
Abstract
Amyotrophic lateral sclerosis (ALS) is a multifactorial neurodegenerative disease. Inflammatory processes are among the mechanisms that are implicated in ALS pathogenesis. The TREM2 rs75932628 T variant may influence the regulatory effect of TREM2 on inflammation. Studies regarding the role of the rs75932628 variant in ALS have yielded inconsistent results, so far. To assess the role of TREM2 rs75932628 on ALS risk. We genotyped 155 patients with sporadic ALS and 155 healthy controls for TREM2 rs75932628. We also merged and meta-analyzed our data with data from previous studies (with a total of 7524 ALS cases and 14,675 controls), regarding TREM2 rs75932628 and ALS. No ALS or healthy subjects carried the TREM2 rs75932628-T variant. Results from meta-analyses (overall approach and sensitivity analyses) yielded no significant results for possible connection between TREM2 rs75932628-T variant and ALS. Based on our results, TREM2 rs75932628 does not seem to play a determining role to the pathophysiology of ALS.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Amyotrophic lateral sclerosis (ALS) is a syndrome with diverse manifestations, and is considered as the commonest motor neuron disease of adulthood [1]. ALS is slightly more frequent among males, while its prevalence is considered to be almost 0.003 cases per 100 human individuals [2]. However, frequency, incidence and age at onset, vary between population with different ancestry [3].
The clinical manifestations of ALS is outlined from symptomatology stemming from both lower and upper motor neuron dysfunction, and is characterized by a progressive course and heterogeneous phenotypes, as any skeletal muscle can be affected [4]. Moreover, non-motor deficits have been described in ALS, as it may also coexist and overlap with frontotemporal degeneration (FTD) regarding both molecular and clinical aspects [5].
The main pathophysiological mechanisms driving the emergence of ALS remain widely elusive [6]. Various possible mechanisms implicated in ALS have been reported, including, but not limited to, mitochondrial dysfunction, excitotoxicity, RNA deficits and toxicity, oxidative stress, inflammation, defected protein degradation and toxic protein aggregation [3, 7].
Again, the pathways via which the aforementioned processes are activated and lead to ALS are poorly understood. However, there is accumulating evidence, that from one side the environment, and from another the genetics can be implicated [7]. In fact, a substantial part of ALS pathogenesis may result from the interaction between environmental and genetic risk factors [8].
The involvement of genetics in ALS is indisputable, as over 120 genetic variants have been reported to be linked with ALS [9]. From a genetic point of view, ALS was traditionally dichotomized as familial ALS (fALS), (1 in 10 of all ALS cases) with a great burden of genes having been associated with the disease, and sporadic ALS (sALS) (9 in 10 of all ALS cases), towards which a few genes have been reported to confer susceptibility [10]. Concerning fALS, the commonest gene mutations reported are in C9orf72, SOD1, FUS, and TARBDP genes [10]. However, recent evidence suggests that similarities in the genetic architecture of familial and sporadic ALS exist, as relatives of patients with sALS are at higher risk of developing ALS [11] whereas mutations involved in fALS can be found in 10% of patients with sALS [10]. Additionally, few genetic risk factors have been associated with sALS as well [10], though the conferred risk from rare genetic variants indicates that ALS may follow an oligogenic disease pattern [3, 12].
Triggering receptor expressed on myeloid cells 2 (TREM2), is a cell surface receptor, member of the TREM transmembrane glycoprotein family, and it mainly located on dendritic cells, microglia, osteoclast precursors and activated macrophages [13, 14]. Its main action is the negative regulation of the inflammation by acting as an immunomodulatory receptor [15, 16]. Additionally, mutations of the TREM2 gene have been reported in patients with Nasu-Hakola disease (NHD) [17]. The concept of the possible implication of TREM2 to neurodegeneration derives from the hypothesis that TREM2 may be an important molecule for microglia phagocytosis and the resolution of inflammation [18]. As such, impaired TREM2 function may lead to defective immune response, extensive inflammatory processes, deficient neuroprotective microglial activation, ultimately resulting in neurodegeneration [19]. This hypothesis was further reinforced, after the identification of the TREM2 rs75932628 (p.R47H) variant, as a possible risk factor for Alzheimer’s disease (AD), the most common neurodegenerative disease [20].
Taking into consideration that inflammatory processes are pivotal perpetuators implicated in ALS pathogenesis [3], that TREM2 rs75932628 variant may influence the regulatory effect of TREM2 in inflammation [20], and that studies regarding the role of the rs75932628 variant to ALS have yielded inconsistent results so far [21], we performed a case-control study aiming to survey the existence or not of association of TREM2 rs75932628 with ALS. We also merged and meta-analyzed our data with data from previous studies concerning TREM2 rs75932628 and ALS.
Case–control study
Methods
Participants
Three hundred and ten individuals were drafted for the current study. More precisely 155 patients with sporadic ALS [78 (50.3%) male, mean age ± standard deviation (SD) = 63.74 ± 11.30 years], and 155 healthy controls, were gathered from the University Hospital of Larissa, in Greece (Neurology Department). Diagnoses of ALS were made by consultant neurologists, according to the El Escorial criteria [22]. Patient characteristics are presented in Table 1. The study protocol got the approval by the local ethics committee. Also, all the participants granted a written informed consent. Additional information concerning sample characteristics has been previously described in great detail [23, 24].
Molecular genetics
Peripheral blood was collected form all the participants and the genomic DNA was isolated from leucocytes via the method of salting out [25]. All the ALS cases and the controls were genotyped for the TREM2 rs75932628 with the TaqMan allele specific discrimination assays on an ABI PRISM 7900 Sequence Detection System. Results were afterwards analyzed with the SDS software (Applied Biosystems, Foster City, California, USA). The genotype call rate was 99.68%.
Results
The genotype call rate was calculated equal to 99.68% (99.35% for ALS cases and 100% for healthy controls) leading to 154 cases with ALS and 155 controls for comparison. A total of 309 samples were genotyped; though the rs75932628-T allele variant was not carried by any of the participants.
Meta-analysis
Methods
Prior to its implementation, the current meta-analysis was not registered in any database. The present meta-analysis was conducted in accordance with the preferred reporting items systematic reviews and meta-analyses (PRISMA) guidelines (Supplementary File 1) [26]. The entire process was independently carried out by two authors (VS and IL), while any divergences were unraveled by a 3rd author (ED).
Literature search
Literature review was performed on MEDLINE (via PubMed). We searched for articles from the inception of PubMed up to July 2020, for studies in humans, regarding ALS and rs75932628. The terms “amyotrophic lateral sclerosis” and “TREM2” were used as free words. The last PubMed searched was carried out on July 21st, 2020. We manually screened all the titles and the abstracts for eligibility. From the articles that passed the initial screening, full texts were evaluated. Moreover, the reference lists of the retrieved articles were scanned for supplementary eligible articles.
Eligibility criteria
Studies were included according to the following criteria: (1) published before July 21st, 2020; (2) the absolute number of carriers of the rs75932628, were accessible for both ALS cases and controls; (3) the absolute number of both ALS cases and controls were available and (4) written in English language.
Data extraction
From each article that fulfilled the eligibility criteria, the following data were collected when possible: (1) author, (2) year of publication, (3) ethnicity of the population, (4) screening patients for causative ALS mutation, (5) diagnosis assessment, (6) main demographic characteristics (age, sex, number of participants) of ALS cases and controls, (7) main results.
Statistical analysis
The MetaXL (www.epigear.com) statistical software [27], as add-in for Microsoft Excel, was used in order for the ORs and the 95% CIs for the effect of the rs75932628 variant, be calculated. The Statistical heterogeneity was calculated with the Q-statistic [28] (homogeneity rejected if PQ < 0.1) and I2 statistic [29] (with the value I2 > 75% as indicative for heterogeneity). Both random effects (RE) and Mantel–Haenszel (MH) [fixed effect (FE)] models were applied [30,31,32]. The detection of publication bias was made visually with the DOI plot and numerically with the LFK index [33]. Continuity correction (add 0.5 to the number of events and non-events) was used for studies with zero events [34]. Sensitivity analyses by omitting one study at a time, aiming to examine the effect of each individual study, were also performed. Further sensitivity analysis was carried out, by excluding studies in which continuity correction was applied (studies with no observed events), aiming to minimize the possible bias effect of this method [34].
Results
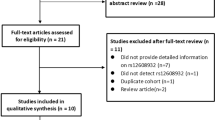
Twenty-eight articles were retrieved from the MEDLINE (through PubMed) search, published between 2013 and 2020. The manual screening of the reference lists yielded no additional eligible articles. Six full texts were assessed for possible eligibility, after the initial evaluation of both titles and abstracts. Seven studies (6 retrieved articles along with our case-control study) were finally included in this meta-analysis, which finally consisted of 11 datasets, with a total of 7524 ALS cases and 14,675 controls [18, 35,36,37,38,39]. Forty-three carriers of TREM2 rs75932628 were counted in ALS cases and 73 in healthy controls. The main traits of the eligible studies are shown in Table 2. The process of study selection is presented in Supplementary File 2 as a PRISMA flow diagram.
Analysis for publication bias was indicative for only minor asymmetry, with LFK index equal to 1.41 (Fig. 1). The statistical analysis revealed no significant results for possible connection between TREM2 rs75932628 and ALS (RE model OR 1.32; 95% CI 0.74–2.35; and ΜΗ model OR 1.17; 95% CI 0.78–1.74). The Forest plots can be accessed in Fig. 2. In the sensitivity analysis, after omitting one study at a time, the pooled ORs (95% CIs) ranged from 0.91 (ΜΗ; 95% CI 0.59–1.41) to 1.57 (RE; 95% CI 0.89–2.75). The additional sensitivity analysis, with the exclusion of studies in which continuity correction was applied, also yielded negative results (RE OR 1.39; 95% CI 0.71–2.72, and ΜΗ model OR 1.17; 95% CI 0.78–1.76). The results from the sensitivity analyses are summarized at Table 3, while the respective forest plots can be found at Supplementary File 3.

Doi plots presenting the publication bias from meta-analysis

Forest Plots presenting the results from overall meta-analysis
Discussion
The purpose of the current case–control study and meta-analysis was to gather and analyze the available published data regarding TREM2 rs75932628, and to evaluate its role in ALS. We did not find any carriers of the rs75932628 T allele in either ALS patients or in healthy controls. Moreover, the results from meta-analysis in a sample consisting of 7524 ALS cases and 14,675 healthy controls did not reveal any association, either, with several sensitivity analyses having been performed. Therefore, based on our results it appears rather unlikely that the TREM2 rs75932628 T allele is among the major risk factors for ALS.
In 2013 Rayaprolu et al. [18].hypothesized that a defective function of TREM2, which appear to be an important element in the proper phagocytic processing of apoptotic neuronal cells by microglia, may be implicated in neurodegenerative processes [18]. Consequently, they conducted the first study exploring the role of TREM2 rs75932628 (a non-synonymous variant) in ALS. However, no evidence of association emerged. Since then, few studies tried to further examine the role of rs75932628 in ALS [35,36,37,38,39]. Only in one of them, the study of Cady et al. [36] was the rs75932628 associated with ALS (OR 2.40; 95% CI 1.29–4.15 and p-value = 4.1 × 10−3) [36]. Moreover, in this study, they only detected 10 rs75932628 carriers in a sample of 920 ALS patients. The rest of the studies produced negative results [35, 37,38,39], and in two of them, no carrier of the rs75932628 was identified [37, 39]. The latter is in accordance with our results. Given the low frequency of T allele of the rs75932628 in European populations [40, 41], it is evident that studies with even larger samples are needed in order for the adequate statistical power to be achieved.
Over the last decade, TREM2 has drawn scientific attention, due to its possible application as a biomarker of neurodegeneration, especially in AD [15]. Therefore, a number of studies have been performed examining the contribution of TREM2 rs75932628 in various neurological diseases (besides ALS), such as AD [20], corticobasal syndrome mild cognitive impairment, FTD, progressive supranuclear palsy syndrome [35], Parkinson’s disease (PD) [42], essential tremor (ET) [43], ischemic stroke [18], and multiple sclerosis [40]. However, the strongest association, and one having high reproducibility rates, appears in the context of AD [21]. On the other hand, for ET, PD and FTD only modest or ancestry specific effects have been described, without stable repeatability [15, 43].
TREM2 expression has been shown to be elevated in the spinal cord of patients with ALS, and SOD1G93A mice, and also with higher levels of TREM2 negatively correlating with survival, suggesting a dysregulation of TREM2 in ALS [36]. Apart from the full-length TREM2, the soluble form of TREM2 (sTREM2) (a product the either from the proteolytic cleavage of the TREM2 receptor or from the alternative splicing of TREM2 lacking the transmembrane domain), has also been the subject of research [15, 44, 45]. Soluble TREM2 (sTREM2) is of great importance for AD, as it has been reported to be increased in the cerebrospinal fluid (CSF) of patients even from the early disease stages [15, 45, 46]. Furthermore, its levels have been positively correlated with both total tau and phosphorylated-tau (p-tau), denoting is importance to neurodegeneration [15, 45, 46]. Concerning ALS, Cooper-Knock et al. [47], found that sTREM2 protein was elevated in the CSF of patients with ALS [47]. However, in a recent study by Huang et al. [48] sTREM2 levels were found increased only in the plasma but not in the CSF of patients with ALS, while, it was also elevated in the CSF of a cluster of ALS patients carrying either C9orf72 or with patients with fast disease progression [48], possibly suggesting that this protein’s levels are linked to worse prognosis.
While the survival of ALS patients varies, patients usually die within 3–4 years from disease onset due to respiratory failure [3]. Therapeutic approaches are very limited, as riluzole remains the only widely available regiment that seems to prolong the survival of ALS patients [49]. These facts highlight that ongoing research, also including genetic studies, that facilitates the understanding of the molecular basis of the disease, can lead to the discovery of much-needed biomarkers [50] and therapeutic agents, which in turn will considerably ameliorate the quality of life of ALS patients.
Among the strengths of our study is the high homogeneity of the ALS sample, as it was drafted from the same geographical area and participants had homogenous ancestry. Moreover, we gathered data form a total of 7524 ALS cases and 14,675 controls, which consist, to the best of our knowledge, the most populous sample to date to be examined for TREM2 rs75932628. The main limitation is that patients with ALS were not screened for major ALS-linked genes, such as C9orf72, SOD1, NEK1, and TARDBP [23].
To conclude, in the current case-control study, we found neither ALS cases nor controls carrying the TREM2 rs75932628 T allele. Whether this variant consists a risk factor for ALS is still debatable, though the evidence so far suggests otherwise; indeed our meta-analysis, which included several sensitivity metrics to enhance its accuracy, found no association between the variant and ALS. We believe that future collaborative studies investigating the carriage of this variant in larger multicenter samples with different ancestry backgrounds, perhaps alongside the study of gene variants with stronger evidence, will be helpful, as ALS may follow an oligogenic disease pattern and rare genetic variants might confer risk in patients with ALS.
References
Picher-Martel V, Valdmanis PN, Gould PV, Julien JP, Dupré N (2016) From animal models to human disease: a genetic approach for personalized medicine in ALS. Acta Neuropathol Commun 4(1):70. https://doi.org/10.1186/s40478-016-0340-5
Al-Chalabi A, Hardiman O (2013) The epidemiology of ALS: a conspiracy of genes, environment and time. Nat Rev Neurol 9(11):617–628. https://doi.org/10.1038/nrneurol.2013.203
van Es MA, Hardiman O, Chio A, Al-Chalabi A, Pasterkamp RJ, Veldink JH, van den Berg LH (2017) Amyotrophic lateral sclerosis. Lancet 390(10107):2084–2098. https://doi.org/10.1016/S0140-6736(17)31287-4
Swinnen B, Robberecht W (2014) The phenotypic variability of amyotrophic lateral sclerosis. Nat Rev Neurol 10(11):661–670. https://doi.org/10.1038/nrneurol.2014.184
Liscic RM, Alberici A, Cairns NJ, Romano M, Buratti E (2020) From basic research to the clinic: innovative therapies for ALS and FTD in the pipeline. Mol Neurodegener 15(1):31. https://doi.org/10.1186/s13024-020-00373-9
van den Bos MAJ, Geevasinga N, Higashihara M, Menon P, Vucic S (2019) Pathophysiology and diagnosis of ALS: insights from advances in neurophysiological techniques. Int J Mol Sci 20(11):2818. https://doi.org/10.3390/ijms20112818
Dardiotis E, Aloizou AM, Siokas V, Patrinos GP, Deretzi G, Mitsias P, Aschner M, Tsatsakis A (2018) The role of microRNAs in patients with amyotrophic lateral sclerosis. J Mol Neurosci 66(4):617–628. https://doi.org/10.1007/s12031-018-1204-1
Bennett SA, Tanaz R, Cobos SN, Torrente MP (2019) Epigenetics in amyotrophic lateral sclerosis: a role for histone post-translational modifications in neurodegenerative disease. Transl Res 204:19–30. https://doi.org/10.1016/j.trsl.2018.10.002
Taylor JP, Brown RH Jr, Cleveland DW (2016) Decoding ALS: from genes to mechanism. Nature 539(7628):197–206. https://doi.org/10.1038/nature20413
Brown RH, Al-Chalabi A (2017) Amyotrophic lateral sclerosis. N Engl J Med 377(2):162–172. https://doi.org/10.1056/NEJMra1603471
Al-Chalabi A, Lewis CM (2011) Modelling the effects of penetrance and family size on rates of sporadic and familial disease. Hum Hered 71(4):281–288. https://doi.org/10.1159/000330167
van Rheenen W, Shatunov A, Dekker AM, McLaughlin RL, Diekstra FP, Pulit SL, van der Spek RA, Võsa U, de Jong S, Robinson MR, Yang J, Fogh I, van Doormaal PT, Tazelaar GH, Koppers M, Blokhuis AM, Sproviero W, Jones AR, Kenna KP, van Eijk KR, Harschnitz O, Schellevis RD, Brands WJ, Medic J, Menelaou A, Vajda A, Ticozzi N, Lin K, Rogelj B, Vrabec K, Ravnik-Glavač M, Koritnik B, Zidar J, Leonardis L, Grošelj LD, Millecamps S, Salachas F, Meininger V, de Carvalho M, Pinto S, Mora JS, Rojas-García R, Polak M, Chandran S, Colville S, Swingler R, Morrison KE, Shaw PJ, Hardy J, Orrell RW, Pittman A, Sidle K, Fratta P, Malaspina A, Topp S, Petri S, Abdulla S, Drepper C, Sendtner M, Meyer T, Ophoff RA, Staats KA, Wiedau-Pazos M, Lomen-Hoerth C, Van Deerlin VM, Trojanowski JQ, Elman L, McCluskey L, Basak AN, Tunca C, Hamzeiy H, Parman Y, Meitinger T, Lichtner P, Radivojkov-Blagojevic M, Andres CR, Maurel C, Bensimon G, Landwehrmeyer B, Brice A, Payan CA, Saker-Delye S, Dürr A, Wood NW, Tittmann L, Lieb W, Franke A, Rietschel M, Cichon S, Nöthen MM, Amouyel P, Tzourio C, Dartigues JF, Uitterlinden AG, Rivadeneira F, Estrada K, Hofman A, Curtis C, Blauw HM, van der Kooi AJ, de Visser M, Goris A, Weber M, Shaw CE, Smith BN, Pansarasa O, Cereda C, Del Bo R, Comi GP, D’Alfonso S, Bertolin C, Sorarù G, Mazzini L, Pensato V, Gellera C, Tiloca C, Ratti A, Calvo A, Moglia C, Brunetti M, Arcuti S, Capozzo R, Zecca C, Lunetta C, Penco S, Riva N, Padovani A, Filosto M, Muller B, Stuit RJ, Blair I, Zhang K, McCann EP, Fifita JA, Nicholson GA, Rowe DB, Pamphlett R, Kiernan MC, Grosskreutz J, Witte OW, Ringer T, Prell T, Stubendorff B, Kurth I, Hübner CA, Leigh PN, Casale F, Chio A, Beghi E, Pupillo E, Tortelli R, Logroscino G, Powell J, Ludolph AC, Weishaupt JH, Robberecht W, Van Damme P, Franke L, Pers TH, Brown RH, Glass JD, Landers JE, Hardiman O, Andersen PM, Corcia P, Vourc’h P, Silani V, Wray NR, Visscher PM, de Bakker PI, van Es MA, Pasterkamp RJ, Lewis CM, Breen G, Al-Chalabi A, van den Berg LH, Veldink JH (2016) Genome-wide association analyses identify new risk variants and the genetic architecture of amyotrophic lateral sclerosis. Nat Genet 48(9):1043–1048. https://doi.org/10.1038/ng.3622
Klesney-Tait J, Turnbull IR, Colonna M (2006) The TREM receptor family and signal integration. Nat Immunol 7(12):1266–1273. https://doi.org/10.1038/ni1411
Ulland TK, Colonna M (2018) TREM2—a key player in microglial biology and Alzheimer disease. Nat Rev Neurol 14(11):667–675. https://doi.org/10.1038/s41582-018-0072-1
Zhong L, Chen X-F (2019) The emerging roles and therapeutic potential of soluble TREM2 in Alzheimer’s disease. Front Aging Neurosci 11:328–328. https://doi.org/10.3389/fnagi.2019.00328
Zhu Z, Zhang X, Dong W, Wang X, He S, Zhang H, Wang X, Wei R, Chen Y, Liu X, Guo C (2020) TREM2 suppresses the proinflammatory response to facilitate PRRSV infection via PI3K/NF-κB signaling. PLoS Pathog 16(5):e1008543–e1008543. https://doi.org/10.1371/journal.ppat.1008543
Dardiotis E, Siokas V, Pantazi E, Dardioti M, Rikos D, Xiromerisiou G, Markou A, Papadimitriou D, Speletas M, Hadjigeorgiou GM (2017) A novel mutation in TREM2 gene causing Nasu-Hakola disease and review of the literature. Neurobiol Aging 53:194.e113-194.e122. https://doi.org/10.1016/j.neurobiolaging.2017.01.015
Rayaprolu S, Mullen B, Baker M, Lynch T, Finger E, Seeley WW, Hatanpaa KJ, Lomen-Hoerth C, Kertesz A, Bigio EH, Lippa C, Josephs KA, Knopman DS, White CL, Caselli R, Mackenzie IR, Miller BL, Boczarska-Jedynak M, Opala G, Krygowska-Wajs A, Barcikowska M, Younkin SG, Petersen RC, Ertekin-Taner N, Uitti RJ, Meschia JF, Boylan KB, Boeve BF, Graff-Radford NR, Wszolek ZK, Dickson DW, Rademakers R, Ross OA (2013) TREM2 in neurodegeneration: evidence for association of the p.R47H variant with frontotemporal dementia and Parkinson’s disease. Mol Neurodegener 8(1):19. https://doi.org/10.1186/1750-1326-8-19
Guerreiro R, Hardy J (2013) TREM2 and neurodegenerative disease. N Engl J Med 369(16):1569–1570. https://doi.org/10.1056/NEJMc1306509
Jonsson T, Stefansson H, Steinberg S, Jonsdottir I, Jonsson PV, Snaedal J, Bjornsson S, Huttenlocher J, Levey AI, Lah JJ, Rujescu D, Hampel H, Giegling I, Andreassen OA, Engedal K, Ulstein I, Djurovic S, Ibrahim-Verbaas C, Hofman A, Ikram MA, van Duijn CM, Thorsteinsdottir U, Kong A, Stefansson K (2013) Variant of TREM2 associated with the risk of Alzheimer’s disease. N Engl J Med 368(2):107–116. https://doi.org/10.1056/NEJMoa1211103
Zhou SL, Tan CC, Hou XH, Cao XP, Tan L, Yu JT (2019) TREM2 variants and neurodegenerative diseases: a systematic review and meta-analysis. J Alzheimer’s Dis 68(3):1171–1184. https://doi.org/10.3233/jad-181038
Ludolph A, Drory V, Hardiman O, Nakano I, Ravits J, Robberecht W, Shefner J (2015) A revision of the El Escorial criteria—2015. Amyotroph Lateral Scler Frontotemp Degener 16(5–6):291–292. https://doi.org/10.3109/21678421.2015.1049183
Dardiotis E, Karampinis E, Siokas V, Aloizou AM, Rikos D, Ralli S, Papadimitriou D, Bogdanos DP, Hadjigeorgiou GM (2019) ERCC6L2 rs591486 polymorphism and risk for amyotrophic lateral sclerosis in Greek population. Neurol Sci 40(6):1237–1244. https://doi.org/10.1007/s10072-019-03825-3
Siokas V, Karampinis E, Aloizou AM, Mentis AA, Liakos P, Papadimitriou D, Liampas I, Nasios G, Bogdanos DP, Hadjigeorgiou GM, Dardiotis E (2020) CYP1A2 rs762551 polymorphism and risk for amyotrophic lateral sclerosis. Neurol Sci. https://doi.org/10.1007/s10072-020-04535-x
Siokas V, Kardaras D, Aloizou AM, Asproudis I, Boboridis KG, Papageorgiou E, Hadjigeorgiou GM, Tsironi EE, Dardiotis E (2019) BDNF rs6265 (Val66Met) polymorphism as a risk factor for blepharospasm. Neuro Mol Med 21(1):68–74. https://doi.org/10.1007/s12017-018-8519-5
Liberati A, Altman DG, Tetzlaff J, Mulrow C, Gøtzsche PC, Ioannidis JP, Clarke M, Devereaux PJ, Kleijnen J, Moher D (2009) The PRISMA statement for reporting systematic reviews and meta-analyses of studies that evaluate healthcare interventions: explanation and elaboration. BMJ 339:b2700. https://doi.org/10.1136/bmj.b2700
Clarke NE, Clements AC, Doi SA, Wang D, Campbell SJ, Gray D, Nery SV (2017) Differential effect of mass deworming and targeted deworming for soil-transmitted helminth control in children: a systematic review and meta-analysis. Lancet 389(10066):287–297. https://doi.org/10.1016/s0140-6736(16)32123-7
Cochran WG (1954) The combination of estimates from different experiments. Biometrics 10:101–129. https://doi.org/10.2307/3001666
Higgins JP, Thompson SG, Deeks JJ, Altman DG (2003) Measuring inconsistency in meta-analyses. BMJ 327(7414):557–560. https://doi.org/10.1136/bmj.327.7414.557
DerSimonian R, Laird N (1986) Meta-analysis in clinical trials. Control Clin Trials 7(3):177–188. https://doi.org/10.1016/0197-2456(86)90046-2
Mantel N, Haenszel W (1959) Statistical aspects of the analysis of data from retrospective studies of disease. JNCI 22(4):719–748. https://doi.org/10.1093/jnci/22.4.719
Banerjee I (2009) CD14 C260T promoter polymorphism and the risk of cerebrovascular diseases: a meta-analysis. J Appl Genet 50(2):153–157. https://doi.org/10.1007/BF03195667
Furuya-Kanamori L, Barendregt JJ, Doi SAR (2018) A new improved graphical and quantitative method for detecting bias in meta-analysis. Int J Evid-Based Healthc 16(4):195–203. https://doi.org/10.1097/xeb.0000000000000141
Efthimiou O (2018) Practical guide to the meta-analysis of rare events. Evid Based Ment Health 21(2):72–76. https://doi.org/10.1136/eb-2018-102911
Ayer AH, Wojta K, Ramos EM, Dokuru D, Chen JA, Karydas AM, Papatriantafyllou JD, Agiomyrgiannakis D, Kamtsadeli V, Tsinia N, Sali D, Gylys KH, Agosta F, Filippi M, Small GW, Bennett DA, Gearing M, Juncos JL, Kramer J, Lee SE, Yokoyama JS, Mendez MF, Chui H, Zarow C, Ringman JM, Kilic U, Babacan-Yildiz G, Levey A, DeCarli CS, Cotman CW, Boxer AL, Miller BL, Coppola G (2019) Frequency of the TREM2 R47H variant in various neurodegenerative disorders. Alzheimer Dis Assoc Disord 33(4):327–330. https://doi.org/10.1097/wad.0000000000000339
Cady J, Koval ED, Benitez BA, Zaidman C, Jockel-Balsarotti J, Allred P, Baloh RH, Ravits J, Simpson E, Appel SH, Pestronk A, Goate AM, Miller TM, Cruchaga C, Harms MB (2014) TREM2 variant p.R47H as a risk factor for sporadic amyotrophic lateral sclerosis. JAMA Neurol 71(4):449–453. https://doi.org/10.1001/jamaneurol.2013.6237
Chen X, Chen Y, Wei Q, Guo X, Cao B, Ou R, Zhao B, Shang HF (2015) Assessment of TREM2 rs75932628 association with amyotrophic lateral sclerosis in a Chinese population. J Neurol Sci 355(1–2):193–195. https://doi.org/10.1016/j.jns.2015.05.010
Lill CM, Rengmark A, Pihlstrøm L, Fogh I, Shatunov A, Sleiman PM, Wang LS, Liu T, Lassen CF, Meissner E, Alexopoulos P, Calvo A, Chio A, Dizdar N, Faltraco F, Forsgren L, Kirchheiner J, Kurz A, Larsen JP, Liebsch M, Linder J, Morrison KE, Nissbrandt H, Otto M, Pahnke J, Partch A, Restagno G, Rujescu D, Schnack C, Shaw CE, Shaw PJ, Tumani H, Tysnes OB, Valladares O, Silani V, van den Berg LH, van Rheenen W, Veldink JH, Lindenberger U, Steinhagen-Thiessen E, Teipel S, Perneczky R, Hakonarson H, Hampel H, von Arnim CAF, Olsen JH, Van Deerlin VM, Al-Chalabi A, Toft M, Ritz B, Bertram L (2015) The role of TREM2 R47H as a risk factor for Alzheimer’s disease, frontotemporal lobar degeneration, amyotrophic lateral sclerosis, and Parkinson’s disease. Alzheimer’s Dement 11(12):1407–1416. https://doi.org/10.1016/j.jalz.2014.12.009
Peplonska B, Berdynski M, Mandecka M, Barczak A, Kuzma-Kozakiewicz M, Barcikowska M, Zekanowski C (2018) TREM2 variants in neurodegenerative disorders in the polish population. Homozygosity and compound heterozygosity in FTD patients. Amyotroph Later Scler Frontotemp Degener 19(5–6):407–412. https://doi.org/10.1080/21678421.2018.1451894
Rikos D, Siokas V, Aloizou AM, Tsouris Z, Aslanidou P, Koutsis G, Anagnostouli M, Bogdanos DP, Grigoriadis N, Hadjigeorgiou GM, Dardiotis E (2019) TREM2 R47H (rs75932628) variant is unlikely to contribute to multiple sclerosis susceptibility and severity in a large Greek MS cohort. Mult Scler Relat Disord 35:116–118. https://doi.org/10.1016/j.msard.2019.07.007
Sherry ST, Ward MH, Kholodov M, Baker J, Phan L, Smigielski EM, Sirotkin K (2001) dbSNP: the NCBI database of genetic variation. Nucleic Acids Res 29(1):308–311. https://doi.org/10.1093/nar/29.1.308
Dardiotis E, Rikos D, Siokas V, Aloizou AM, Tsouris Z, Sakalakis E, Brotis AG, Bogdanos DP, Hadjigeorgiou GM (2020) Assessment of TREM2 rs75932628 variant’s association with Parkinson’s disease in a Greek population and meta-analysis of current data. Int J Neurosci. https://doi.org/10.1080/00207454.2020.1750388
Ortega-Cubero S, Lorenzo-Betancor O, Lorenzo E, Agúndez JA, Jiménez-Jiménez FJ, Ross OA, Wurster I, Mielke C, Lin JJ, Coria F, Clarimon J, Ezquerra M, Brighina L, Annesi G, Alonso-Navarro H, García-Martin E, Gironell A, Marti MJ, Yueh KC, Wszolek ZK, Sharma M, Berg D, Krüger R, Pastor MA, Pastor P (2015) TREM2 R47H variant and risk of essential tremor: a cross-sectional international multicenter study. Parkinsonism Relat Disord 21(3):306–309. https://doi.org/10.1016/j.parkreldis.2014.12.010
Suárez-Calvet M, Morenas-Rodríguez E, Kleinberger G, Schlepckow K, Araque Caballero MÁ, Franzmeier N, Capell A, Fellerer K, Nuscher B, Eren E, Levin J, Deming Y, Piccio L, Karch CM, Cruchaga C, Shaw LM, Trojanowski JQ, Weiner M, Ewers M, Haass C, for the Alzheimer’s Disease Neuroimaging I (2019) Early increase of CSF sTREM2 in Alzheimer’s disease is associated with tau related-neurodegeneration but not with amyloid-β pathology. Mol Neurodegener 14(1):1. https://doi.org/10.1186/s13024-018-0301-5
Zhong L, Xu Y, Zhuo R, Wang T, Wang K, Huang R, Wang D, Gao Y, Zhu Y, Sheng X, Chen K, Wang N, Zhu L, Can D, Marten Y, Shinohara M, Liu CC, Du D, Sun H, Wen L, Xu H, Bu G, Chen XF (2019) Soluble TREM2 ameliorates pathological phenotypes by modulating microglial functions in an Alzheimer’s disease model. Nat Commun 10(1):1365. https://doi.org/10.1038/s41467-019-09118-9
Suárez-Calvet M, Kleinberger G, Araque Caballero M, Brendel M, Rominger A, Alcolea D, Fortea J, Lleó A, Blesa R, Gispert JD, Sánchez-Valle R, Antonell A, Rami L, Molinuevo JL, Brosseron F, Traschütz A, Heneka MT, Struyfs H, Engelborghs S, Sleegers K, Van Broeckhoven C, Zetterberg H, Nellgård B, Blennow K, Crispin A, Ewers M, Haass C (2016) sTREM2 cerebrospinal fluid levels are a potential biomarker for microglia activity in early-stage Alzheimer’s disease and associate with neuronal injury markers. EMBO Mol Med 8(5):466–476. https://doi.org/10.15252/emmm.201506123
Cooper-Knock J, Green C, Altschuler G, Wei W, Bury JJ, Heath PR, Wyles M, Gelsthorpe C, Highley JR, Lorente-Pons A, Beck T, Doyle K, Otero K, Traynor B, Kirby J, Shaw PJ, Hide W (2017) A data-driven approach links microglia to pathology and prognosis in amyotrophic lateral sclerosis. Acta Neuropathol Commun 5(1):23. https://doi.org/10.1186/s40478-017-0424-x
Huang F, Zhu Y, Hsiao-Nakamoto J, Tang X, Dugas JC, Moscovitch-Lopatin M, Glass JD, Brown RH Jr, Ladha SS, Lacomis D, Harris JM, Scearce-Levie K, Ho C, Bowser R, Berry JD (2020) Longitudinal biomarkers in amyotrophic lateral sclerosis. Ann Clin Transl Neurol 7(7):1103–1116. https://doi.org/10.1002/acn3.51078
Fang T, Al Khleifat A, Meurgey J-H, Jones A, Leigh PN, Bensimon G, Al-Chalabi A (2018) Stage at which riluzole treatment prolongs survival in patients with amyotrophic lateral sclerosis: a retrospective analysis of data from a dose-ranging study. Lancet Neurol 17(5):416–422. https://doi.org/10.1016/S1474-4422(18)30054-1
Yang B, Wu Y, Wang Y, Yang H, Du B, Di W, Xu X, Shi X (2020) Cerebrospinal fluid MFG-E8 as a promising biomarker of amyotrophic lateral sclerosis. Neurol Sci. https://doi.org/10.1007/s10072-020-04416-3
Funding
This study was supported in part by a research Grant from the Research Committee of the University of Thessaly, Greece (code: 5287).
Author information
Authors and Affiliations
Contributions
Conceptualization: VS, GMH and ED; methodology: VS, AA, IL and ZT; formal analysis and investigation: VS, AA, IL and ZT; writing—original draft preparation: VS and AA; writing—review and editing: VS, AA, IL, ZT, AFAM, GN, DP, DPB, GMH and ED; funding acquisition: ED; resources: ED; supervision: ED.
Corresponding author
Ethics declarations
Conflict of interest
The authors declare no conflict of interest.
Informed consent
Informed consent was obtained from all individual participants included in the study.
Consent to participate
All the authors listed have approved the manuscript that is enclosed.
Consent for publication
The manuscript is approved by all authors for publication.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Below is the link to the electronic supplementary material.
Rights and permissions
About this article

Cite this article
Siokas, V., Aloizou, AM., Liampas, I. et al. Lack of association between TREM2 rs75932628 variant and amyotrophic lateral sclerosis. Mol Biol Rep 48, 2601–2610 (2021). https://doi.org/10.1007/s11033-021-06312-1
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s11033-021-06312-1