007/s11033-021-06514-7
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s11033-021-06514-7