
Abstract
Chronic liver diseases are attributed to liver injury. Development of fibrosis from chronic liver diseases is a dynamic process that involves multiple molecular and cellular processes. As the first to be impacted by injury, liver sinusoidal endothelial cells (LSECs) are involved in the pathogenesis of liver diseases caused by a variety of etiologies. Moreover, capillarization of LSECs has been recognized as an important event in the development of chronic liver diseases and fibrosis. Studies have reported that various cytokines (such as vascular endothelial growth factor, transforming growth factor-β), and pathways (such as hedgehog, and Notch), as well as epigenetic and metabolic factors are involved in the development of LSEC-mediated liver fibrosis. This review describes the complexity and plasticity of LSECs in fibrotic liver diseases from several perspectives, including the cross-talk between LSECs and other intra-hepatic cells. Moreover, it summarizes the mechanisms of several kinds of LSECs-targeting anti-fibrosis chemicals, and provides a theoretical basis for future studies.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Liver sinusoidal endothelial cells (LSECs) encompass approximately 50% of hepatic non-parenchymal cells, and constitute the wall of the hepatic sinusoid. LSECs exhibit unique biological and functional characteristics, and are phenotypically different from vascular endothelial cells. They have small pores (fenestrae), while lack diaphragm and basal lamina, therefore, they are more permeable. By adjusting the diameter and number of fenestrae, LSECs regulate the bidirectional transport of macromolecules and solutes between hepatic cells and the perisinusoidal space (Disse space). Regarding their ultrastructural characteristics, LSECs have numerous bristle-coated micropinocytotic vesicles and lysosome-like vacuoles in the perikaryon, indicating a special endocytotic activity. In addition, LSECs membranes have high affinity endocytosis receptors, including scavenger receptors (SR-A, SR-B and SR-H), Fc gamma receptor IIb2 (FcγR IIb2, also known as CD32b) and mannose receptors (MRs). Through endocytic vesicles and receptor-mediated endocytosis, LSECs can eliminate antigens, cell debris and immune complexes [1, 2]. In addition, they can regulate inflammation, leukocyte recruitment and immune responses to pathogens, thereby maintaining liver homeostasis and normal immune tolerance [3].
Chronic liver injury can be caused by various factors, including viral infections, alcohol intake, cholestasis and abnormal fat accumulation. Uncontrolled chronic liver disease can progress to fibrosis and even cirrhosis, which enhances the risk of decompensation and hepatocellular carcinoma (HCC). Through a variety of mechanisms, LSECs are involved in various stages of liver fibrosis. Therefore, it is important to elucidate on the roles of LSECs in chronic liver diseases, fibrosis and cirrhosis. After hepatic injury, LSECs rapidly lose their highly specialized phenotype and become capillaried. Capillarization refers to the disappearance of fenestrae (defenestration) and formation of continuous basement membranes, which transforms LSECs into ordinary non-specific or microvascular endothelial cells. These alterations impair the specific functions of LSECs, leading to impaired filtration and endocytosis. Moreover, capillarized LSECs interact with other hepatic cells to mediate the adhesion of immune cells and to modulate inflammatory responses in chronic liver diseases. They also activate parenchymal and non-parenchymal cells, particularly hepatic stellate cells (HSCs), leading to accumulation of the extracellular matrix (ECM). This process can be partially modeled in vitro by culturing LSECs on plastic dishes in serum-containing medium, a process referred to as differentiation [4]. Figure 1 depicts a basic overview about comparing LSEC morphology and function in health versus fibrosis. In summary, capillarization of LSECs is a key step in the development of chronic liver diseases; thus maintaining normal LSECs phenotypes and functions can inhibit liver fibrosis and the development of cirrhosis.

Morphological and functional changes between fenestrated and capillarized LSECs. During capillarization, the number and diameter of LSEC fenestration decrease, a continuous basement membrane is formed. At the same time, markers of LSECs also change. Makers in the blue arrow is highly expressed in fenestrated LESCs, while markers in the pink arrow is highly expressed in capillarized LSECs. The corresponding functions also change accordingly. LYVE-1 lymphatic vessel endothelial hyaluronic acid receptor 1, VCAM-1 vascular cell adhesion molecule-1, vWF von Willebrand Factor, ECM extracellular matrix
Cytokines and pathways involved in the regulation of LSEC capillarization
Vascular endothelial growth factor (VEGF)
VEGF, derived from either hepatocytes (HCs) or HSCs, has been shown to be closely associated with LSEC phenotypes and functions, both in vivo and in vitro [5]. VEGF regulates LSEC capillarization through both NO-dependent and NO-independent pathways. In this process, after binding to its receptors (VEGFR), VEGF stimulates the release of endogenous nitric oxide synthase (eNOS)-induced NO in LSECs. Then, NO participates in the conversion of guanosine triphosphate (GTP) to 3′,5′-cyclic guanosine monophosphate (cGMP) as well as in the activation of protein kinase G (PKG) through the soluble guanylate cyclase (sGC) thereby, phosphorylating target proteins. Activation of the eNOS-NO-sGC pathway enhances LSEC differentiation and it inhibits capillarization [5]. However, NO-independent pathways have not been clearly elucidated. Luo et al. [6] reported that VEGF suppresses the caveolin-1 (CAV-1) autophagy on cell membranes, which maintains the fenestrae of LSECs. However, the role of VEGF in liver fibrosis is complex, because it is associated with the angiogenesis of liver sinusoids. Studies have documented the associations between angiogenesis and the progression of fibrosis, with the involvement of VEGF in this process [7]. Notably, Kantari-Mimoun et al. [8] documented that myeloid cells, particularly macrophages, are constantly recruited to the fibrotic liver. Myeloid cell-derived VEGF up-regulates the expression of matrix metalloproteases while down-regulating the expression of tissue inhibitor of metalloproteases in LSECs. These processes promote collagen degradation, which resolves liver fibrosis.
In conclusion, the role of VEGF in LSECs to the development of fibrosis is multifaceted, and it is thought to be a “double-edged sword”. VEGF from different sources play multiple roles through several pathways.
Transforming growth factor-β (TGF-β) superfamily
Members of the TGF-β superfamily play important roles in liver fibrosis. This family has 33 members, including TGF-βs, activins and bone morphogenetic proteins (BMPs). TGF-β is secreted as part of a large latent complex that is then stored in the ECM, remaining inert under physiological conditions. Activation of latent TGF-β, a precursor of most fibrogenic TGF-β cytokines, is localized to sites where it is released [9]. This process is at least partially induced through integral interactions between αv integrins, ECM, and the actin cytoskeleton. Briefly, under cellular contraction-induced mechanical force, αv integrins attached to the actin cytoskeleton bind the latent complexes attached to ECM, thereby activating TGF-β, which binds its receptor [9, 10]. Moreover, LSEC-derived latent TGF-β is activated on HSCs membrane through plasma kallikrein, which is also released by LSECs during the process of fibrosis-related pathological angiogenesis [11]. Even though partially latent TGF-β is secreted by LSECs, it functions in HSCs. Activation of latent TGF-β promotes HSC transformation to myofibroblast and regulates remodeling of the extracellular matrix by stimulating the synthesis of ECM proteins, such as collagen and fibronectin [12]. Endothelial interstitial differentiation (EndMT) is involved in the development of some fibrotic diseases [13]. EndMT, a dynamic process in which endothelial cells evolve into myofibroblasts, is characterized by the loss of cell–cell junctions and endothelial markers, while acquiring mesenchymal phenotypes. Ribera et al. [14]. reported that a small subpopulation of LSECs in their study exhibited the EndMT process in a liver fibrosis mouse model and cirrhosis patients. TGF-β1 is one of the three isoforms of TGF-β proteins, it is a crucial molecular promoter of LSEC EndMT. BMP7, another member of the TGF-β superfamily, has been shown to antagonize EndMT in vivo [14]. However, the proportion of these transformed cells in endothelial cells is very low (about 4%) [14]. This study provides descriptive evidences that rely on co-localization of cell markers by IHC in cirrhotic patients, which is not a proof that EndMT occurs in humans. Therefore, the role of EndMT in liver fibrosis should be elucidated. Specific treatments of EndMT may have little effects on liver fibrosis. Additionally, HSCs-produced BMP9 occurs in blood as a BMP9-BMP10 dimer [15, 16], and plays an important role in maintaining endothelial cell quiescence through activin receptor-like kinase (ALK) [17]. In LSECs from BMP9-knockout mice, the number of fenestrae was shown to be significantly suppressed. Consistently, the expression of GATA-binding protein 4 (GATA4), a specific transcription regulator that maintains LSECs differentiation, were significantly inhibited. Endothelial differentiation markers, including lymphatic vessel endothelial hyaluronic acid receptor 1 (LYVE-1) and stabilin-1/2 were also shown to be down-regulated. Plasmalemmal vesicle-associated protein (PLVAP), is the main integral protein that is correlated with fenestrae. Addition of BMP9 to the LSECs medium maintains fenestrae and elevates the expression of GATA4 and PLVAP in LSECs. However, one in vitro study involving human LSECs reported dissimilar findings, in which co-stimulation of LPS and BMP9 inhibited the expression of stabilin-1 and LYVE-1 in LSECs [18]. Based on these findings, the overall impact of BMP9 on LSEC capillarization has not been fully elucidated, and should be studied further.
Compared to control rats, it was reported that bone marrow (BM)-derived endothelial progenitor cells (EPCs) were significantly recruited and migrated to the livers of thioacetamide (TAA)-induced fibrotic rats, implying the positive correlation between engraftment and fibrosis. However, EPCs did not upregulate ALK1 and TGF-β signaling, but mediated by noninhibited ALK5 which up-regulates thrombospondin. Up-regulation of thrombospondin effectively down-regulates NO production, and inhibits the maturation of LSEC progenitors [19]. In addition, an in vitro study showed that BM-derived EPCs isolated from patients with cirrhosis can stimulate profuse angiogenesis when co-cultured with rat resident LSECs [20], which presents a new perspective on the role of LSECs dedifferentiation in liver fibrosis.
Hedgehog (Hh)
The hedgehog signaling pathway is a complex system composed of ligands, receptors and transcription factors. The binding of Hh ligands to their transmembrane receptors (Patched, Ptc), activates Smoothened (Smo) to act as a co-receptor-like molecule in initiating the intracellular Hh-induced signaling. Hh signaling is important in the regulation of vasculogenesis. After chronic liver injury, its activation regulates the functions of several types of liver cells, including LSECs, cholangiocytes, HCs, HSCs, myofibroblastic hepatic stellate cells (MF-HSCs), and some immune cells [21]. Specifically, Pereira et al. [22] reported that, compared to healthy people, macrophages in schistosomiasis patients over-express Hh ligands. In response, Hh-responsive LSECs up-regulate the expression levels of Hh target genes, and the intensity of this response is positively correlated with fibrosis stage. In addition, in a BDL mice model, cholestasis enhanced the secretion of Hh-rich microparticles by cholangiocytes and myofibroblastic hepatic stellate cells. As a result, Ptc is up-regulated in LSECs, while the expression levels of CD31, and iNOS also increase [23]. Xie et al. [24] cultured freshly isolated primary LSECs to further evaluate the roles of Hh in the cellular phenotypic changes. They found that Hh induces LSEC capillarization and formation of vascular tubes in vitro. Hh-related gene expression in culture-induced capillaried LSECs was found to have increased. Moreover, Hh agonists further promoted LSEC capillarization, a process that was further shown to be suppressed in Smo-knocked out LSECs. Consistently, LSEC capillarization was found to be suppressed in chronic and acute liver injury mice models treated with a Hh signaling antagonist [24]. In general, activation of the Hh pathway in LSECs promotes the cirrhosis-related sinusoidal remodeling process.
Notch
Notch signaling is an evolutionarily highly conserved pathway in vertebrates. It encompasses four transmembrane receptors (Notch1-4), and five canonical Delta-like ligands (DLL), 1 3, and 4 and Jagged (JAG) 1 and 2. The notch pathway is involved in multiple cellular physiological processes including angiogenesis and vascular remodeling [25]. Duan et al. [26] reported that endothelial Notch activation induced LSEC capillarization and deranged liver homeostasis in mice. This process enhances eNOS-sGC pathway dependent CCL4-induced liver fibrosis. Consistently, activated Notch pathways in LSECs have also been reported in liver cirrhosis patients [26]. Regarding Notch ligands, DLL4 is primarily expressed in endothelial cells. Shen et al. [27] found that DLL4 was highly expressed in LSECs and kupffer cells (KCs) of HBV-associated active cirrhosis patients, when compared to those without cirrhosis. They reported that intraperitoneal injection of recombinant DLL4 in mice suppressed liver inflammation by inhibiting CCL2, thereby alleviating CCL4-induced liver injury. However, their study did not establish the role of LSEC-specific DLL4 during fibrosis, and the positive correlation between DLL4 and liver injury may be attributed to other notch-responsive cells such as KCs. Chen et al. [28] found that the expression levels of DLL4 in LSECs were up-regulated in both cirrhotic patients and in CCL4-induced fibrotic mice. Moreover, DLL4 and basement membrane proteins were found to be elevated in culture-induced dedifferentiated murine primary LSECs. Generation of basement membrane proteins was inhibited after DLL4 knockout. The same effect on basement membrane proteins was also shown to be caused by the overexpression, or deletion of DLL4 in an immortalized LSEC cell line (TMNK-1). This DLL4-induced upregulation of basement membrane proteins may be associated with ECM accumulation, which is correlated with liver fibrosis.
Mechanotransduction
Mechanical forces are determined by physical properties of the extracellular matrix, as well as by the pressure of the circulating blood flow. This force is detected and transmitted by mechanosensors on the plasma membrane, thereby affecting cellular functions. This process is called mechanotransduction.
Shear stress
Shear stress is a key factor in the regulation of the endothelial phenotypes, including those of LSECs. Mechanosensors, such as integrins, G protein receptors, tyrosine kinase receptors, and ion channels, are localized on endothelial cells, and respond to changes in wall flow shear stress [29]. KLF2 is an important transcription factor that is involved in the maintenance of endothelial cell quiescence. In addition, it exhibits vasoprotective effects under shear stress. Under healthy conditions, laminar flow-induced shear stress activates the transcription of myocyte enhancing factor 5 (MEF5), which subsequently induces a highly flow-induced factor, extracellular signal regulated kinase 5 (ERK5) [30]. Myocyte enhancing factor 2 (MEF2) is a characteristic target of EKR5 that can bind the KLF2 promoter [31]. Therefore, shear stress activates KLF2 expression through mechanosensory complexes on endothelial membranes. KLF2 expression was shown to be upregulated in the liver of cirrhotic rat models, especially in hepatic endothelial cells [32]. Elevated expression levels of KLF2 induces the expression of eNOS to mediate vasodilatation, which compensates for the dysregulated microcirculation and portal pressure. Marrone et al. [33] reported that overexpression of KLF2 in LSECs reduces endothelial dysfunction and inhibits the activation of HSCs co-cultured with LSECs. KLF2 inhibits the proliferation, migration, and angiogenesis of LSECs through the protein kinases 1/2 (ERK1/2) signaling pathway [34]. Endogenous KLF2 upregulation is not sufficient to maintain the normal phenotype and function of LSECs under pathological sinusoidal blood flow, neither is it sufficient to resist the progression of liver fibrosis. Animal model studies have shown that high perfusion pressure in the portal vein fuses with and enlarges the LSEC fenestrae leading to an abnormal transport of chylomicron to the liver parenchyma [35]. In addition, Hilscher et al. [36] mechanically stretched primary mice LSECs to mimic the pulsatile forces induced by congestion. This treatment activates integrins and piezo channels on LSEC membranes, thereby promoting the expression and secretion of CXCL1 from LSECs, culminating in the formation of neutrophil extracellular traps and microthrombi pivotal. This process is involved in pressure adjustment in the sinusoidal lumen and portal hypertension.
Extracellular matrix (ECM)
Studies have shown that HSCs are not the only ones involved in the formation of extracellular matrix (ECM) as LSECs also play an important role in the excessive deposition of ECM in fibrotic liver by secreting collagen type I, type IV, and fibronectin, which are important units of the ECM [37, 38]. In the early stages of liver fibrosis, LSECs-dependent angiogenesis promotes the condensation of collagen fibrils. Remodeled collagen-mediated mechanotransduction activates HSCs, thereby increasing the stiffness of ECM [4]. In effect, stiff ECM plays an important role in regulating the LSEC phenotype. Juin and colleagues [39] found that a harder ECM matrix increases the number of LSECs-formed podosomes. Podosomes are actin-rich structures that are involved in the migration, proteolysis, and cytoskeletal remodeling of LSECs. Consistently, in vitro studies revealed that the harder the ECM, the earlier the cells lose their fenestrae [40]. In summary, LSECs fenestrae is directly involved in ECM accumulation, which is maintained by complex matrix components.
Metabolic regulation of LSECs
Autophagy
Liver fibrosis refers to the scarring of liver tissues, a process that is mediated by integral participation of liver cells, including HSCs, KCs and LSECs, and HCs. Autophagy is central to fibrotic progression, however, it performs different, or even opposite functions in specific cell types. Autophagy in HCs and macrophages promotes anti-fibrosis [41]. However, it enhances the activation of HSCs, which exacerbates fibrotic liver disease [42].
In the early stages of liver injury, autophagy is rapidly up-regulated to maintain the LSECs phenotype and hepatic homeostasis [43]. However, the protective functions of autophagy decrease as the damage continues [43]. Accordingly, dedifferentiation of LSECs and development of liver fibrosis continues [43]. Autophagy inhibits fibrosis by increasing NO bioavailability and through the excretion of reactive oxygen species (ROS) in LSECs [43]. Moreover, it up-regulates transcription factor KLF2, which has a protective role in endothelial cells [44]. A recent study [41] reported that endothelial autophagic defects enhances susceptibility to liver damage, liver cell apoptosis and perisinusoidal fibrosis in mice fed on a high-fat diet and treated with CCL4. In an immortalized LSEC line, deficiency in autophagy enhanced inflammation, EndMT, and apoptosis. In general, up-regulation of autophagy confers protective effects on LSECs, in both early acute injury and advanced fibrosis.
Notably, due to different cell types and disease stages, autophagy has a context-dependent impact on liver fibrosis, which complicates autophagy-targeted therapy for fibrosis. Selective enhancement of autophagy in LSECs in the early stages of liver disease may prevent fibrotic progression.
Lipids
Non-alcoholic fatty liver disease (NAFLD) can develop from a simple degeneration of fatty acids in the liver to cirrhosis, one of the main causes of chronic liver disease. Sinusoidal endothelial cell dysfunction is an early event in the development of NASH [45]. In vitro, excessive lipid exposure led to a decrease in the diameter and number of fenestrae. In addition, NO bioavailability in LSECs was suppressed after exposure to ox-LDL and free fatty acids (FFA) [46]. Using a nutrition-induced NAFLD model, Miyao et al. [45] revealed that sinusoidal capillarization occurs in the early stages of NAFLD, and it can be partially reversed by discontinuing the high-fat diet. Consistently, Cogger et al. [47] used mice models to demonstrate that fenestrae frequency and overall porosity are negatively correlated with dietary fat intake and FFA levels.
Adipocytokines
Adipocytokines are involved in the pathogenesis of obesity and NAFLD-induced liver fibrosis. Among them, adiponectin has a strong anti-fibrosis effect, whereas leptin exhibits a pro-fibrosis effect. Leptin promotes fibrosis by up-regulating TGF levels in LSECs and KCs, as well as by promoting angiogenesis [46]. Using a toxin-induced NASH rodent model, Pourhoseini et al. [48] found that leptin impairs functional activities of nitric oxide synthase 3 (NOS3) through miR-21. Since NOS3 activation is important for increased NO bioavailability, miR-21 induces LSEC capillarization. However, the functionality of NOS3 can be restored by deleting leptin or its receptors.
Ethanol
In vitro and in vivo, chronic stimulation of LSECs using ethanol was shown to lead to early defenestration and formation of a basement membrane. Over time it may prevent fluid and macromolecular exchange as well as immune cell migration, which eventually promotes tissue fibrosis [49, 50]. Due to the ability of LSECs to clear hyaluronic acid (HA), circulating HA levels are a hallmark of LSECs function. Chronic alcoholic exposure upregulates circulating HA levels. Deaciuc et al. [51] reported that elevated circulating HA levels are due to apoptosis, which impairs LSECs functions, and that this process is preceded by alcoholic hepatocyte injury. In LSEC-specific STAT3-knockout mice, it was confirmed that during chronic alcohol consumption, STAT3 in endothelial cells inhibit LSEC apoptosis and hepatic inflammation, thereby reducing alcoholic liver injury [52]. Consistently, alcohol exacerbates acetaminophen-induced LSECs injury, which occurs prior to liver parenchymal injury [53]. In general, alcohol-induced LSEC injury is the initial step in chronic alcoholic liver damage, and restoring LSEC functions may alleviate alcoholic cirrhosis.
Age
The phenotype, and functions of LSECs change with age. LSECs exhibit fewer fenestrae, more basal lamina deposition, and less endocytosis during aging, a process referred to as pseudocapillarization. In this process, the expression of eNOS and sGC, as well as NO bioavailability in aging LSECs is suppressed, resulting in decreased liver blood flow [54]. Aging LSECs are in an inflammatory status, and elevated intercellular inflammatory molecule 1 expression mediates a substantial increase in leukocyte adhesion, further reducing the sinusoidal blood flow [55]. Age-associated plastic roles and peculiar characteristics of LSECs have a special significance in elderly liver injury patients.
Epigenetic changes in LSECs during liver fibrosis
lncRNAs and miRNAs
Changes of several lncRNAs or miRNAs are involved in LSEC injury during fibrosis as shown in Table 1. miR-199 was found to be involved in alcohol-induced improvement of endothelin-1 (ET-1) and HIF-α in rat LSECs and in human endothelial cells. However, miR-155 was shown to induce the expression of ET-1 and HIF-α in rat LSECs. Elevated ET-1 and HIF-α levels are associated with microcirculatory failure and portal hypertension in liver fibrosis [56]. In addition, in patients and mice with liver cirrhosis, the expression levels of miR-322 in mice and its human rodent homolog miR-424 were found to be elevated. miR-322/424 promotes cirrhosis-related angiogenesis by targeting the Cullin-2/HIF-1α pathway [57]. Transcriptional changes in some non-coding RNAs (lncRNA) regulate the LSEC phenotype. H19, a lncRNA located on human chromosome 11p15.5, regulates microvascular formation [58] as well as tumors development [59]. Zhu et al. [60] reported that H19 negatively regulates miRNA-148b-3p, while NADPH oxidases (NOX4) were negatively regulated by miR-148b-3p. In patients with cirrhosis or in human LSECs under hypoxia conditions, H19 exacerbates hypoxic stress of LSECs through indirect positive regulation of endothelial NOX4 and negative regulation of eNOS/NO signals. Moreover, lncRNA taurine-upregulated gene 1 (TUG1) is another lncRNA associated with LSEC-related liver fibrosis. TUG1, a 7.6-kb gene located on chromosome 22q12.2, is involved in several biological processes (e.g., cell proliferation, apoptosis, EMT) and disorders (e.g., liver fibrosis, tumors including HCC) [61]. Zhang et al. [62] reported that TUG1 is up-regulated, while miR-142-3p is down-regulated in patients with cirrhosis. They treated LSECs with LPS or starvation to partially simulate a fibrotic microenvironment. After LPS or starvation exposure upregulates TUG1 and competitively inhibits miR-142-3p. As a result, the expression of autophagy-related gene ATG5, a miR-142-3p-targeted gene, is upregulated, which induces autophagy-mediated EndMT in LSECs.
DNA methylation
Brahma-related gene 1 (BRG1) is a chromatin remodeling protein that participates in hepatic fibrosis by regulating transcription in endothelial cells. Shao et al. [63] reported that a lack of endothelial-specific BRG1 alleviates TAA-induced liver fibrosis in mice. Their study confirmed that deleting endothelial BRG1 is accompanied by the up-regulation of eNOS activity and NO bioavailability, which together resolves fibrosis. Further analysis showed that Sp1 recruits BRG1 to the CAV-1 promoter, in which BRG1 interacts and regulates the H3K4 trimethyltransferase MLL1. This cascade of events activates CAV-1 transcription, and further inhibits eNOS activity. Therefore, BRG1-induced DNA methylation regulates endothelial functions and plays an anti-fibrosis role.
Histone modification
Lysine residue acetylation, catalyzed by histone acetyltransferase (HAT), is one of the most common forms of histone modification. Conversly, histone deacetylase (HDAC) catalyzes deacetylation. Silent information regulator 1 (SIRT1) is an NAD+-depended HDAC. Down-regulation of SIRT1 expression causes the contraction and capillarization of LSECs, thereby increasing oxidative stress and ultimately activating of HSCs [64].
Receptor-mediated regulation of LSECs
Nuclear receptors
Farnesoid X receptor (FXR), known as bile acid receptor, is an essential factor in bile and lipid metabolism. FXR deficiency aggravates liver inflammation and fibrosis. Obeticholic acid (OCA), a steroidal FXR agonist, reversed the impaired vasodilation in TAA and BDL-treated rats by up-regulating eNOS expression in endothelial cells [65]. Verbeke et al. reported that OCA inhibits the expression of MCP-1 in LPS or TGF-α treated LSECs. MCP-1 suppresses chemotactic and fibrogenic potential of HSCs [66]. In addition, the non-steroidal FXR agonist (PX20606), was shown to elevate eNOS expression and NO production in human LSECs and rat tissues, thereby ameliorating portal hypertension by reducing vascular remodeling, sinusoidal dysfunction, and liver fibrosis in CCL4-treated rats [67].
Liver X receptors (LXRs), including LXRα and LXRβ, are members of the ligand-excited transcription factor family. LXRs agonists were shown resolve CCL4-induced liver fibrosis in mice. However, LXRs gene knockdown impairs this protective effect. The roles of LXRs in inhibiting the Hh signaling pathway and the subsequent capillarization of LSECs have been reported [68]. In hepatitis C virus infected human liver tissues, Baiocchini et al. [69] reported that LSECs capillarization was only observed in a few patients with stage S2 fibrosis. Lack of capillarization may be associated with indirect LXRs-up-regulation by HCV cores and NS5A proteins.
Peroxisome proliferation-activated receptors (PPARs) are nuclear receptors of the steroid hormone receptors superfamily. PPARα, PPARβ and PPARγ are all expressed in endothelial cells. During cirrhosis, PPARα is significantly up-regulated in LSECs, and PPARs agonists can reverses liver fibrosis by preventing vasoconstriction, thereby, inhibiting angiogenesis as well as reducing undesirable inflammatory responses [70, 71].
These nuclear receptors negatively modulate LSECs-related angiogenesis and fibrosis progression.
G protein-coupled receptors
Sphingosine-1-phosphate receptor-1 (S1P1) is widely expressed in endothelial cells. Its ligand is commonly bound to a high-density lipoprotein (HDL) to form a HDL-S1P complex, which regulates lipid transport. The binding of S1P1 with its ligand was shown to maintains the normal phenotype and functions of LSECs after partial hepatectomy in mice, thereby inhibiting fibrosis [72].
TGR5 is a G protein-coupled cell surface receptor that responds to activation by bile acids and multiple steroid hormones in LSECs, KCs and cholangiocytes. Activation of TGR5 in LSECs upregulates eNOS expression of and NO production [73], and further reduced the expression and secretion of ET-1 in primary murine LSECs cultured in vitro [74].
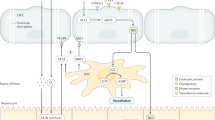
GPR182, a novel marker for LSEC differentiation, is potentially an LSEC-specific orphan G-protein coupled receptor. Expression of GPR182 in LSECs during liver fibrosis and cirrhosis is suppressed [75]. In addition, GPR14, a urotensin II receptor, is mainly found in LSECs and KCs. It is highly expressed in patients with portal hypertension and cirrhosis. However, its role in cirrhosis is not well understood, even though it is thought to be associated with pro-vasoconstriction [76]. Several nuclear receptors and G protein-coupled receptors involved in fibrosis-related LSEC injury are shown in Table 2. In addition, above signaling molecules and pathways is depicted in Fig. 2.

Signalling molecules and pathways involved in LSECs capillarization. Key fibrogenic cytokines and adipocytokines contribute to capillarization of LSECs, include transforming growth factor‑β (TGFβ), vascular endothelial growth factor (VEGF), and leptin. Hedgehog (Hh) ligand and its receptor (Patched, Ptc) and/or smoothened (Smo) promote LSECs capillarization. The Notch signaling pathway also induces capillarization of LSECs. G protein-coupled receptors expressed by LSECs can either negatively or positively affect LSECs capillarization. Autophagy maintains phenotype of LSECs. Nuclear receptors negatively modulate LSECs capillarization. Epigenetic signals including microRNAs/lncRNAs, DNA methylation and histone modification either induce or inhibit capillarization of LSECs. Stimulation of mechanotransduction, including upregulation of shear stress and stiffness of extracellular matrix (ECM), promotes LSECs capillarization. DLL4 delta-like canonical notch ligand 4, S1P1 sphingosine-1-phosphate receptor-1, TGR5, takeda G protein-coupled receptor 5, GPR182 G protein-coupled receptor 182, GPR14 G protein-coupled receptor 14, KLF2 kruppel-like factor 2
Interactions between LSECs and other hepatic cells during liver injury
LSECs-HSCs
The extracellular matrix in fibrotic liver is mainly produced by activated HSCs, and is involved in the development of liver fibrosis. Differentiated LSECs maintain the nonproliferative quiescent phenotype of HSCs. Loss of the normal LSECs phenotype is permissive for the activation of HSCs in vivo [5]. Several cytokines involved in LSECs-HSCs crosstalk have been mentioned above. After capillarization, LSECs can secrete exosomes containing sphingosine kinase-1 (SK1) and sphingosine 1-phosphate, which activate HSCs [77]. Stromal cell-derived factor 1 (SDF-1), also known as CXCL12, is an LSEC and HSC derivative. HSCs express its downstream receptor, CXCR4. In chronic liver injury, SDF-1 binds to CXCR4, thereby activating HSCs, while also recruiting mesenchymal cells from the bone marrow that in effect, promotes liver fibrosis progression [78, 79]. CXCR7, expressed on LSECs, is another chemokine receptor for SDF-1. During chronic liver injury, SDF-1-CXCR7 interactions promote liver cell regeneration [80]. In addition, LSECs, HSCs and KCs can all secrete platelet derived growth factor (PDGF) [81,82,83], which, upon binding to its homologous receptor (PDGFR-β) on HSCs, induces the activation, proliferation and migration of HSCs and subsequent sinusoidal remodeling.
On the other hand, activated HSCs act on LSECs through VEGF and large amounts of ECM. This process leads to the defenestration of LSECs, thereby promoting fibrosis-related angiogenesis. Activation of HSCs can also induce the production of thrombospondin-1 (TSP-1), which enhances LSEC capillarization by blocking the NO-dependent pathway. In addition, multiple in vitro studies have shown that TSP-1 promotes dedifferentiation of LSECs through the Rho/Rho-kinase K pathway [84]. The cross-talk between LSECs and HSCs is depicted in Supplementary Fig. 1.
LSECs-hepatocytes (HCs)
LSECs secrete Wnt2a, Wnt9b and a small amounts of hepatocyte growth factor (HGF), which together, act as key hepatocyte mitogens that induce the self-regeneration of hepatocytes in the early stages of CCL4/BDL-induced liver fibrosis [85].
In contrast, HCs secrete VEGF to regulate the LSECs phenotype [5]. In addition, HC-derived membrane-shed microparticles contents are high in cirrhosis patients, playing a regulated role in endothelial cells [86]. One study [35] showed that under adipotoxicity, HCs release VNN1-rich exosomes, which mediates the transformation into angiogenic phenotypes after uptaken by endothelial cells.
LSECs-liver macrophages
Through the cross-talk with liver macrophages, LSECs are involved in the regulation of liver immunity, among liver diseases. Liver macrophages include liver resident macrophages (KCs) and monocyte-derived macrophages (MoMFs), which play a dual role in the process of liver fibrosis. LSECs interact with both KCs and MoMFs.
LSECs-KCs
KCs adhere to the sinusoidal endothelial layer, where they closely interact with LSECs. In an organotypic three-dimensional (3D) hepatic culture, KCs maintained the dedifferentiated phenotype of LSECs by down-regulating the FAK pathway [87]. However, in severe liver injuries, LSECs secrete proinflammatory mediators such as TNF-α、IL-6、IL-1 and CCL2, which activates KCs [88]. Capillarization of LSECs in the early stages of NASH is necessary for KCs activation [45]. In return, activated KCs impair fenestrae of LSECs while increase CD31 expression [89].
LSECs-MoMFs
During liver injury, LSECs promote the migration and adhesion of circulating monocytes through surface ICAM-1, VCAM-1, and vascular protein-1 (VAP-1), thereby mediating inflammation and fibrosis in the liver [88]. However, little is known about the pathophysiological roles of MoMFs toward LSECs, therefore, further studies are needed to discern the complex immune microenvironment in the liver and the interaction mechanisms between LSECs and liver macrophages.
Anti-fibrosis chemicals for LSECs
LSEC injury occurs at the onset of liver fibrosis and dysfunctions of LSECs lead to hepatic sinus microcirculation disorders, which affect the role of anti-fibrosis drugs in the liver. Drugs targeting LSECs have been shown to effectively ameliorate chronic liver disease or fibrosis in rodent models or in vitro. (as shown in Supplementary Table 1).
Increasing NO availability
Statins promote eNOS phosphorylation, thereby increasing NO bioavailability. This effect is achieved by inhibiting the RhoA/Rho-kinase signaling pathway, which down-regulates the expression and activity of eNOS. Statins can also increase Akt/protein kinase B activity, which phosphorylates and activates eNOS [90]. In addition, statins were shown to directly protect LSECs and inactivate HSCs in cirrhotic rats by up-regulating autophagy and the KLF2-related pathway [33]. Using simvastatin and atorvastatin to treat early-stage NASH rat without fibrosis, Bravo et al. [91] showed that statins can maintain the differentiated phenotype and significantly reverse the dysfunctional LSEC phenotype, thereby inducing activated HSCs to quiescent cells.
5-aminoimidazole-4-carboxyamide ribonucleoside (AICAR) is an agonist of the adenosine 5′-monophosphate-activated protein kinase (AMPK). By activating the AMPK/eNOS pathway in LSECs, AICAR enhances NO synthesis in the liver without changing systemic hemodynamics, thereby ameliorating cell functions as well as reducing portal pressure. AICAR has also been shown to directly alleviate HSCs contraction in vitro [92]. Consistently, metformin improves the age-related pseudocapillarization of LSECs by activating the AMPK/eNOS pathway [93].
Angiogenesis
Hepatic angiogenesis is highly associated with liver fibrosis progression. Fibrosis-induced hypoxia and immature LSEC angiogenesis interact as both cause and effect. Some drugs target key angiogenic-related factors to restore the normal functions of injured LSECs. Oral administrations of vatalanib, a small molecule tyrosine kinase inhibitor, has been shown to be effective against all VEGF proteins. In CCL4-induced mouse liver fibrosis, vatalanib reduced LSECs capillarization and resolved fibrosis by regulating VEGF and TGF-β [94]. In addition, temsirolimus and everolimus are rapamycin analogues that can maintain the normal phenotype and functions of LSECs by inhibiting the PI3K-AKT-mTOR signaling, which also prevents fibrosis-related angiogenesis [95]. Vatalanib, temsirolimus and everolimus also inhibit the activation and proliferation of HSCs [95]. Therefore, besides LESCs, these drugs also alleviate liver fibrosis through HSCs.
Adrenergic receptor blocker
Carvedilol, an adrenergic receptor blocker, inhibits sympathetic activation by antagonizing β1-, β2- and α1-adrenoreceptors. Ling et al. [96] reported that carvedilol alleviates PH in rats by inhibiting neovascularization and reconstruction of the hepatic sinusoid in the liver. Separate in vitro studies in cultured human umbilical vein endothelial cells (HUVECs) showed that it can also impair fibronectin synthesis. Consistently, treatment of CCL4-induced liver fibrosis with carvedilol showed that this drug inhibits LSECs capillarization [97]. In addition, it induces apoptosis but inhibits the activation of HSCs, which is another anti-fibrosis mechanism.
Miscellaneous
As a dual PPARα-PPARγ agonist, Aleglitazar (Ale) alleviates liver fibrosis by inhibiting inflammatory responses and neovascularization [70]. Similarly, the Pan-peroxisome proliferator-activated receptors (pan-PPAR) agonist (lanifibranor) protects LSECs in cirrhotic rats [71]. Moreover, the two chemicals potentially reduce portal pressure, while avoiding the side effects associated with one single PPAR agonist. Actin, a cytoskeleton component, has been implicated in the regulation of size and formation of fenestrae in LSECs. Actin-targeted chemicals such as cytochalasin D (cyto-D) and Latrunculin A, bind actin filaments and inhibit actin monomer polymerization, thereby promoting the formation of fenestrae in differentiated LSECs [98]. These chemicals regulate other hepatic cells, especially HSCs, through similar effects as in LSECs [70, 71, 99]
In general, clinical trials of drugs targeting LSECs, specifically for the treatment of chronic liver diseases or liver fibrosis, are limited. Since most drugs act on several types of hepatic cells, the therapeutic effects of most drugs may be multi-dimensional. In-depth investigations of LSEC targeting drugs and their interactions with other hepatic cells are needed.
Conclusions
Elucidation of the roles of LSECs have informed the diagnosis and treatment of chronic liver diseases or fibrosis. Cellular signal regulation and cellular interactions have been documented. Besides, a variety of anti-fibrosis chemicals targeted at LSECs have been developed, which lays a concrete foundation for resolving liver fibrosis.
References
Poisson J, Lemoinne S, Boulanger C et al (2017) Liver sinusoidal endothelial cells: physiology and role in liver diseases. J Hepatol 66(1):212–227. https://doi.org/10.1016/j.jhep.2016.07.009
Steffan AM, Gendrault JL, McCuskey RS, McCuskey PA, Kirn A (1986) Phagocytosis, an unrecognized property of murine endothelial liver cells. Hepatology 6(5):830–836
Wilkinson AL, Qurashi M, Shetty S (2020) The role of sinusoidal endothelial cells in the axis of inflammation and cancer within the liver. Front Physiol 11:990. https://doi.org/10.3389/fphys.2020.00990
Liu L, You Z, Yu H et al (2017) Mechanotransduction-modulated fibrotic microniches reveal the contribution of angiogenesis in liver fibrosis. Nat Mater 16(12):1252–1261. https://doi.org/10.1038/nmat5024
Xie G, Wang X, Wang L et al. Role of differentiation of liver sinusoidal endothelial cells in progression and regression of hepatic fibrosis in rats. Gastroenterology. 2012;142(4). doi:https://doi.org/10.1053/j.gastro.2011.12.017.
Leo CH, Jelinic M, Ng HH et al (2017) Vascular actions of relaxin: nitric oxide and beyond. Br J Pharmacol 174(10):1002–1014. https://doi.org/10.1111/bph.13614
Yan Z, Qu K, Zhang J et al (2015) CD147 promotes liver fibrosis progression via VEGF-A/VEGFR2 signalling-mediated cross-talk between hepatocytes and sinusoidal endothelial cells. Clin Sci (Lond) 129(8):699–710. https://doi.org/10.1042/CS20140823
Kantari-Mimoun C, Castells M, Klose R et al (2015) Resolution of liver fibrosis requires myeloid cell-driven sinusoidal angiogenesis. Hepatology (Baltimore, MD) 61(6):2042–2055. https://doi.org/10.1002/hep.27635
Shi M, Zhu J, Wang R et al (2011) Latent TGF-β structure and activation. Nature 474(7351):343–349. https://doi.org/10.1038/nature10152
Henderson NC, Arnold TD, Katamura Y et al (2013) Targeting of αv integrin identifies a core molecular pathway that regulates fibrosis in several organs. Nat Med 19(12):1617–1624. https://doi.org/10.1038/nm.3282
Sakata K, Eda S, Lee E-S, Hara M, Imoto M, Kojima S (2014) Neovessel formation promotes liver fibrosis via providing latent transforming growth factor-β. Biochem Biophys Res Commun 443(3):950–956. https://doi.org/10.1016/j.bbrc.2013.12.074
Caja L, Dituri F, Mancarella S et al (2018) TGF-β and the tissue microenvironment: relevance in fibrosis and cancer. Int J Mol Sci. https://doi.org/10.3390/ijms19051294
Pardali E, Sanchez-Duffhues G, Gomez-Puerto MC, Ten Dijke P (2017) TGF-β-induced endothelial-mesenchymal transition in fibrotic diseases. Int J Mol Sci. https://doi.org/10.3390/ijms18102157
Ribera J, Pauta M, Melgar-Lesmes P et al (2017) A small population of liver endothelial cells undergoes endothelial-to-mesenchymal transition in response to chronic liver injury. Am J Physiol Gastrointest Liver Physiol 313(5):G492–G504. https://doi.org/10.1152/ajpgi.00428.2016
Tillet E, Ouarné M, Desroches-Castan A et al (2018) A heterodimer formed by bone morphogenetic protein 9 (BMP9) and BMP10 provides most BMP biological activity in plasma. J Biol Chem 293(28):10963–10974. https://doi.org/10.1074/jbc.RA118.002968
Breitkopf-Heinlein K, Meyer C, König C et al (2017) BMP-9 interferes with liver regeneration and promotes liver fibrosis. Gut 66(5):939–954. https://doi.org/10.1136/gutjnl-2016-313314
Desroches-Castan A, Tillet E, Ricard N et al (2019) Bone morphogenetic protein 9 is a paracrine factor controlling liver sinusoidal endothelial cell fenestration and protecting against hepatic fibrosis. Hepatology (Baltimore, MD) 70(4):1392–1408. https://doi.org/10.1002/hep.30655
Gaitantzi H, Karch J, Germann L et al (2020) BMP-9 modulates the hepatic responses to LPS. Cells. https://doi.org/10.3390/cells9030617
Maretti-Mira AC, Wang X, Wang L, DeLeve LD (2019) Incomplete differentiation of engrafted bone marrow endothelial progenitor cells initiates hepatic fibrosis in the rat. Hepatology 69(3):1259–1272. https://doi.org/10.1002/hep.30227
Kaur S, Tripathi D, Dongre K et al (2012) Increased number and function of endothelial progenitor cells stimulate angiogenesis by resident liver sinusoidal endothelial cells (SECs) in cirrhosis through paracrine factors. J Hepatol 57(6):1193–1198. https://doi.org/10.1016/j.jhep.2012.07.016
Choi SS, Omenetti A, Syn W-K, Diehl AM (2011) The role of Hedgehog signaling in fibrogenic liver repair. Int J Biochem Cell Biol 43(2):238–244. https://doi.org/10.1016/j.biocel.2010.10.015
Pereira TA, Xie G, Choi SS et al (2013) Macrophage-derived Hedgehog ligands promotes fibrogenic and angiogenic responses in human schistosomiasis mansoni. Liver Int 33(1):149–161. https://doi.org/10.1111/liv.12016
Witek RP, Yang L, Liu R et al (2009) Liver cell-derived microparticles activate hedgehog signaling and alter gene expression in hepatic endothelial cells. Gastroenterology. https://doi.org/10.1053/j.gastro.2008.09.066
Xie G, Choi SS, Syn W-K et al (2013) Hedgehog signalling regulates liver sinusoidal endothelial cell capillarisation. Gut 62(2):299–309. https://doi.org/10.1136/gutjnl-2011-301494
Dill MT, Rothweiler S, Djonov V et al (2012) Disruption of Notch1 induces vascular remodeling, intussusceptive angiogenesis, and angiosarcomas in livers of mice. Gastroenterology. https://doi.org/10.1053/j.gastro.2011.12.052
Duan J-L, Ruan B, Yan X-C et al (2018) Endothelial Notch activation reshapes the angiocrine of sinusoidal endothelia to aggravate liver fibrosis and blunt regeneration in mice. Hepatology 68(2):677–690. https://doi.org/10.1002/hep.29834
Shen Z, Liu Y, Dewidar B et al (2016) Delta-like ligand 4 modulates liver damage by down-regulating chemokine expression. Am J Pathol 186(7):1874–1889. https://doi.org/10.1016/j.ajpath.2016.03.010
Chen L, Gu T, Li B et al (2019) Delta-like ligand 4/DLL4 regulates the capillarization of liver sinusoidal endothelial cell and liver fibrogenesis. Biochim Biophys Acta Mol Cell Res 1866(10):1663–1675. https://doi.org/10.1016/j.bbamcr.2019.06.011
Resnick N, Yahav H, Shay-Salit A et al (2003) Fluid shear stress and the vascular endothelium: for better and for worse. Prog Biophys Mol Biol 81(3):177–199
Yoshizumi M, Abe J-I, Tsuchiya K, Berk BC, Tamaki T (2003) Stress and vascular responses: atheroprotective effect of laminar fluid shear stress in endothelial cells: possible role of mitogen-activated protein kinases. J Pharmacol Sci 91(3):172–176
Kumar A, Lin Z, SenBanerjee S, Jain MK (2005) Tumor necrosis factor alpha-mediated reduction of KLF2 is due to inhibition of MEF2 by NF-kappaB and histone deacetylases. Mol Cell Biol 25(14):5893–5903
Gracia-Sancho J, Russo L, García-Calderó H, García-Pagán JC, García-Cardeña G, Bosch J (2011) Endothelial expression of transcription factor Kruppel-like factor 2 and its vasoprotective target genes in the normal and cirrhotic rat liver. Gut 60(4):517–524. https://doi.org/10.1136/gut.2010.220913
Marrone G, Maeso-Díaz R, García-Cardena G et al (2015) KLF2 exerts antifibrotic and vasoprotective effects in cirrhotic rat livers: behind the molecular mechanisms of statins. Gut 64(9):1434–1443. https://doi.org/10.1136/gutjnl-2014-308338
Zeng X-Q, Li N, Pan D-Y et al (2015) Kruppel-like factor 2 inhibit the angiogenesis of cultured human liver sinusoidal endothelial cells through the ERK1/2 signaling pathway. Biochem Biophys Res Commun 464(4):1241–1247. https://doi.org/10.1016/j.bbrc.2015.07.113
Natarajan V, Harris EN, Kidambi S (2017) SECs (sinusoidal endothelial cells), liver microenvironment, and fibrosis. Biomed Res Int 2017:4097205. https://doi.org/10.1155/2017/4097205
Hilscher MB, Sehrawat T, Arab JP et al (2019) Mechanical stretch increases expression of CXCL1 in liver sinusoidal endothelial cells to recruit neutrophils, generate sinusoidal microthombi, and promote portal hypertension. Gastroenterology. https://doi.org/10.1053/j.gastro.2019.03.013
Maher JJ, McGuire RF (1990) Extracellular matrix gene expression increases preferentially in rat lipocytes and sinusoidal endothelial cells during hepatic fibrosis in vivo. J Clin Invest 86(5):1641–1648
Neubauer K, Krüger M, Quondamatteo F, Knittel T, Saile B, Ramadori G (1999) Transforming growth factor-beta1 stimulates the synthesis of basement membrane proteins laminin, collagen type IV and entactin in rat liver sinusoidal endothelial cells. J Hepatol 31(4):692–702
Juin A, Planus E, Guillemot F et al (2013) Extracellular matrix rigidity controls podosome induction in microvascular endothelial cells. Biol Cell 105(1):46–57. https://doi.org/10.1111/boc.201200037
Marrone G, Shah VH, Gracia-Sancho J (2016) Sinusoidal communication in liver fibrosis and regeneration. J Hepatol 65(3):608–617. https://doi.org/10.1016/j.jhep.2016.04.018
Hammoutene A, Biquard L, Lasselin J et al (2020) A defect in endothelial autophagy occurs in patients with non-alcoholic steatohepatitis and promotes inflammation and fibrosis. J Hepatol 72(3):528–538. https://doi.org/10.1016/j.jhep.2019.10.028
Hernández-Gea V, Ghiassi-Nejad Z, Rozenfeld R et al (2012) Autophagy releases lipid that promotes fibrogenesis by activated hepatic stellate cells in mice and in human tissues. Gastroenterology 142(4):938–946. https://doi.org/10.1053/j.gastro.2011.12.044
Ruart M, Chavarria L, Campreciós G et al (2019) Impaired endothelial autophagy promotes liver fibrosis by aggravating the oxidative stress response during acute liver injury. J Hepatol 70(3):458–469. https://doi.org/10.1016/j.jhep.2018.10.015
Guixé-Muntet S, de Mesquita FC, Vila S et al (2017) Cross-talk between autophagy and KLF2 determines endothelial cell phenotype and microvascular function in acute liver injury. J Hepatol 66(1):86–94. https://doi.org/10.1016/j.jhep.2016.07.051
Miyao M, Kotani H, Ishida T et al (2015) Pivotal role of liver sinusoidal endothelial cells in NAFLD/NASH progression. Lab Invest 95(10):1130–1144. https://doi.org/10.1038/labinvest.2015.95
Hammoutene A, Rautou P-E (2019) Role of liver sinusoidal endothelial cells in non-alcoholic fatty liver disease. J Hepatol 70(6):1278–1291. https://doi.org/10.1016/j.jhep.2019.02.012
Cogger VC, Mohamad M, Solon-Biet SM et al (2016) Dietary macronutrients and the aging liver sinusoidal endothelial cell. Am J Physiol Heart Circ Physiol 310(9):H1064–H1070. https://doi.org/10.1152/ajpheart.00949.2015
Pourhoseini S, Seth RK, Das S et al (2015) Upregulation of miR21 and repression of Grhl3 by leptin mediates sinusoidal endothelial injury in experimental nonalcoholic steatohepatitis. PLoS ONE 10(2):e0116780. https://doi.org/10.1371/journal.pone.0116780
Wang BY, Ju XH, Fu BY, Zhang J, Cao YX (2005) Effects of ethanol on liver sinusoidal endothelial cells-fenestrae of rats. Hepatobiliary Pancreat Dis Int 4(3):422–426
Witte MH, Borgs P, Way DL, Ramirez G, Bernas MJ, Witte CL (1992) Alcohol, hepatic sinusoidal microcirculation, and chronic liver disease. Alcohol 9(6):473–480
Deaciuc D, Fortunato H, Sarphie McClain (2001) Alcohol-induced sinusoidal endothelial cell dysfunction in the mouse is associated with exacerbated liver apoptosis and can be reversed by caspase inhibition. Hepatol Res 19(1):85–97
Miller AM, Wang H, Park O et al (2010) Anti-inflammatory and anti-apoptotic roles of endothelial cell STAT3 in alcoholic liver injury. Alcohol Clin Exp Res 34(4):719–725. https://doi.org/10.1111/j.1530-0277.2009.01141.x
McCuskey RS, Bethea NW, Wong J et al (2005) Ethanol binging exacerbates sinusoidal endothelial and parenchymal injury elicited by acetaminophen. J Hepatol 42(3):371–377
Maeso-Díaz R, Ortega-Ribera M, Fernández-Iglesias A et al (2018) Effects of aging on liver microcirculatory function and sinusoidal phenotype. Aging Cell 17(6):e12829. https://doi.org/10.1111/acel.12829
Ito Y, Sørensen KK, Bethea NW et al (2007) Age-related changes in the hepatic microcirculation in mice. Exp Gerontol 42(8):789–797
Yeligar S, Tsukamoto H, Kalra VK (2009) Ethanol-induced expression of ET-1 and ET-BR in liver sinusoidal endothelial cells and human endothelial cells involves hypoxia-inducible factor-1alpha and microrNA-199. J Immunol 183(8):5232–5243. https://doi.org/10.4049/jimmunol.0901084
Wang Q, Zhang F, Lei Y, Liu P, Liu C, Tao Y (2020) microRNA-322/424 promotes liver fibrosis by regulating angiogenesis through targeting CUL2/HIF-1α pathway. Life Sci 266:118819. https://doi.org/10.1016/j.lfs.2020.118819
Zhu A, Chu L, Ma Q, Li Y (2018) WITHDRAWN: Long non-coding RNA H19 promotes angiogenesis in microvascular endothelial cells by down-regulating miR-181a. Int J Biol Macromol. https://doi.org/10.1016/j.ijbiomac.2018.08.091
Ye Y, Shen A, Liu A (2019) Long non-coding RNA H19 and cancer: a competing endogenous RNA. Bull Cancer 106(12):1152–1159. https://doi.org/10.1016/j.bulcan.2019.08.011
Zhu Y, Ni T, Lin J, Zhang C, Zheng L, Luo M (2019) Long non-coding RNA H19, a negative regulator of microRNA-148b-3p, participates in hypoxia stress in human hepatic sinusoidal endothelial cells via NOX4 and eNOS/NO signaling. Biochimie 163:128–136. https://doi.org/10.1016/j.biochi.2019.04.006
Guo C, Qi Y, Qu J, Gai L, Shi Y, Yuan C (2020) Pathophysiological functions of the lncRNA TUG1. Curr Pharm Des 26(6):688–700. https://doi.org/10.2174/1381612826666191227154009
Zhang R, Huang X-Q, Jiang Y-Y, Li N, Wang J, Chen S-Y (2020) LncRNA TUG1 regulates autophagy-mediated endothelial-mesenchymal transition of liver sinusoidal endothelial cells by sponging miR-142-3p. Am J Transl Res 12(3):758–772
Shao J, Xu Y, Fang M (2020) BRG1 deficiency in endothelial cells alleviates thioacetamide induced liver fibrosis in mice. Biochem Biophys Res Commun 521(1):212–219. https://doi.org/10.1016/j.bbrc.2019.10.109
Sun L-J, Yu J-W, Shi Y-G, Zhang X-Y, Shu M-N, Chen M-Y (2018) Hepatitis C virus core protein induces dysfunction of liver sinusoidal endothelial cell by down-regulation of silent information regulator 1. J Med Virol 90(5):926–935. https://doi.org/10.1002/jmv.25034
Verbeke L, Farre R, Trebicka J et al (2014) Obeticholic acid, a farnesoid X receptor agonist, improves portal hypertension by two distinct pathways in cirrhotic rats. Hepatology 59(6):2286–2298. https://doi.org/10.1002/hep.26939
Verbeke L, Mannaerts I, Schierwagen R et al (2016) FXR agonist obeticholic acid reduces hepatic inflammation and fibrosis in a rat model of toxic cirrhosis. Sci Rep 6:33453. https://doi.org/10.1038/srep33453
Schwabl P, Hambruch E, Seeland BA et al (2017) The FXR agonist PX20606 ameliorates portal hypertension by targeting vascular remodelling and sinusoidal dysfunction. J Hepatol 66(4):724–733. https://doi.org/10.1016/j.jhep.2016.12.005
Xing Y, Zhao T, Gao X, Wu Y (2016) Liver X receptor α is essential for the capillarization of liver sinusoidal endothelial cells in liver injury. Sci Rep 6:21309. https://doi.org/10.1038/srep21309
Baiocchini A, Del Nonno F, Taibi C et al (2019) Liver sinusoidal endothelial cells (LSECs) modifications in patients with chronic hepatitis C. Sci Rep 9(1):8760. https://doi.org/10.1038/s41598-019-45114-1
Tsai H-C, Li T-H, Huang C-C et al (2018) Beneficial effects of the peroxisome proliferator-activated receptor α/γ agonist aleglitazar on progressive hepatic and splanchnic abnormalities in cirrhotic rats with portal hypertension. Am J Pathol 188(7):1608–1624. https://doi.org/10.1016/j.ajpath.2018.03.018
Boyer-Diaz Z, Aristu-Zabalza P, Andrés-Rozas M et al (2020) Pan-PPAR agonist lanifibranor improves portal hypertension and hepatic fibrosis in experimental advanced chronic liver disease. J Hepatol. https://doi.org/10.1016/j.jhep.2020.11.045
Ding B-S, Liu CH, Sun Y et al (2016) HDL activation of endothelial sphingosine-1-phosphate receptor-1 (S1P) promotes regeneration and suppresses fibrosis in the liver. JCI insight 1(21):e87058. https://doi.org/10.1172/jci.insight.87058
Keitel V, Reinehr R, Gatsios P et al (2007) The G-protein coupled bile salt receptor TGR5 is expressed in liver sinusoidal endothelial cells. Hepatology 45(3):695–704
Klindt C, Reich M, Hellwig B et al (2019) The G protein-coupled bile acid receptor TGR5 (Gpbar1) modulates endothelin-1 signaling in liver. Cells. https://doi.org/10.3390/cells8111467
Schmid CD, Schledzewski K, Mogler C et al (2018) GPR182 is a novel marker for sinusoidal endothelial differentiation with distinct GPCR signaling activity in vitro. Biochem Biophys Res Commun 497(1):32–38. https://doi.org/10.1016/j.bbrc.2018.01.185
Liu D, Chen J, Wang J et al (2010) Increased expression of urotensin II and GPR14 in patients with cirrhosis and portal hypertension. Int J Mol Med 25(6):845–851
Wang R, Ding Q, Yaqoob U et al (2015) Exosome adherence and internalization by hepatic stellate cells triggers sphingosine 1-phosphate-dependent migration. J Biol Chem 290(52):30684–30696. https://doi.org/10.1074/jbc.M115.671735
Hong F, Tuyama A, Lee TF et al (2009) Hepatic stellate cells express functional CXCR4: role in stromal cell-derived factor-1alpha-mediated stellate cell activation. Hepatology 49(6):2055–2067. https://doi.org/10.1002/hep.22890
Liu Y, Yang X, Jing Y et al (2015) Contribution and mobilization of mesenchymal stem cells in a mouse model of carbon tetrachloride-induced liver fibrosis. Sci Rep 5:17762. https://doi.org/10.1038/srep17762
Liepelt A, Tacke F (2016) Stromal cell-derived factor-1 (SDF-1) as a target in liver diseases. Am J Physiol Gastrointest Liver Physiol 311(2):G203–G209. https://doi.org/10.1152/ajpgi.00193.2016
Ramirez-Pedraza M, Fernández M (2019) Interplay between macrophages and angiogenesis: a double-edged sword in liver disease. Front Immunol 10:2882. https://doi.org/10.3389/fimmu.2019.02882
Wu M-H, Chen Y-L, Lee K-H et al (2017) Glycosylation-dependent galectin-1/neuropilin-1 interactions promote liver fibrosis through activation of TGF-β- and PDGF-like signals in hepatic stellate cells. Sci Rep 7(1):11006. https://doi.org/10.1038/s41598-017-11212-1
Semela D, Das A, Langer D, Kang N, Leof E, Shah V (2008) Platelet-derived growth factor signaling through ephrin-b2 regulates hepatic vascular structure and function. Gastroenterology 135(2):671–679. https://doi.org/10.1053/j.gastro.2008.04.010
Venkatraman L, Tucker-Kellogg L (2013) The CD47-binding peptide of thrombospondin-1 induces defenestration of liver sinusoidal endothelial cells. Liver Int 33(9):1386–1397. https://doi.org/10.1111/liv.12231
Lao Y, Li Y, Zhang P et al (2018) Targeting endothelial Erk1/2-Akt axis as a regeneration strategy to bypass fibrosis during chronic liver injury in mice. Mol Ther 26(12):2779–2797. https://doi.org/10.1016/j.ymthe.2018.08.016
Rautou P-E, Bresson J, Sainte-Marie Y et al (2012) Abnormal plasma microparticles impair vasoconstrictor responses in patients with cirrhosis. Gastroenterology. https://doi.org/10.1053/j.gastro.2012.03.040
Tegge AN, Rodrigues RR, Larkin AL, Vu L, Murali TM, Rajagopalan P (2018) Transcriptomic analysis of hepatic cells in multicellular organotypic liver models. Sci Rep 8(1):11306. https://doi.org/10.1038/s41598-018-29455-x
Lafoz E, Ruart M, Anton A, Oncins A, Hernández-Gea V (2020) The endothelium as a driver of liver fibrosis and regeneration. Cells. https://doi.org/10.3390/cells9040929
Ford AJ, Jain G, Rajagopalan P (2015) Designing a fibrotic microenvironment to investigate changes in human liver sinusoidal endothelial cell function. Acta Biomater 24:220–227. https://doi.org/10.1016/j.actbio.2015.06.028
Abraldes JG, Rodríguez-Vilarrupla A, Graupera M et al (2007) Simvastatin treatment improves liver sinusoidal endothelial dysfunction in CCl4 cirrhotic rats. J Hepatol 46(6):1040–1046
Bravo M, Raurell I, Hide D et al (2019) Restoration of liver sinusoidal cell phenotypes by statins improves portal hypertension and histology in rats with NASH. Sci Rep 9(1):20183. https://doi.org/10.1038/s41598-019-56366-2
Hu L, Su L, Dong Z et al (2019) AMPK agonist AICAR ameliorates portal hypertension and liver cirrhosis via NO pathway in the BDL rat model. J Mol Med (Berl) 97(3):423–434. https://doi.org/10.1007/s00109-019-01746-4
Hunt NJ, Lockwood GP, Kang SWS et al (2020) The effects of metformin on age-related changes in the liver sinusoidal endothelial cell. J Gerontol A Biol Sci Med Sci 75(2):278–285. https://doi.org/10.1093/gerona/glz153
Kong L-J, Li H, Du Y-J et al (2017) Vatalanib, a tyrosine kinase inhibitor, decreases hepatic fibrosis and sinusoidal capillarization in CCl4-induced fibrotic mice. Mol Med Rep 15(5):2604–2610. https://doi.org/10.3892/mmr.2017.6325
Piguet A-C, Majumder S, Maheshwari U et al (2014) Everolimus is a potent inhibitor of activated hepatic stellate cell functions in vitro and in vivo, while demonstrating anti-angiogenic activities. Clin Sci (Lond) 126(11):775–784. https://doi.org/10.1042/CS20130081
Ling L, Li G, Meng D, Wang S, Zhang C (2018) Carvedilol ameliorates intrahepatic angiogenesis, sinusoidal remodeling and portal pressure in cirrhotic rats. Med Sci Monit 24:8290–8297. https://doi.org/10.12659/MSM.913118
Wu Y, Li Z, Xiu A-Y, Meng D-X, Wang S-N, Zhang C-Q (2019) Carvedilol attenuates carbon tetrachloride-induced liver fibrosis and hepatic sinusoidal capillarization in mice. Drug Des Dev Ther 13:2667–2676. https://doi.org/10.2147/DDDT.S210797
Di Martino J, Mascalchi P, Legros P et al (2019) Actin depolymerization in dedifferentiated liver sinusoidal endothelial cells promotes fenestrae re-formation. Hepatol Commun 3(2):213–219. https://doi.org/10.1002/hep4.1301
Bi Y, Mukhopadhyay D, Drinane M et al (2014) Endocytosis of collagen by hepatic stellate cells regulates extracellular matrix dynamics. Am J Physiol, Cell Physiol 307(7):C622–C633. https://doi.org/10.1152/ajpcell.00086.2014
Funding
This manuscript is supported by no funding.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
The authors have no conflicts of interest to disclose.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Below is the link to the electronic supplementary material.
11033_2021_6269_MOESM1_ESM.png
Supplementary Fig. 1 The LSECs-HSCs cross-talk and changes of markers in Normal or Fibrotic Livers. Several cytokines and pathways are involved in the cross-talk between LSECs and HSCs. ECM, extracellular matrix; ET-1, endothelin-1; NO, nitric oxide; KLF2, Kruppel-like factor 2; PDGF, Platelet derived growth factor; SDF-1, stromal cell derived factor 1; SK-1, sphingosine kinase-1; S1P, sphingosine 1-phosphate; TSP-1, thrombospondin-1; VEGF, vascular endothelial growth factor. (PNG 218 kb)
Rights and permissions
About this article

{kind=link}
Cite this article
Ma, H., Liu, X., Zhang, M. et al. Liver sinusoidal endothelial cells are implicated in multiple fibrotic mechanisms. Mol Biol Rep 48, 2803–2815 (2021). https://doi.org/10.1007/s11033-021-06269-1
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s11033-021-06269-1